
A FRET Approach to Detect Paraoxon among Organophosphate Pesticides Using a Fluorescent Biosensor

 ,
,  ,
,  ,
,  and
and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Over-Expression and Purification of EST2-S35C
2.3. EST2-S35C Labelling and Inhibition Efficiency
2.4. Förster Resonance Energy Transfer Measurements
2.5. Interference in FRET by pH and Chemicals
2.6. Detection of Paraoxon by FRET
2.7. Data Analysis
2.8. In Silico Preparation of Mutated and Labelled EST2-S35C 3D Structure
2.9. Docking Analysis
3. Results and Discussion
3.1. EST2-S35C Purification and Labelling
3.2. Fluorescence Resonance Energy Transfer (FRET) Measurements
3.3. pH Influence on FRET Measurements
3.4. Effects of Organic Compounds on FRET Measurements
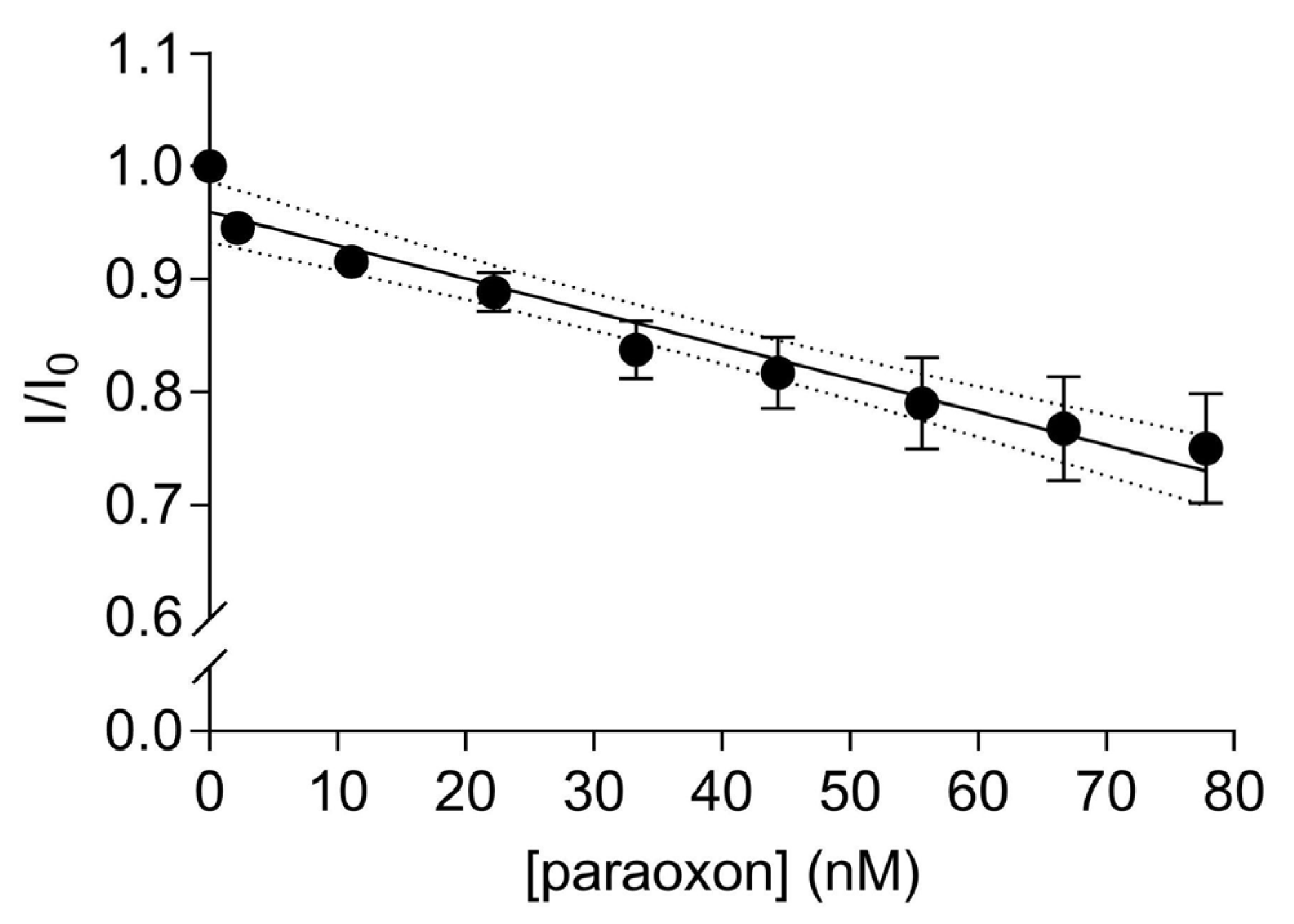
3.5. FRET Quenching by Paraoxon Addition
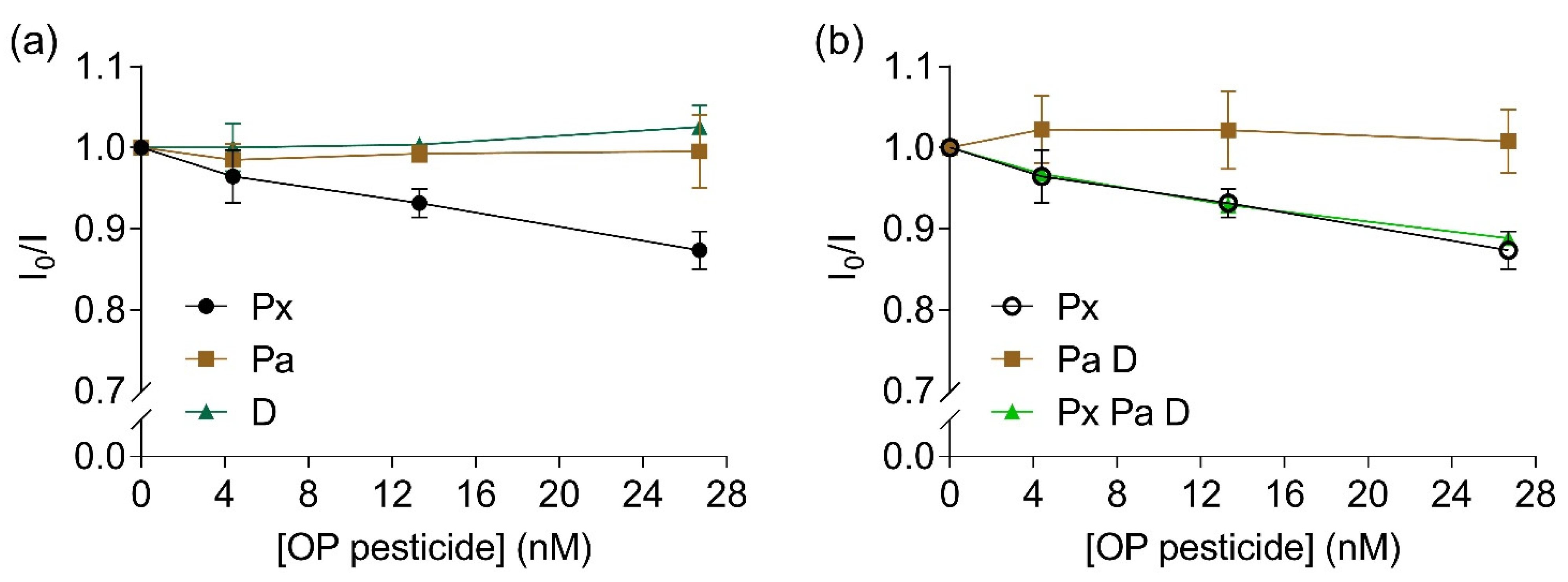
3.6. EST2-S35C Specificity towards Paraoxon Detection by FRET Measurements
3.7. In Silico Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jeyaratnam, J.; Maroni, M. Organophosphorous Compounds. Toxicology 1994, 91, 15–27. [Google Scholar] [CrossRef]
- Babu, V.; Unnikrishnan, P.; Anu, G.; Nair, S.M. Distribution of Organophosphorus Pesticides in the Bed Sediments of a Backwater System Located in an Agricultural Watershed: Influence of Seasonal Intrusion of Seawater. Arch. Environ. Contam. Toxicol. 2011, 60, 597–609. [Google Scholar] [CrossRef]
- Kanzari, F.; Syakti, A.D.; Asia, L.; Malleret, L.; Piram, A.; Mille, G.; Doumenq, P. Distributions and Sources of Persistent Organic Pollutants (Aliphatic Hydrocarbons, PAHs, PCBs and Pesticides) in Surface Sediments of an Industrialized Urban River (Huveaune), France. Sci. Total Environ. 2014, 478, 141–151. [Google Scholar] [CrossRef]
- Fadaei, A.; Dehghani, M.H.; Nasseri, S.; Mahvi, A.H.; Rastkari, N.; Shayeghi, M. Organophosphorous Pesticides in Surface Water of Iran. Bull. Environ. Contam. Toxicol. 2012, 88, 867–869. [Google Scholar] [CrossRef]
- Gao, J.; Liu, L.; Liu, X.; Zhou, H.; Lu, J.; Huang, S.; Wang, Z. The Occurrence and Spatial Distribution of Organophosphorous Pesticides in Chinese Surface Water. Bull. Environ. Contam. Toxicol. 2009, 82, 223–229. [Google Scholar] [CrossRef]
- Costa, L.G.; Giordano, G.; Guizzetti, M.; Vitalone, A. Neurotoxicity of Pesticides: A Brief Review. Front. Biosci. 2008, 13, 1240–1249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naughton, S.X.; Terry, A.V. Neurotoxicity in Acute and Repeated Organophosphate Exposure. Toxicology 2018, 408, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Aman, S.; Paul, S.; Chowdhury, F.R. Management of Organophosphorus Poisoning: Standard Treatment and Beyond. Crit. Care Clin. 2021, 37, 673–686. [Google Scholar] [CrossRef]
- Pascale, A.; Laborde, A. Impact of Pesticide Exposure in Childhood. Rev. Environ. Health 2020, 35, 221–227. [Google Scholar] [CrossRef]
- Dórea, J.G. Exposure to Environmental Neurotoxic Substances and Neurodevelopment in Children from Latin America and the Caribbean. Environ. Res. 2021, 192, 110199. [Google Scholar] [CrossRef]
- Pszczolińska, K.; Michel, M. The QuEChERS Approach for the Determination of Pesticide Residues in Soil Samples: An Overview. J. AOAC Int. 2016, 99, 1403–1414. [Google Scholar] [CrossRef]
- Pasupuleti, R.R.; Tsai, P.-C.; Lin, P.-I.D.; Wu, M.-T.; Ponnusamy, V.K. Rapid and Sensitive Analytical Procedure for Biomonitoring of Organophosphate Pesticide Metabolites in Human Urine Samples Using a Vortex-Assisted Salt-Induced Liquid-Liquid Microextraction Technique Coupled with Ultra-High-Performance Liquid Chromatography/Tandem Mass Spectrometry. Rapid Commun. Mass Spectrom. 2020, 34 (Suppl. 1), e8565. [Google Scholar] [CrossRef]
- Nasiri, M.; Ahmadzadeh, H.; Amiri, A. Organophosphorus Pesticides Extraction with Polyvinyl Alcohol Coated Magnetic Graphene Oxide Particles and Analysis by Gas Chromatography-Mass Spectrometry: Application to Apple Juice and Environmental Water. Talanta 2021, 227, 122078. [Google Scholar] [CrossRef]
- Febbraio, F. Biochemical Strategies for the Detection and Detoxification of Toxic Chemicals in the Environment. World J. Biol. Chem. 2017, 8, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Wang, M.; Yu, H.; She, Y.; Cao, Z.; Ye, J.; Abd El-Aty, A.M.; Hacımüftüoğlu, A.; Wang, J.; Lao, S. An Overview on the Mechanisms and Applications of Enzyme Inhibition-Based Methods for Determination of Organophosphate and Carbamate Pesticides. J. Agric. Food Chem. 2020, 68, 7298–7315. [Google Scholar] [CrossRef]
- Levine, M. Fluorescence-Based Sensing of Pesticides Using Supramolecular Chemistry. Front. Chem. 2021, 9, 27. [Google Scholar] [CrossRef] [PubMed]
- Febbraio, F.; D’Andrea, S.E.; Mandrich, L.; Merone, L.; Rossi, M.; Nucci, R.; Manco, G. Irreversible Inhibition of the Thermophilic Esterase EST2 from Alicyclobacillus acidocaldarius. Extremophiles 2008, 12, 719–728. [Google Scholar] [CrossRef] [PubMed]
- Febbraio, F.; Merone, L.; Cetrangolo, G.P.; Rossi, M.; Nucci, R.; Manco, G. Thermostable Esterase 2 from Alicyclobacillus acidocaldarius as Biosensor for the Detection of Organophosphate Pesticides. Anal. Chem. 2011, 83, 1530–1536. [Google Scholar] [CrossRef]
- Cetrangolo, G.P.; Gori, C.; Rusko, J.; Terreri, S.; Manco, G.; Cimmino, A.; Febbraio, F. Determination of Picomolar Concentrations of Paraoxon in Human Urine by Fluorescence-Based Enzymatic Assay. Sensors 2019, 19, 4852. [Google Scholar] [CrossRef] [Green Version]
- Cetrangolo, G.P.; Rusko, J.; Gori, C.; Carullo, P.; Manco, G.; Chino, M.; Febbraio, F. Highly Sensitive Detection of Chemically Modified Thio-Organophosphates by an Enzymatic Biosensing Device: An Automated Robotic Approach. Sensors 2020, 20, 1365. [Google Scholar] [CrossRef] [Green Version]
- Carullo, P.; Cetrangolo, G.P.; Mandrich, L.; Manco, G.; Febbraio, F. Fluorescence Spectroscopy Approaches for the Development of a Real-Time Organophosphate Detection System Using an Enzymatic Sensor. Sensors 2015, 15, 3932–3951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hawe, A.; Sutter, M.; Jiskoot, W. Extrinsic Fluorescent Dyes as Tools for Protein Characterization. Pharm. Res. 2008, 25, 1487–1499. [Google Scholar] [CrossRef] [Green Version]
- Carullo, P.; Chino, M.; Cetrangolo, G.P.; Terreri, S.; Lombardi, A.; Manco, G.; Cimmino, A.; Febbraio, F. Direct Detection of Organophosphate Compounds in Water by a Fluorescence-Based Biosensing Device. Sens. Actuators B Chem. 2018, 255, 3257–3266. [Google Scholar] [CrossRef]
- Bhatt, P.; Zhou, X.; Huang, Y.; Zhang, W.; Chen, S. Characterization of the Role of Esterases in the Biodegradation of Organophosphate, Carbamate, and Pyrethroid Pesticides. J. Hazard. Mater. 2021, 411, 125026. [Google Scholar] [CrossRef]
- Manco, G.; Adinolfi, E.; Pisani, F.M.; Ottolina, G.; Carrea, G.; Rossi, M. Overexpression and Properties of a New Thermophilic and Thermostable Esterase from Bacillus acidocaldarius with Sequence Similarity to Hormone-Sensitive Lipase Subfamily. Biochem. J. 1998, 332, 203–212. [Google Scholar] [CrossRef] [Green Version]
- Bradford, M.M. A Rapid and Sensitive Method for the Quantitation of Microgram Quantities of Protein Utilizing the Principle of Protein-Dye Binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Medintz, I.L.; Hildebrandt, N. FRET—Förster Resonance Energy Transfer: From Theory to Applications; John Wiley & Sons: Hoboken, NJ, USA, 2013; ISBN 978-3-527-65604-2. [Google Scholar]
- De Simone, G.; Galdiero, S.; Manco, G.; Lang, D.; Rossi, M.; Pedone, C. A Snapshot of a Transition State Analogue of a Novel Thermophilic Esterase Belonging to the Subfamily of Mammalian Hormone-Sensitive Lipase. J. Mol. Biol. 2000, 303, 761–771. [Google Scholar] [CrossRef]
- Schrödinger, LLC. The PyMOL Molecular Graphics System, Version 1.8; Schrödinger, LLC: New York, NY, USA, 2015. [Google Scholar]
- Brooks, B.R.; Brooks, C.L.; Mackerell, A.D.; Nilsson, L.; Petrella, R.J.; Roux, B.; Won, Y.; Archontis, G.; Bartels, C.; Boresch, S.; et al. CHARMM: The Biomolecular Simulation Program. J. Comput. Chem. 2009, 30, 1545–1614. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated Docking with Selective Receptor Flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodrigues, A.C.M.; Barbieri, M.V.; Chino, M.; Manco, G.; Febbraio, F. A 3D Printable Adapter for Solid-State Fluorescence Measurements: The Case of an Immobilized Enzymatic Bioreceptor for Organophosphate Pesticides Detection. Anal. Bioanal. Chem. 2022. [Google Scholar] [CrossRef]
- Wu, X.; Song, Y.; Yan, X.; Zhu, C.; Ma, Y.; Du, D.; Lin, Y. Carbon Quantum Dots as Fluorescence Resonance Energy Transfer Sensors for Organophosphate Pesticides Determination. Biosens. Bioelectron. 2017, 94, 292–297. [Google Scholar] [CrossRef]
- Zhang, H.; Li, Z.; Snyder, A.; Xie, J.; Stanciu, L.A. Functionalized Graphene Oxide for the Fabrication of Paraoxon Biosensors. Anal. Chim. Acta 2014, 827, 86–94. [Google Scholar] [CrossRef] [PubMed]
- Lang, Q.; Han, L.; Hou, C.; Wang, F.; Liu, A. A Sensitive Acetylcholinesterase Biosensor Based on Gold Nanorods Modified Electrode for Detection of Organophosphate Pesticide. Talanta 2016, 156–157, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Arduini, F.; Forchielli, M.; Scognamiglio, V.; Nikolaevna, K.A.; Moscone, D. Organophosphorous Pesticide Detection in Olive Oil by Using a Miniaturized, Easy-to-Use, and Cost-Effective Biosensor Combined with QuEChERS for Sample Clean-Up. Sensors 2016, 17, 34. [Google Scholar] [CrossRef] [Green Version]
- Zhai, R.; Chen, G.; Liu, G.; Huang, X.; Xu, X.; Li, L.; Zhang, Y.; Wang, J.; Jin, M.; Xu, D.; et al. Enzyme Inhibition Methods Based on Au Nanomaterials for Rapid Detection of Organophosphorus Pesticides in Agricultural and Environmental Samples: A Review. J. Adv. Res. 2021. [Google Scholar] [CrossRef]
- Yan, X.; Song, Y.; Zhu, C.; Li, H.; Du, D.; Su, X.; Lin, Y. MnO2 Nanosheet-Carbon Dots Sensing Platform for Sensitive Detection of Organophosphorus Pesticides. Anal. Chem. 2018, 90, 2618–2624. [Google Scholar] [CrossRef]
- Xue, G.; Yue, Z.; Bing, Z.; Yiwei, T.; Xiuying, L.; Jianrong, L. Sensitive Fluorescence Assay of Organophosphorus Pesticides Based on the Fluorescence Resonance Energy Transfer between CdTe Quantum Dots and Porphyrin. Analyst 2016, 141, 4941–4946. [Google Scholar] [CrossRef]
- Arvand, M.; Mirroshandel, A.A. An Efficient Fluorescence Resonance Energy Transfer System from Quantum Dots to Graphene Oxide Nano Sheets: Application in a Photoluminescence Aptasensing Probe for the Sensitive Detection of Diazinon. Food Chem. 2019, 280, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Rong, Y.; Li, H.; Ouyang, Q.; Ali, S.; Chen, Q. Rapid and Sensitive Detection of Diazinon in Food Based on the FRET between Rare-Earth Doped Upconversion Nanoparticles and Graphene Oxide. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2020, 239, 118500. [Google Scholar] [CrossRef]
- Wang, M.; Su, K.; Cao, J.; She, Y.; Abd El-Aty, A.M.; Hacımüftüoğlu, A.; Wang, J.; Yan, M.; Hong, S.; Lao, S.; et al. “Off-On” Non-Enzymatic Sensor for Malathion Detection Based on Fluorescence Resonance Energy Transfer between β-Cyclodextrin@Ag and Fluorescent Probe. Talanta 2019, 192, 295–300. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Sheng, R.; Wang, P.; Ouyang, Q.; Wang, A.; Ali, S.; Zareef, M.; Hassan, M. Ultra-Sensitive Detection of Malathion Residues Using FRET-Based Upconversion Fluorescence Sensor in Food. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2020, 241, 118654. [Google Scholar] [CrossRef] [PubMed]
- De Simone, G.; Mandrich, L.; Menchise, V.; Giordano, V.; Febbraio, F.; Rossi, M.; Pedone, C.; Manco, G. A Substrate-Induced Switch in the Reaction Mechanism of a Thermophilic Esterase—Kinetic Evidences and Structural Basis. J. Biol. Chem. 2004, 279, 6815–6823. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound Name | Affinity (kcal/mol) | Pocket Binding |
|---|---|---|
| Paraoxon | −6.5 to −6.1 | acyl |
| −6.1 to −5.6 | alcohol | |
| Parathion | n.d. | acyl |
| −6.2 to −6.1 | alcohol | |
| Diazinon | n.d. | acyl |
| −6.2 to −5.2 | alcohol |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rodrigues, A.C.M.; Barbieri, M.V.; Chino, M.; Manco, G.; Febbraio, F. A FRET Approach to Detect Paraoxon among Organophosphate Pesticides Using a Fluorescent Biosensor. Sensors 2022, 22, 561. https://doi.org/10.3390/s22020561
Rodrigues ACM, Barbieri MV, Chino M, Manco G, Febbraio F. A FRET Approach to Detect Paraoxon among Organophosphate Pesticides Using a Fluorescent Biosensor. Sensors. 2022; 22(2):561. https://doi.org/10.3390/s22020561
Chicago/Turabian StyleRodrigues, Andreia C. M., Maria Vittoria Barbieri, Marco Chino, Giuseppe Manco, and Ferdinando Febbraio. 2022. "A FRET Approach to Detect Paraoxon among Organophosphate Pesticides Using a Fluorescent Biosensor" Sensors 22, no. 2: 561. https://doi.org/10.3390/s22020561
APA StyleRodrigues, A. C. M., Barbieri, M. V., Chino, M., Manco, G., & Febbraio, F. (2022). A FRET Approach to Detect Paraoxon among Organophosphate Pesticides Using a Fluorescent Biosensor. Sensors, 22(2), 561. https://doi.org/10.3390/s22020561