
A Micro-Platinum Wire Biosensor for Fast and Selective Detection of Alanine Aminotransferase

Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Instrumentation
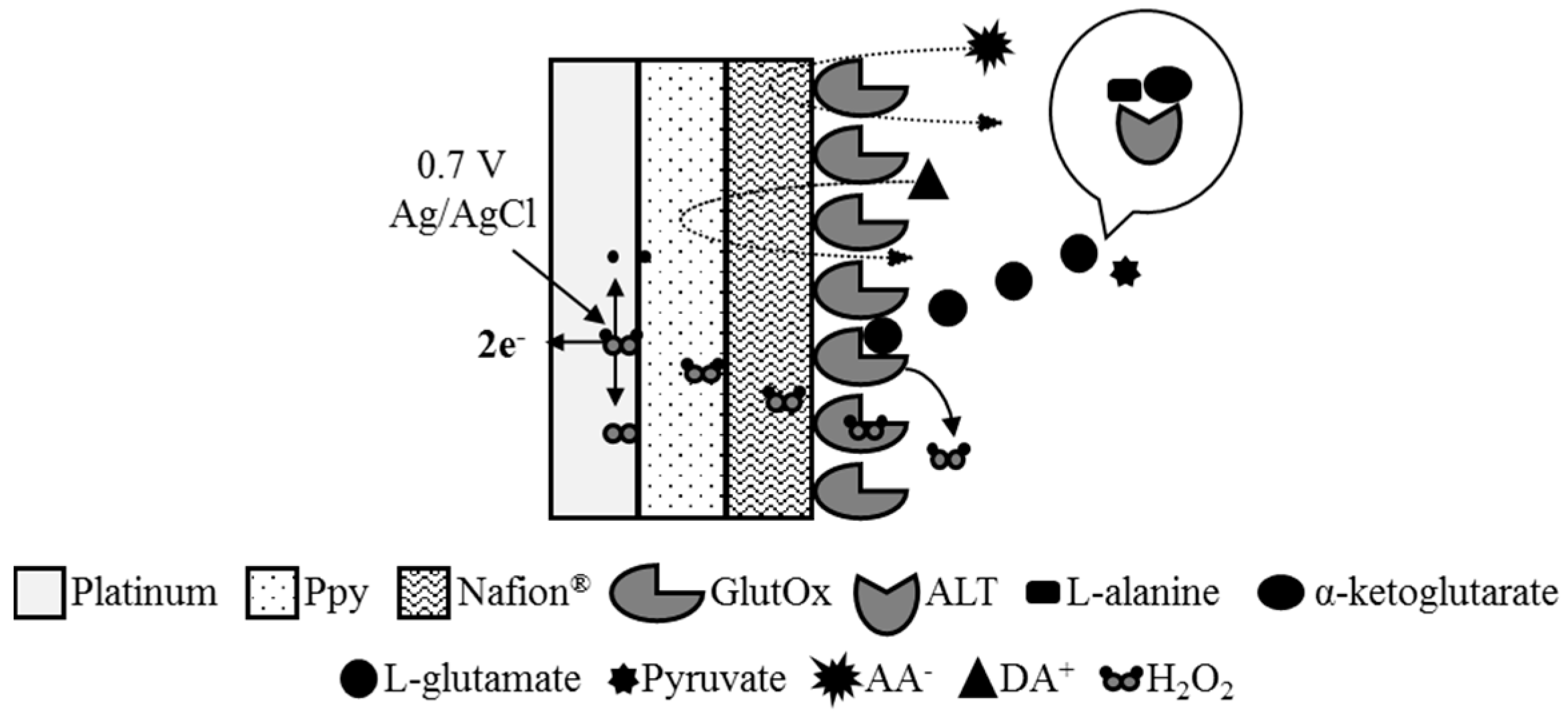
2.3. Preparation of ALT Biosensors
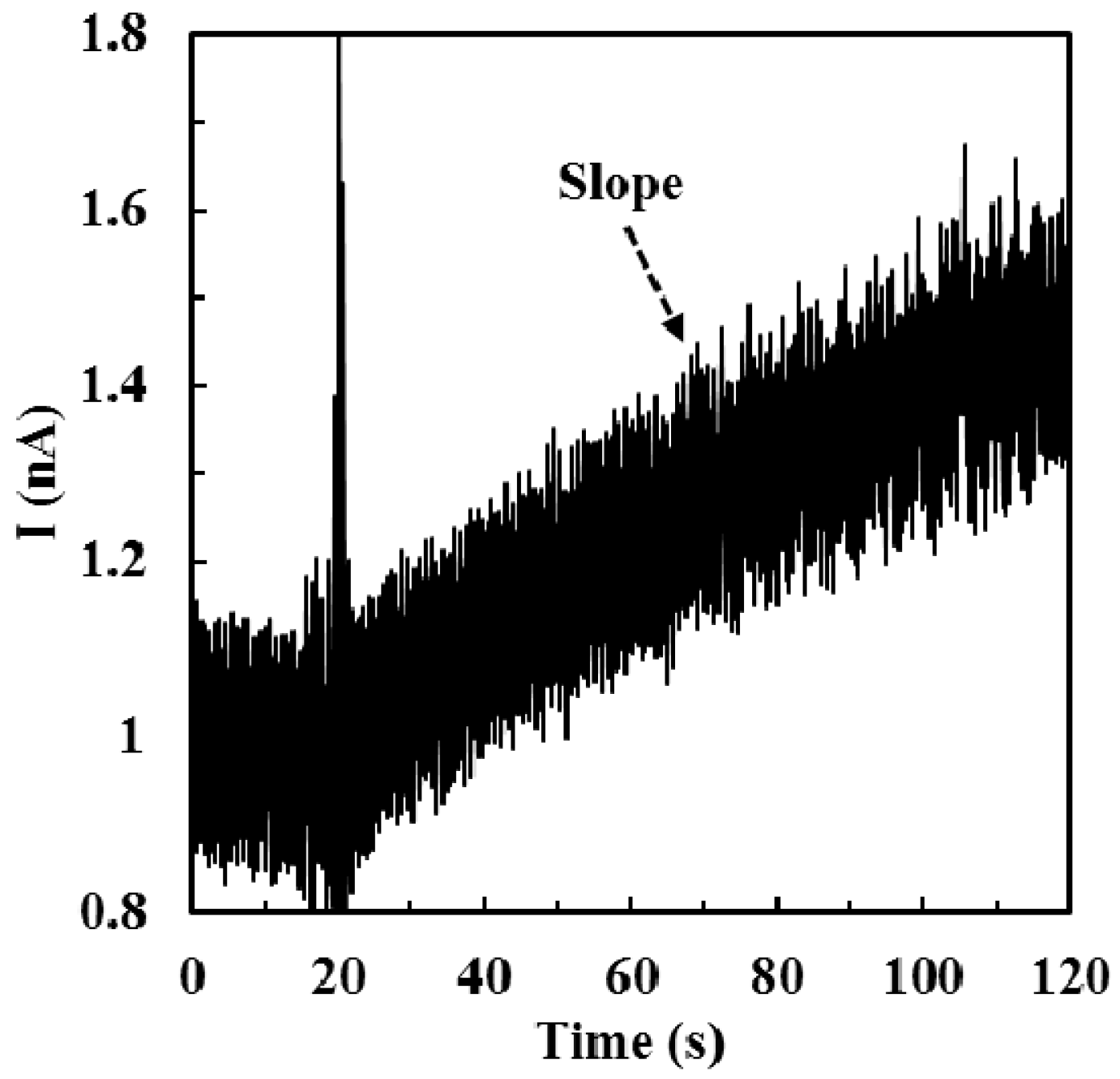
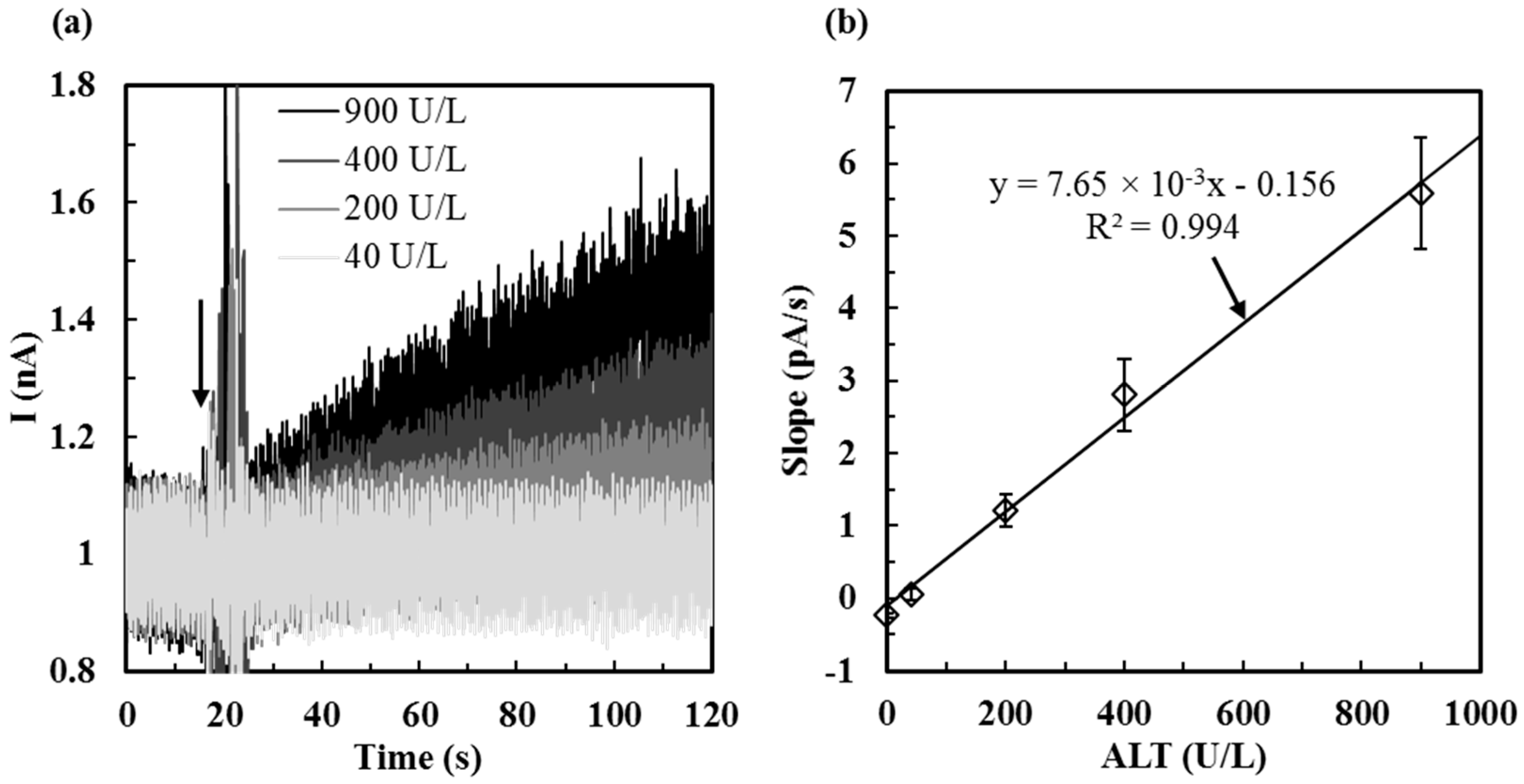
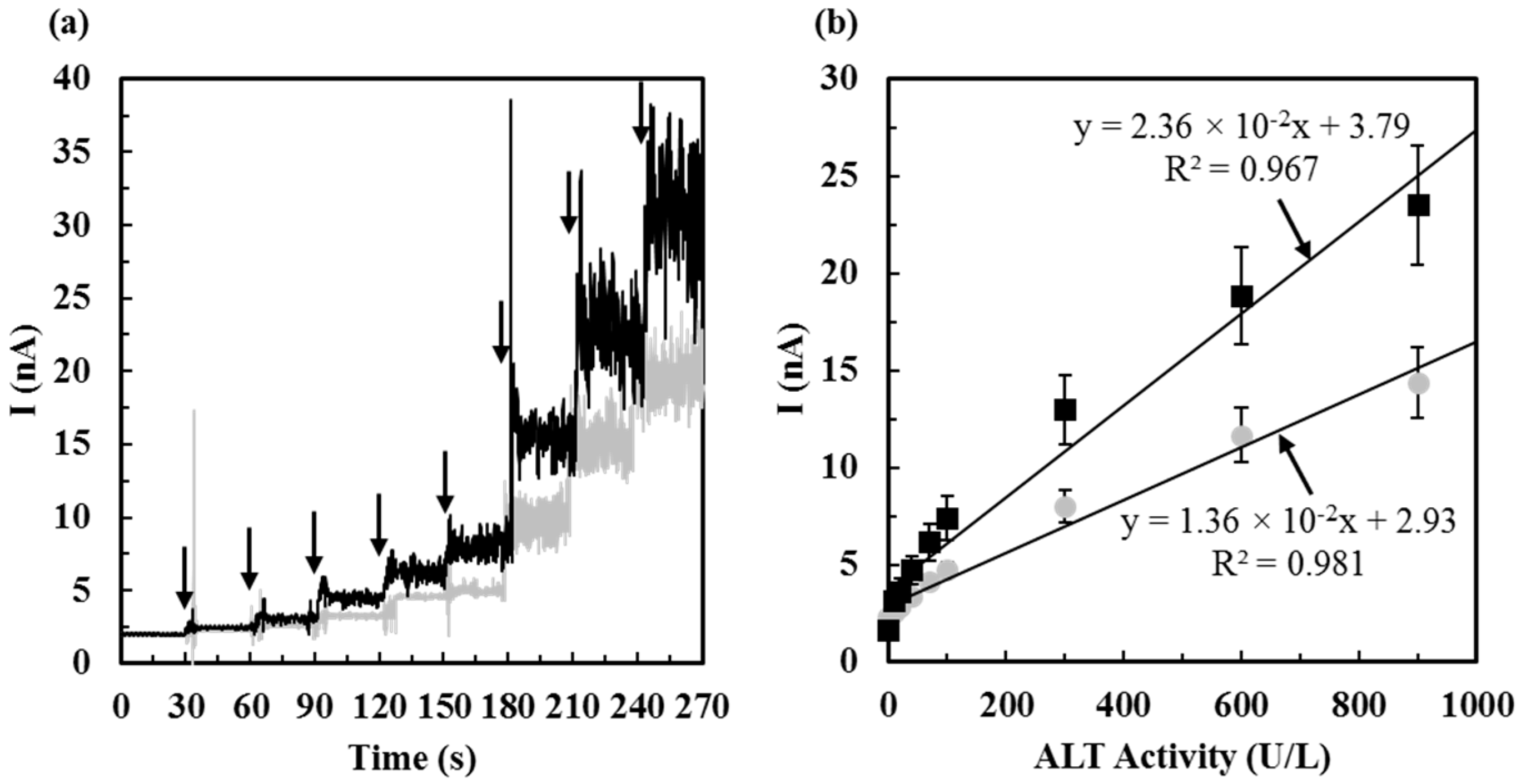
2.4. Calibration of ALT Biosensors by Method 1
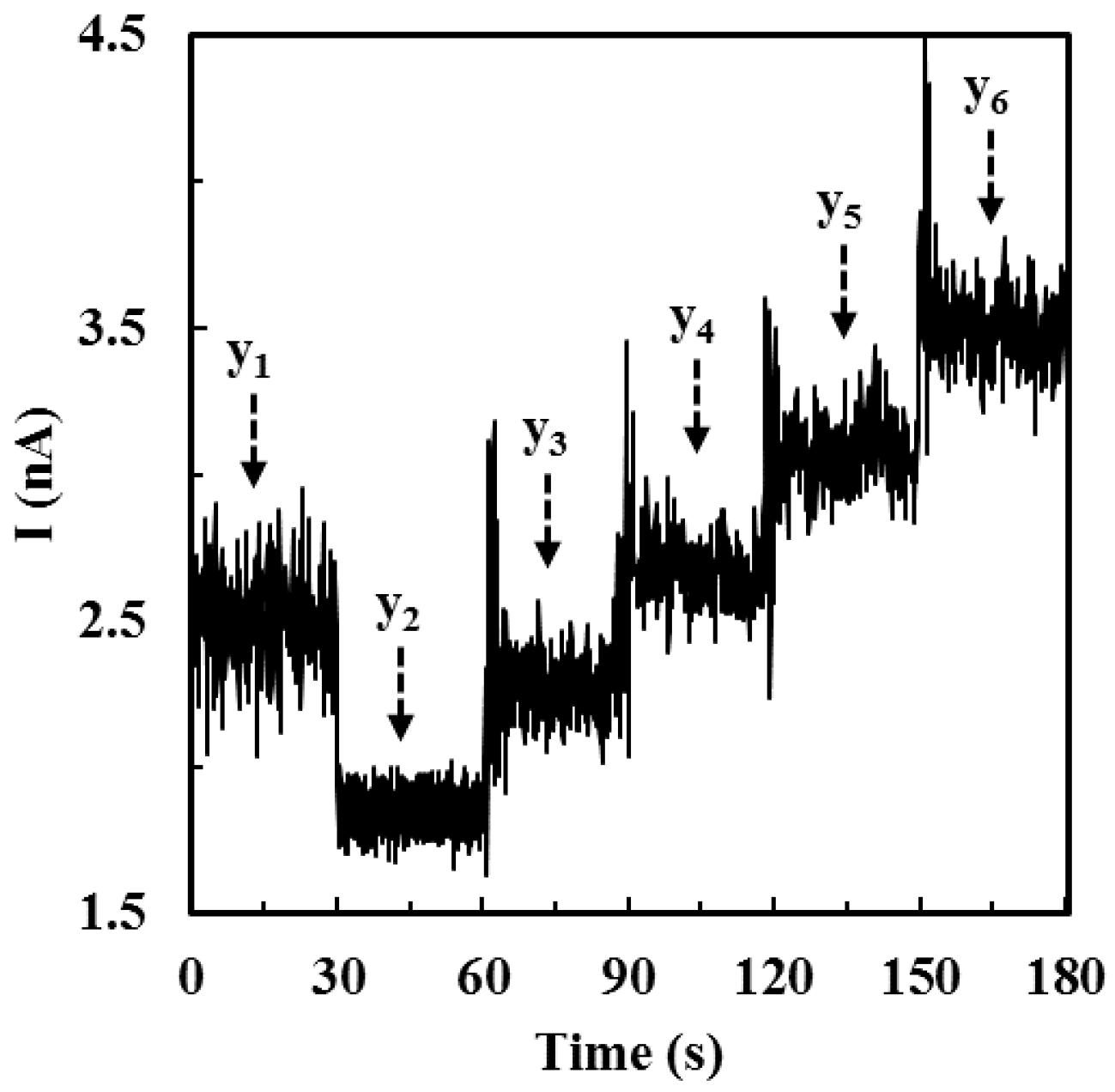
2.5. Calibration of ALT Biosensors by Method 2
2.6. Determination of ALT Activities from Spiked Samples
3. Results and Discussion
3.1. Calibration of ALT Biosensors
3.1.1. Sensitivity, Detection Limit, Sampling Time, and Detection Range of ALT Biosensors Tested by Method 1
3.1.2. Sensitivity, Detection Limit, Sampling Time, and Detection Range of ALT Biosensors Tested by Method 2
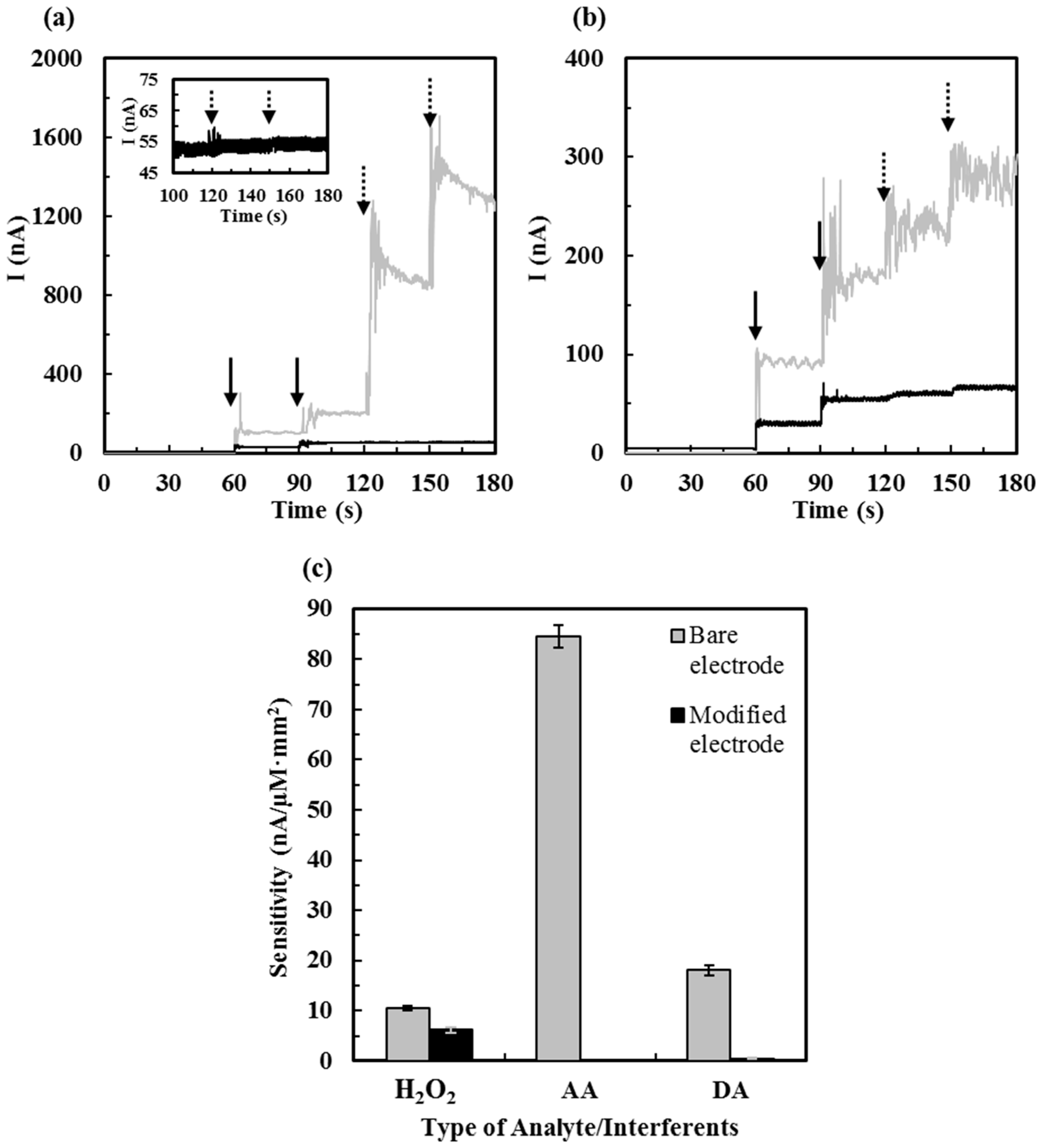
3.2. Effect of Interference
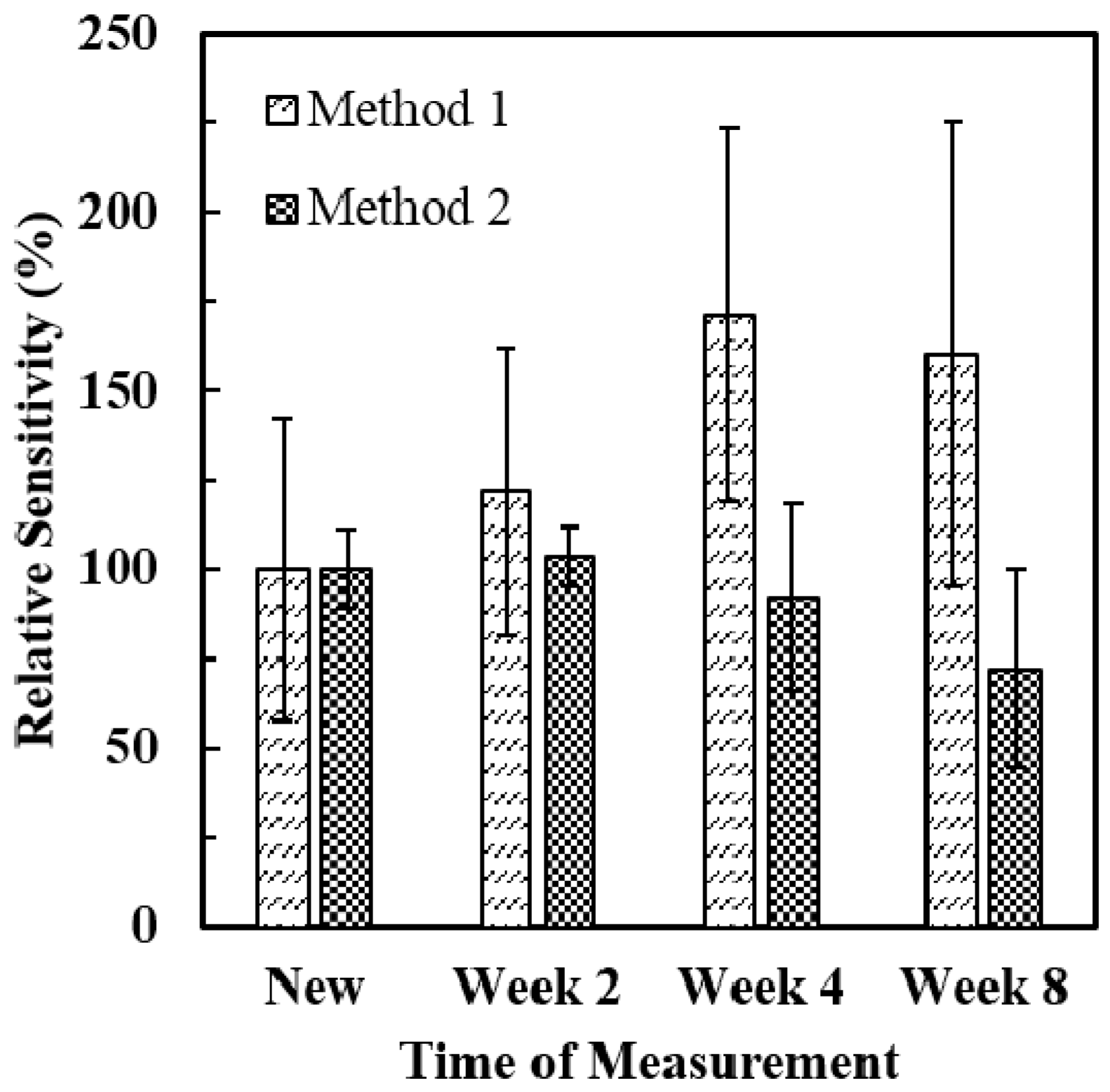
3.3. Stability of ALT Biosensors
3.4. Determination of ALT Activities in Spiked Samples
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Heidelbaugh, J.J.; Bruderly, M. Cirrhosis and chronic liver failure: Part I. Diagnosis and evaluation. Am. Fam. Phys. 2006, 74, 756–762. [Google Scholar]
- Huang, X.J.; Choi, Y.K.; Im, H.S.; Yarimaga, O.; Yoon, E.; Kim, H.S. Aspartate aminotransferase (AST/GOT) and alanine aminotransferase (ALT/GPT) detection techniques. Sensors 2006, 6, 756–782. [Google Scholar] [CrossRef]
- Jamal, M.; Worsfold, O.; McCormac, T.; Demsey, E. A stable and selective electrochemical biosensor for the liver enzyme alanine aminotransferase (ALT). Biosens. Bioelectron. 2009, 24, 2926–2930. [Google Scholar] [CrossRef] [PubMed]
- Matsuzawa, T.; Katunuma, N. Colorimetric assays for serum alanine transaminase and lactic dehydrogenase using diazonium zinc salt. Anal. Biochem. 1966, 17, 143–153. [Google Scholar] [CrossRef]
- Whitaker, J.F. A general colorimetric procedure for the estimation of enzymes which are linked to the NADH/NAD+ system. Clin. Chim. Acta 1969, 24, 23–37. [Google Scholar] [CrossRef]
- Lippi, U.; Guidi, G. A new colorimetric ultramicromethod for serum glutamicoxalacetic and glutamic-pyruvic transaminase determination. Clin. Chim. Acta 1970, 28, 431–437. [Google Scholar] [CrossRef]
- Giusti, G.; Ruggiero, G.; Cacciatore, L. A comparative study of some spectrophotometric and colorimetric procedures for the determination of serum glutamic-oxaloacetic and glutamic-pyruvic transaminase in hepatic diseases. Enzymol. Biol. Clin. 1969, 10, 17–38. [Google Scholar]
- Rodgerson, D.O.; Osberg, I.M. Sources of error in spectrophotometric measurement of aspartate aminotransferase and alanine aminotransferase activities in serum. Clin. Chem. 1974, 20, 43–50. [Google Scholar] [PubMed]
- Janasek, D.; Spohn, U. A chemiluminometric FIA procedure for the enzymatic determination of l-aspartate. Sens. Actuator B Chem. 2001, 74, 163–167. [Google Scholar] [CrossRef]
- Ohmori, S.; Tanaka, Y.; Ikeda, M.; Hirota, K. A sensitive determination of α-keto acids by gas-liquid chromatography and its application to the assay of l-glutamate dehydrogenase and aminotransferases. Anal. Biochem. 1981, 112, 204–212. [Google Scholar] [CrossRef]
- Rietz, B.; Guilbault, G.G. Fluorimetric assay of serum glutamate oxaloacetate transaminase, glutamate pyruvate transaminase and α-hydroxybutyrate dehydrogenase by solution and solid surface fluorescent methods. Anal. Chim. Acta 1975, 77, 191–198. [Google Scholar] [CrossRef]
- Khampha, W.; Yakovleva, J.; Isarangkul, D.; Wiyakrutta, S.; Meevootisom, V.; Emnéus, J. Specific detection of L-glutamate in food using flow-injection analysis and enzymatic recycling of substrate. Anal. Chim. Acta 2004, 518, 127–135. [Google Scholar] [CrossRef]
- Bonizzoni, M.; Fabbrizzi, L.; Piovani, G.; Taglietti, A. Fluorescent detection of glutamate with a dicopper(II) polyamine cage. Tetrahedron 2004, 60, 11159–11162. [Google Scholar] [CrossRef]
- Peguin, S.; Coulet, P.R.; Bardeletti, G. Pyruvate oxidase and oxaloacetate decarboxylase enzyme electrodes: Simultaneous determination of transaminases with a two-electrode-based analyzer. Anal. Chim. Acta 1989, 222, 83–93. [Google Scholar] [CrossRef]
- Kihara, K.; Yasukawa, E.; Hirose, S. Sequential determination of glutamate oxalacetate transaminase and glutamate pyruvate transaminase activities in serum using an immobilized bienzyme-poly(viny1 chloride) membrane electrode. Anal. Chem. 1984, 56, 1876–1880. [Google Scholar] [CrossRef] [PubMed]
- Kihara, K.; Yasukawa, E.; Hayashi, M.; Hirose, S. Determination of glutamate-pyruvate transaminase activity in blood serum with a pyruvate oxidase/poly(vinyl chloride) membrane sensor. Anal. Chim. Acta 1984, 159, 81–86. [Google Scholar] [CrossRef]
- Cooper, J.M.; McNeil, C.J.; Spoors, J.A. Amperometric enzyme electrode for the determination of aspartate aminotransferase and alanine aminotransferase in serum. Anal. Chim. Acta 1991, 245, 57–62. [Google Scholar] [CrossRef]
- Compagnone, D.; Federici, G.; Massoud, R.; Santoro, L.; Anichini, M.; Palleschi, G. Analysis for transaminases in serum with an amperometric glutamate electrode. Clin. Chem. 1992, 38, 2306–2310. [Google Scholar] [PubMed]
- Catz, S.D.; Carreras, M.C.; Poderoso, J.J. Nitric oxide synthase inhibitors decrease human polymorphonuclear leukocyte luminol-dependent chemiluminescence. Free Radic. Biol. Med. 1995, 19, 741–748. [Google Scholar] [CrossRef]
- Li, X.J.; Nie, Z.H.; Cheng, C.M.; Goodale, A.B.; Whitesides, G.M. Paper-based Electrochemical ELISA. In Proceedings of the 14th International Conference on Miniaturized Systems for Chemistry and Life Sciences (MicroTAS 2010), Groningen, The Netherlands, 3–7 October 2010; pp. 1487–1489.
- He, Y.N.; Chen, H.Y. The kinetics-based electrochemical determination of serum glutamate pyruvate transaminase activity with a gold microelectrode. Anal. Chim. Acta 1997, 353, 319–323. [Google Scholar] [CrossRef]
- Xuan, G.S.; Oh, S.W.; Choi, E.Y. Development of an electrochemical immunosensor for alanine aminotransferase. Biosens. Bioelectron. 2003, 19, 365–371. [Google Scholar] [CrossRef]
- Paraiso, L.F.; Paula, L.F.; Franco, D.L.; Madurro, J.M.; Brito-Madurro, A.G. Bioelectrochemical detection of alanine aminotransferase for molecular diagnostic of the liver disease. Int. J. Electrochem. Sci. 2014, 9, 1286–1297. [Google Scholar]
- Mizutani, F.; Tsuda, K. Determination of glutamate pyruvate transaminase and pyruvate with an amperometric pyruvate oxidase sensor. Anal. Chim. Acta 1980, 118, 65–71. [Google Scholar] [CrossRef]
- Upadhyay, S.; Ohgami, N.; Kusakabe, H.; Mizuno, H.; Arima, J.; Tamura, T.; Inagaki, K.; Suzuki, H. Performance characterization of recombinant l-glutamate oxidase in a micro GOT/GPT sensing system. Sens. Actuator B Chem. 2006, 119, 570–576. [Google Scholar] [CrossRef]
- Song, M.J.; Yun, D.H.; Jin, J.H.; Min, N.K.; Hong, S.I. Electrochemical biosensor array for liver diagnosis using silanization technique on nanoporous silicon electrode. J. Biosci. Bioeng. 2007, 103, 32–37. [Google Scholar] [CrossRef] [PubMed]
- Ohgami, N.; Upadhyay, S.; Kabata, A.; Morimoto, K.; Kusakabe, H.; Suzuki, H. Determination of the activities of glutamic oxaloacetic transaminase and glutamic pyruvic transaminase in a microfluidic system. Biosens. Bioelectron. 2007, 22, 1330–1336. [Google Scholar] [CrossRef] [PubMed]
- Chang, K.S.; Chang, C.K.; Chou, S.F.; Chen, C.Y. Sequential measurement of aminotransferase activities by amperometric biosensors. Biosens. Bioelectron. 2007, 22, 2914–2920. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, R.D.; Chang, S.C.; Lowry, J.P.; McNeil, C.J. Comparisons of platinum, gold, palladium and glassy carbon as electrode materials in the design of biosensors for glutamate. Biosens. Bioelectron. 2004, 19, 1521–1528. [Google Scholar] [CrossRef] [PubMed]
- Hamdi, N.; Wang, J.J.; Monbouquette, H.G. Polymer films as permselective coatings for H2O2-sensing electrodes. J. Electroanal. Chem. 2005, 581, 258–264. [Google Scholar] [CrossRef]
- Wassum, K.M.; Tolosa, V.M.; Tseng, T.C.; Balleine, B.W.; Monbouquette, H.G.; Maidment, N.T. Transient extracellular glutamate events in the basolateral amygdala track reward seeking actions. J. Neurosci. 2012, 32, 2734–2746. [Google Scholar] [CrossRef] [PubMed]
- Tseng, T.T.-C.; Monbouquette, H.G. Implantable microprobe with arrayed microsensors for combined amperometric monitoring of the neurotransmitters, glutamate and dopamine. J. Electroanal. Chem. 2012, 682, 141–146. [Google Scholar] [CrossRef] [PubMed]
- Dong, S.; Wang, B.; Liu, B. Amperometric glucose sensor with ferrocene as an electron transfer mediator. Biosens. Bioelectron. 1992, 7, 215–222. [Google Scholar] [CrossRef]
- Tripathi, V.S.; Kandimalla, V.B.; Ju, H. Amperometric biosensor for hydrogen peroxide based on ferrocene-bovine serum albumin and multiwall carbon nanotube modified ormosil composite. Biosens. Bioelectron. 2006, 21, 1529–1535. [Google Scholar] [CrossRef] [PubMed]
- Reiber, H.; Ruff, M.; Uhr, M. Ascorbate concentration in human cerebrospinal fluid (CSF) and serum. Intrathecal accumulation and CSF flow rate. Clin. Chim. Acta 1993, 217, 163–173. [Google Scholar] [CrossRef]
- Xiao, Y.H.; Guo, C.X.; Li, C.M.; Li, Y.B.; Zhang, J.; Xue, R.H.; Zhang, S. Highly sensitive and selective method to detect dopamine in the presence of ascorbic acid by a new polymeric composite film. Anal. Biochem. 2007, 371, 229–237. [Google Scholar] [CrossRef] [PubMed]
- Iniouchine, M.Y.; Sibarov, D.A.; Volnova, A.B.; Jimenez-Rivera, C.A.; Nozdrachev, A.D. Blockers of monoamine transporters influence high dopamine concentration uptake in rat brain slices. Dokl. Biol. Sci. 2008, 419, 80–82. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.B.; Fei, J.J.; Hu, S.S. Simultaneous determination of dopamine and serotonin on a glassy carbon electrode coated with a film of carbon nanotubes. Anal. Biochem. 2003, 318, 100–106. [Google Scholar] [CrossRef]
- Jiang, X.H.; Lin, X.Q. Overoxidized polypyrrole film directed DNA immobilization for construction of electrochemical micro-biosensors and simultaneous determination of serotonin and dopamine. Anal. Chim. Acta 2005, 537, 145–151. [Google Scholar] [CrossRef]
- Goyal, R.N.; Gupta, V.K.; Oyama, M.; Bachheti, N. Gold nanoparticles modified indium tin oxide electrode for the simultaneous determination of dopamine and serotonin: Application in pharmaceutical formulations and biological fluids. Talanta 2007, 72, 976–983. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Electrode/size (mm2) | EC Method | Modification & Enzyme Layers | Selectivity | Response Time (s) | Sensitivity | LOD/range | Ref. |
|---|---|---|---|---|---|---|---|---|
| 1 | O2 electrode/490.9 | CPA | Tf, porous AC & PyOx | NA | 120 | NA | 0.5/0.5–180 U/L | [24] |
| 2 | Pt/50.3 | CPA | PVC, CA & PyOx | NA | 60 | 1.46 × 10−5 nA/(s·U/L·mm2) | 5/5–1600 U/L | [16] |
| 3 | BIA membrane/28.3 | CPA | PyOx only | NA | 240 | NA | 3/6–30000 U/L | [14] |
| 4 | PAC/1.77 | CPA | PC & GlutOx | NA | 300 | 3.77 nA/(s·U/L·mm2) | 5/5–500 U/L | [17] |
| 5 | Pt/0.05 | CPA | CA, immobilon IA membrane, PC & GlutOx | AA, UA | 300 | NA | 2/5–1200 U/L | [18] |
| 6 | Au/7.85 × 10−3 | DPV | LDH only | NA | NA | 0.122 nA/(s·U/L·mm2) | 0.3/0.3–200 U/L | [21] |
| 7 | Au/19.63 | ChA | SAM mediator-coated layer, anti-ALT Ab & PyOx | NA | NA | 26.3 nA/(ng/mL) | 0.01/0.01–1000 ng/mL | [22] |
| 8 | Pt/0.15 | CPA | GlutOx only | NA | NA | NA | 11/11–88 U/L | [25] |
| 9 | Pt/50.5 | CPA | Nanoporous Si & GlutOx | NA | 20 | 2.69 nA/(U/L·mm2) | 1.3/1.3–250 U/L | [26] |
| 10 | Pt/0.15 | CPA | GlutOx only | AA, UA | 600 | NA | 9/ 9–250 U/L | [27] |
| 11 | Pd/4.8 | CPA | Nf & GlutOx | AA, UA | 120 | 2.07 × 10−3 nA/(s·U/L·mm2) | 8/ 8–250 U/L | [28] |
| 12 | Pt/5.5 | CPA | Nf, mediated medium & GlutOx | NA | 180 | 0.153 nA/(U/L·mm2) | 3.29/ 10–1000 U/L | [3] |
| 13 | Graphite/28.3 | CV | P4AP & PyOx | AA, UA, Glut, Glu | 200 | NA | 2.68×10−5/ 3 × 10−5–3 U/L | [23] |
| 14 | Pt/0.401 | CPA | Ppy, Nf & GlutOx | AA, DA | 60 | 1.908 × 10−5 nA/(s·U/L·mm2) | 15.9/40–900 U/L | This work a |
| 15 | Pt/0.401 | CPA | Ppy, Nf & GlutOx | AA, DA | 5 | 0.059 nA/(U/L·mm2) | 8.48/10–900 U/L | This work b |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thuy, T.N.T.; Tseng, T.T.-C. A Micro-Platinum Wire Biosensor for Fast and Selective Detection of Alanine Aminotransferase. Sensors 2016, 16, 767. https://doi.org/10.3390/s16060767
Thuy TNT, Tseng TT-C. A Micro-Platinum Wire Biosensor for Fast and Selective Detection of Alanine Aminotransferase. Sensors. 2016; 16(6):767. https://doi.org/10.3390/s16060767
Chicago/Turabian StyleThuy, Tran Nguyen Thanh, and Tina T.-C. Tseng. 2016. "A Micro-Platinum Wire Biosensor for Fast and Selective Detection of Alanine Aminotransferase" Sensors 16, no. 6: 767. https://doi.org/10.3390/s16060767
APA StyleThuy, T. N. T., & Tseng, T. T. -C. (2016). A Micro-Platinum Wire Biosensor for Fast and Selective Detection of Alanine Aminotransferase. Sensors, 16(6), 767. https://doi.org/10.3390/s16060767