
Meta-Assessment of Metformin Absorption and Disposition Pharmacokinetics in Nine Species

Abstract
:1. Introduction
2. Results
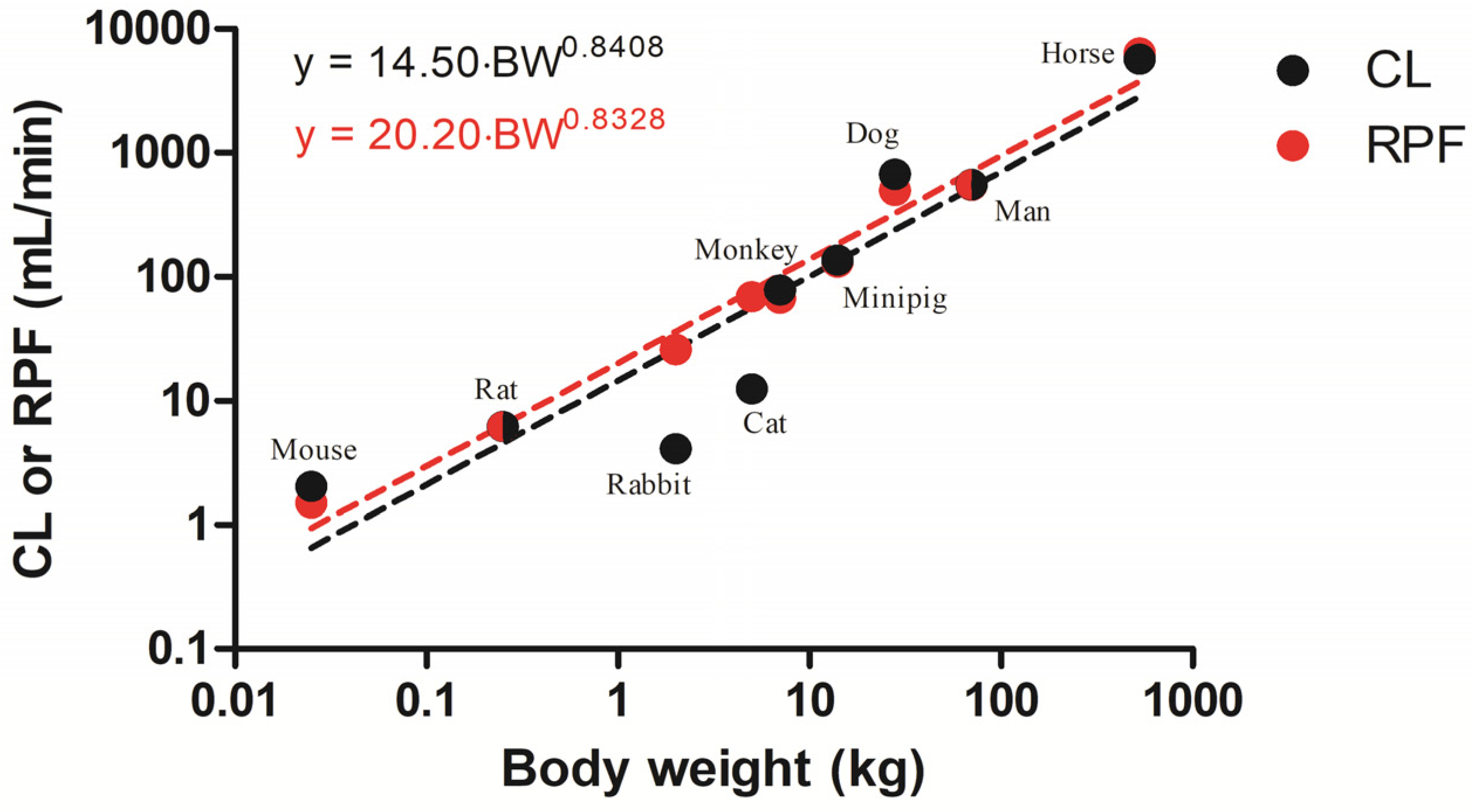
2.1. Allometric Scaling
2.2. Joint Fittings of IV Profiles
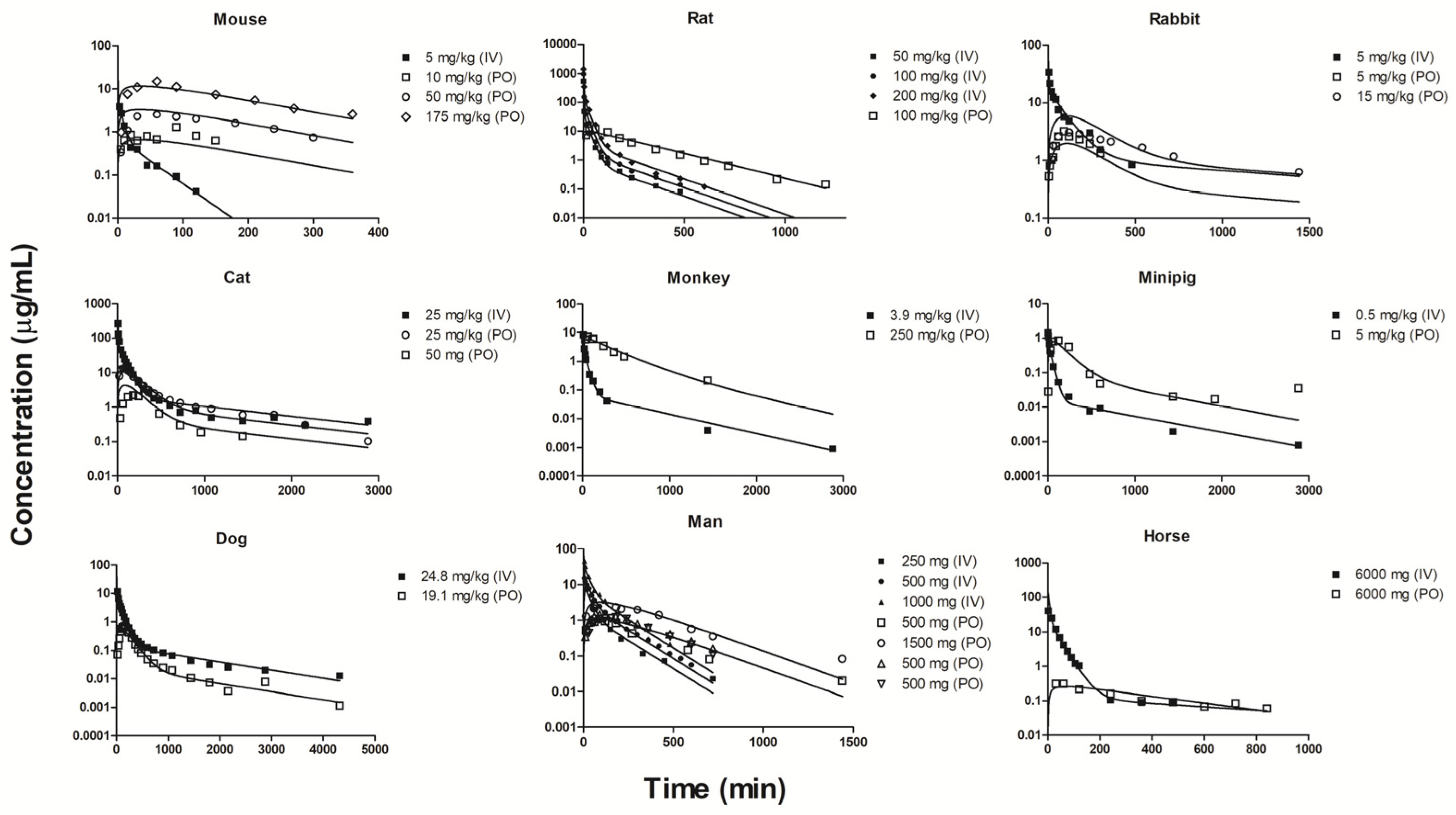
2.3. Individual Oral and IV Fittings
2.4. Tissue Distribution of Metformin
3. Discussion
4. Methods
4.1. Data Collection and Basic Allometric Scaling
4.2. mPBPK Modeling via Joint Fittings
4.3. mPBPK Modeling via Separate Fittings
4.4. Model Fittings
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hill, J. The Vegetable System, or, the Internal Structure and the Life of Plants: Their Classes, Orders, Genera, and Species Ascertained and Described in a Method Altogether New: Comprehending an Artificial Index, and a Natural System: With Figures of all the Plants Designed and Engraved by the Author: The Whole from Nature Only; Bradbury & Evans: London, UK, 1759; p. 54. [Google Scholar]
- Bailey, C.J.; Day, C. Traditional Plant Medicines as Treatments for Diabetes. Diabetes Care 1989, 12, 553–564. [Google Scholar] [CrossRef]
- Thomas, I.; Gregg, B. Metformin; a review of its history and future: From lilac to longevity. Pediatr. Diabetes 2017, 18, 10–16. [Google Scholar] [CrossRef]
- Werner, E.A.; Bell, J. CCXIV—The preparation of methylguanidine, and of ββ-dimethylguanidine by the interaction of dicyanodiamide, and methylammonium and dimethylammonium chlorides respectively. J. Chem. Soc. Trans. 1922, 121, 1790–1794. [Google Scholar] [CrossRef]
- Vecchio, I.; Tornali, C.; Bragazzi, N.L.; Martini, M. The Discovery of Insulin: An Important Milestone in the History of Medicine. Front. Endocrinol. 2018, 9, 613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ungar, G.; Freedman, L.; Shapiro, S.L. Pharmacological Studies of a New Oral Hypoglycemic Drug. Proc. Soc. Exp. Biol. Med. 1957, 95, 190–192. [Google Scholar] [CrossRef] [PubMed]
- Beringer, A. Zur Behandlung der Zuckerkrankheit mit Biguaniden. Wien. Med. Wschr. 1958, 108, 880–882. [Google Scholar]
- Sterne, J. Du nouveau dans les antidiabetiques. La NN dimethylamine guanyl guanide (NNDG). Maroc. Med. 1957, 36, 1295–1296. [Google Scholar]
- Pasik, C. Diabetes and the Biguanides: The Mystery of Each; Pasik, C., Ed.; Glucophage: Serving diabetology for 40 years; Groupe Lipha: Lyon, France, 1997; p. 79. [Google Scholar]
- Nattrass, M.; Alberti, K. Biguanides. Diabetologia 1978, 14, 71–74. [Google Scholar] [CrossRef] [Green Version]
- Nathan, D.M.; Buse, J.B.; Davidson, M.B.; Ferrannini, E.; Holman, R.R.; Sherwin, R.; Zinman, B. Medical Management of Hyperglycemia in Type 2 Diabetes: A Consensus Algorithm for the Initiation and Adjustment of Therapy: A consensus statement of the American Diabetes Association and the European Association for the Study of Diabetes. Diabetes Care 2008, 32, 193–203. [Google Scholar] [CrossRef] [Green Version]
- United Kingdom Prospective Diabetes Study Group. Effect of intensive blood-glucose control with metformin on complications in overweight patients with type 2 diabetes (UKPDS 34). Lancet 1998, 352, 854–865. [Google Scholar] [CrossRef]
- Evans, J.M.M.; Donnelly, L.A.; Emslie-Smith, A.M.; Alessi, D.R.; Morris, A.D. Metformin and reduced risk of cancer in diabetic patients. BMJ 2005, 330, 1304–1305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, D.; Yeung, S.C.J.; Hassan, M.M.; Konopleva, M.; Abbruzzese, J.L. Antidiabetic Therapies Affect Risk of Pancreatic Cancer. Gastroenterology 2009, 137, 482–488. [Google Scholar] [CrossRef] [Green Version]
- Landman, G.W.; Kleefstra, N.; Van Hateren, K.J.; Groenier, K.H.; Gans, R.O.; Bilo, H.J. Metformin Associated with Lower Cancer Mortality in Type 2 Diabetes: ZODIAC-16. Diabetes Care 2009, 33, 322–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crouse, A.B.; Grimes, T.; Li, P.; Might, M.; Ovalle, F.; Shalev, A. Metformin Use Is Associated with Reduced Mortality in a Diverse Population With COVID-19 and Diabetes. Front. Endocrinol. 2021, 11, 600439. [Google Scholar] [CrossRef] [PubMed]
- Bramante, C.T.; Ingraham, N.E.; Murray, T.A.; Marmor, S.; Hovertsen, S.; Gronski, J.; McNeil, C.; Feng, R.; Guzman, G.; Abdelwahab, N.; et al. Metformin and risk of mortality in patients hospitalised with COVID-19: A retrospective cohort analysis. Lancet Health Longev. 2021, 2, e34–e41. [Google Scholar] [CrossRef]
- Bailey, C.J. Metformin: Historical overview. Diabetologia 2017, 60, 1566–1576. [Google Scholar] [CrossRef] [Green Version]
- Le, S.; Lee, G.C. Emerging Trends in Metformin Prescribing in the United States from 2000 to 2015. Clin. Drug Investig. 2019, 39, 757–763. [Google Scholar] [CrossRef]
- Miller, R.A.; Chu, Q.; Xie, J.; Foretz, M.; Viollet, B.; Birnbaum, M.J. Biguanides suppress hepatic glucagon signalling by decreasing production of cyclic AMP. Nature 2013, 494, 256–260. [Google Scholar] [CrossRef] [Green Version]
- Madiraju, A.K.; Erion, D.M.; Rahimi, Y.; Zhang, X.-M.; Braddock, D.T.; Albright, R.A.; Prigaro, B.J.; Wood, J.L.; Bhanot, S.; Macdonald, M.J.; et al. Metformin suppresses gluconeogenesis by inhibiting mitochondrial glycerophosphate dehydrogenase. Nature 2014, 510, 542–546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bailey, C.J.; Puah, J.A. Effect of metformin on glucose metabolism in mouse soleus muscle. Diabete Metab. 1986, 12, 212–218. [Google Scholar] [PubMed]
- Rossetti, L.; De Fronzo, R.A.; Gherzi, R.; Stein, P.; Andraghetti, G.; Falzetti, G.; Shulman, G.I.; Klein-Robbenhaar, E.; Cordera, R. Effect of metformin treatment on insulin action in diabetic rats: In vivo and in vitro correlations. Metab. Clin. Exp. 1990, 39, 425–435. [Google Scholar] [CrossRef]
- Kirpichnikov, D.; McFarlane, S.I.; Sowers, J.R. Metformin: An Update. Ann. Intern. Med. 2002, 137, 25–33. [Google Scholar] [CrossRef]
- Bailey, C.J.; Mynett, K.J.; Page, T. Importance of the intestine as a site of metformin-stimulated glucose utilization. Br. J. Pharmacol. 1994, 112, 671–675. [Google Scholar] [CrossRef] [Green Version]
- Ikeda, T.; Iwata, K.; Murakami, H. Inhibitory effect of metformin on intestinal glucose absorption in the perfused rat intestine. Biochem. Pharmacol. 2000, 59, 887–890. [Google Scholar] [CrossRef]
- Stepensky, D.; Friedman, M.; Raz, I.; Hoffman, A. Pharmacokinetic-Pharmacodynamic Analysis of the Glucose-Lowering Effect of Metformin in Diabetic Rats Reveals First-Pass Pharmacodynamic Effect. Drug Metab. Dispos. 2002, 30, 861–868. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Chen, Y.; Zhao, Z.; Lu, W.; Zhou, T. Pharmacokinetic/Pharmacodynamic Analysis of Metformin using Different Models in Diabetic Rats. Drug Res. 2016, 66, 547–554. [Google Scholar] [CrossRef] [PubMed]
- Graham, G.G.; Punt, J.; Arora, M.; Day, R.O.; Doogue, M.; Duong, J.; Furlong, T.J.; Greenfield, J.R.; Greenup, L.C.; Kirkpatrick, C.M.; et al. Clinical Pharmacokinetics of Metformin. Clin. Pharmacokinet. 2011, 50, 81–98. [Google Scholar] [CrossRef]
- Ray, P. Complex Compounds of Biguanides and Guanylureas with Metallic Elements. Chem. Rev. 1961, 61, 313–359. [Google Scholar] [CrossRef]
- Cheng, C.-L.; Lawrence, X.Y.; Lee, H.-L.; Yang, C.-Y.; Lue, C.-S.; Chou, C.-H. Biowaiver extension potential to BCS Class III high solubility-low permeability drugs: Bridging evidence for metformin immediate-release tablet. Eur. J. Pharm. Sci. 2004, 22, 297–304. [Google Scholar] [CrossRef]
- Schäfer, G.; Bojanowski, D. Interaction of Biguanides with Mitochondrial and Synthetic Membranes. Eur. JBIC J. Biol. Inorg. Chem. 1972, 27, 364–375. [Google Scholar] [CrossRef] [PubMed]
- Song, N.-N.; Li, Q.-S.; Liu, C.-X. Intestinal permeability of metformin using single-pass intestinal perfusion in rats. World J. Gastroenterol. 2006, 12, 4064–4070. [Google Scholar] [CrossRef] [PubMed]
- Proctor, W.R.; Bourdet, D.L.; Thakker, D.R. Mechanisms Underlying Saturable Intestinal Absorption of Metformin. Drug Metab. Dispos. 2008, 36, 1650–1658. [Google Scholar] [CrossRef]
- Liang, X.; Giacomini, K.M. Transporters Involved in Metformin Pharmacokinetics and Treatment Response. J. Pharm. Sci. 2017, 106, 2245–2250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sirtori, C.R.; Franceschini, G.; Galli-Kienle, M.; Cighetti, G.; Galli, G.; Bondioli, A.; Conti, F. Disposition of metformin (N, N-dimethylbiguanide) in man. Clin. Pharmacol. Ther. 1978, 24, 683–693. [Google Scholar] [CrossRef]
- Tucker, G.T.; Casey, C.; Phillips, P.J.; Connor, H.; Ward, J.D.; Woods, H.F. Metformin kinetics in healthy subjects and in patients with diabetes mellitus. Br. J. Clin. Pharmacol. 1981, 12, 235–246. [Google Scholar] [CrossRef]
- Sambol, N.C.; Chiang, J.; Lin, E.T.; Goodman, A.M.; Liu, C.Y.; Benet, L.Z.; Cogan, M.G. Kidney Function and Age Are Both Predictors of Pharmacokinetics of Metformin. J. Clin. Pharmacol. 1995, 35, 1094–1102. [Google Scholar] [CrossRef] [PubMed]
- Robert, F.; Fendri, S.; Hary, L.; Lacroix, C.; Andréjak, M.; Lalau, J. Kinetics of plasma and erythrocyte metformin after acute administration in healthy subjects. Diabetes Metab. 2003, 29, 279–283. [Google Scholar] [CrossRef]
- Pentikäinen, P.; Neuvonen, P.; Penttilä, A. Pharmacokinetics of metformin after intravenous and oral administration to man. Eur. J. Clin. Pharmacol. 1979, 16, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Wilcock, C.; Bailey, C.J. Accumulation of metformin by tissues of the normal and diabetic mouse. Xenobiotica 1994, 24, 49–57. [Google Scholar] [CrossRef]
- Hanke, N.; Türk, D.; Selzer, D.; Ishiguro, N.; Ebner, T.; Wiebe, S.; Müller, F.; Stopfer, P.; Nock, V.; Lehr, T. A Comprehensive Whole-Body Physiologically Based Pharmacokinetic Drug–Drug–Gene Interaction Model of Metformin and Cimetidine in Healthy Adults and Renally Impaired Individuals. Clin. Pharmacokinet. 2020, 59, 1419–1431. [Google Scholar] [CrossRef]
- Cao, Y.; Jusko, W.J. Applications of minimal physiologically-based pharmacokinetic models. J. Pharmacokinet. Pharmacodyn. 2012, 39, 711–723. [Google Scholar] [CrossRef]
- Boxenbaum, H. Interspecies scaling, allometry, physiological time, and the ground plan of pharmacokinetics. J. Pharmacokinet. Biopharm. 1982, 10, 201–227. [Google Scholar] [CrossRef] [PubMed]
- Jansen, K.; Casellas, C.P.; Groenink, L.; Wever, K.E.; Masereeuw, R. Humans are animals, but are animals human enough? A systematic review and meta-analysis on interspecies differences in renal drug clearance. Drug Discov. Today 2020, 25, 706–717. [Google Scholar] [CrossRef]
- Hall, C.; Lueshen, E.; Mošat’, A.; Linninger, A.A. Interspecies Scaling in Pharmacokinetics: A Novel Whole-Body Physiologically Based Modeling Framework to Discover Drug Biodistribution Mechanisms in vivo. J. Pharm. Sci. 2012, 101, 1221–1241. [Google Scholar] [CrossRef]
- Song, D.; Jusko, W.J. Across-species meta-analysis of dexamethasone pharmacokinetics utilizing allometric and scaling modeling approaches. Biopharm. Drug Dispos. 2021, 42, 191–203. [Google Scholar] [CrossRef]
- Zhao, J.; Cao, Y.; Jusko, W.J. Across-Species Scaling of Monoclonal Antibody Pharmacokinetics Using a Minimal PBPK Model. Pharm. Res. 2015, 32, 3269–3281. [Google Scholar] [CrossRef]
- Huang, Q.; Gehring, R.; Tell, L.A.; Li, M.; Riviere, J.E. Interspecies allometric meta-analysis of the comparative pharmacokinetics of 85 drugs across veterinary and laboratory animal species. J. Vet. Pharmacol. Ther. 2015, 38, 214–226. [Google Scholar] [CrossRef]
- Zhang, D. Evaluation of the Allometric Exponents in Prediction of Human Drug Clearance. Ph.D. Thesis, Virginia Commonwealth University, Richmond, VA, USA, 2014. [Google Scholar]
- Johnston, C.A.; Dickinson, V.S.M.; Alcorn, J.; Gaunt, M.C. Pharmacokinetics and oral bioavailability of metformin hydrochloride in healthy mixed-breed dogs. Am. J. Vet. Res. 2017, 78, 1193–1199. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.H.; Kim, S.G.; Lee, M.G. Dose-Independent Pharmacokinetics of Metformin in Rats: Hepatic and Gastrointestinal First-Pass Effects. J. Pharm. Sci. 2006, 95, 2543–2552. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.; Lee, U.; Lee, B.; Lee, M. Pharmacokinetic interaction between itraconazole and metformin in rats: Competitive inhibition of metabolism of each drug by each other via hepatic and intestinal CYP3A1/2. Br. J. Pharmacol. 2010, 161, 815–829. [Google Scholar] [CrossRef] [Green Version]
- Higgins, J.W.; Bedwell, D.W.; Zamek-Gliszczynski, M.J. Ablation of Both Organic Cation Transporter (Oct)1 and Oct2 Alters Metformin Pharmacokinetics but Has No Effect on Tissue Drug Exposure and Pharmacodynamics. Drug Metab. Dispos. 2012, 40, 1170–1177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamichi, N.; Shima, H.; Asano, S.; Ishimoto, T.; Sugiura, T.; Matsubara, K.; Kusuhara, H.; Sugiyama, Y.; Sai, Y.; Miyamoto, K.-I.; et al. Involvement of Carnitine/Organic Cation Transporter OCTN1/SLC22A4 in Gastrointestinal Absorption of Metformin. J. Pharm. Sci. 2013, 102, 3407–3417. [Google Scholar] [CrossRef]
- Bouriche, S.; Alonso-García, A.; Cárceles-Rodríguez, C.M.; Rezgui, F.; Fernández-Varón, E. Potential of sustained release microparticles of metformin in veterinary medicine: An in vivo pharmacokinetic study of metformin microparticles as oral sustained release formulation in rabbits. BMC Vet. Res. 2020. [Google Scholar] [CrossRef]
- Choi, J.S.; Choi, I. Drug interaction of metformin and aspirin in rabbits. Korean J. Clin. Pharm. 2003, 13, 67–71. [Google Scholar]
- Michels, G.M.; Boudinot, F.D.; Ferguson, D.C.; Hoenig, M. Pharmacokinetics of the antihyperglycemic agent metformin in cats. Am. J. Vet. Res. 1999, 60, 738–742. [Google Scholar] [PubMed]
- Nelson, R.; Spann, D.; Elliott, D.; Brondos, A.; Vulliet, R. Evaluation of the oral antihyperglycemic drug metformin in normal and diabetic cats. J. Vet. Intern. Med. 2004, 18, 18–24. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Liu, T.; Jiang, H.; Titsch, C.; Taylor, K.; Kandoussi, H.; Qiu, X.; Chen, C.; Sukrutharaj, S.; Kuit, K.; et al. Cynomolgus Monkey as a Clinically Relevant Model to Study Transport Involving Renal Organic Cation Transporters: In Vitro and In Vivo Evaluation. Drug Metab. Dispos. 2016, 44, 238–249. [Google Scholar] [CrossRef] [Green Version]
- Heinig, K.; Bucheli, F. Fast liquid chromatographic-tandem mass spectrometric (LC–MS–MS) determination of metformin in plasma samples. J. Pharm. Biomed. Anal. 2004, 34, 1005–1011. [Google Scholar] [CrossRef] [PubMed]
- Patel, N.J.; Yumibe, N.; Ruterbories, K.; Huang, N.; Burns, L.; Tan, J.; White, D.; Liu, J.; Brocksmith, D.; Bouchard, G.; et al. Pharmacokinetics of intravenous and oral metformin and r,s-verapamil in Sinclair, Hanford, Yucatan and Göttingen minipigs. Int. J. Pharmacokinet. 2017, 2, 81–91. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.H.; Kwon, K.-I. Pharmacokinetic-pharmacodynamic modeling for the relationship between glucose-lowering effect and plasma concentration of metformin in volunteers. Arch. Pharm. Res. 2004, 27, 806–810. [Google Scholar] [CrossRef] [PubMed]
- Hustace, J.L.; Firshman, A.M.; Mata, J.E. Pharmacokinetics and bioavailability of metformin in horses. Am. J. Vet. Res. 2009, 70, 665–668. [Google Scholar] [CrossRef]
- Thuesen, A.D.; Andersen, H.; Cardel, M.; Toft, A.; Walter, S.; Marcussen, N.; Jensen, B.L.; Bie, P.; Hansen, P.B.L. Differential effect of T-type voltage-gated Ca2+ channel disruption on renal plasma flow and glomerular filtration rate in vivo. Am. J. Physiol. Renal Fluid Electrolyte Physiol. 2014, 307, F445–F452. [Google Scholar] [CrossRef] [Green Version]
- Brown, R.P.; Delp, M.D.; Lindstedt, S.L.; Rhomberg, L.R.; Beliles, R.P. Physiological Parameter Values for Physiologically Based Pharmacokinetic Models. Toxicol. Ind. Health 1997, 13, 407–484. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, L.M.; Kirman, C.R.; Gannon, S.A.; Thrall, K.D.; Gargas, M.L.; Kinzell, J.H. Development of a physiologically based pharmacokinetic (PBPK) model for methyl iodide in rats, rabbits, and humans. Inhal. Toxicol. 2009, 21, 552–582. [Google Scholar] [CrossRef] [PubMed]
- Lindstedt, S.L.; Schaeffer, P.J. Use of allometry in predicting anatomical and physiological parameters of mammals. Lab. Anim. 2002, 36, 1–19. [Google Scholar] [CrossRef]
- Jamei, M.; Marciniak, S.; Feng, K.; Barnett, A.; Tucker, G.; Rostami-Hodjegan, A. The Simcyp®Population-based ADME Simulator. Expert Opin. Drug Metab. Toxicol. 2009, 5, 211–223. [Google Scholar] [CrossRef] [PubMed]
- Suenderhauf, C.; Parrott, N. A Physiologically Based Pharmacokinetic Model of the Minipig: Data Compilation and Model Implementation. Pharm. Res. 2012, 30, 1–15. [Google Scholar] [CrossRef]
- Wesolowski, C.A.; Wanasundara, S.N.; Babyn, P.S.; Alcorn, J. Comparison of the gamma-Pareto convolution with conventional methods of characterising metformin pharmacokinetics in dogs. J. Pharmacokinet. Pharmacodyn. 2019, 47, 19–45. [Google Scholar] [CrossRef] [Green Version]
- Holdstock, N.B.; Ousey, J.C.; Rossdale, P.D. Glomerular filtration rate, effective renal plasma flow, blood pressure and pulse rate in the equine neonate during the first 10 days post partum. Equine Vet. J. 1998, 30, 335–343. [Google Scholar] [CrossRef] [PubMed]
- Beckmann, R. Absorption, distribution in the organism and elimination of metformin. Diabetologia 1969, 5, 318–324. [Google Scholar] [CrossRef] [Green Version]
- Balimane, P.V.; Han, Y.-H.; Chong, S. Current industrial practices of assessing permeability and P-glycoprotein interaction. AAPS J. 2006, 8, E1–E13. [Google Scholar] [CrossRef] [Green Version]
- Nestorov, I.A.; Aarons, L.J.; Arundel, P.A.; Rowland, M. Lumping of whole-body physiologically based pharmacokinetic models. J. Pharmacokinet. Biopharm. 1998, 26, 21–46. [Google Scholar] [CrossRef]
- Björkman, S. Reduction and Lumping of Physiologically Based Pharmacokinetic Models: Prediction of the Disposition of Fentanyl and Pethidine in Humans by Successively Simplified Models. J. Pharmacokinet. Pharmacodyn. 2003, 30, 285–307. [Google Scholar] [CrossRef] [PubMed]
- Gueorguieva, I.; Nestorov, I.A.; Rowland, M. Reducing Whole Body Physiologically Based Pharmacokinetic Models Using Global Sensitivity Analysis: Diazepam Case Study. J. Pharmacokinet. Pharmacodyn. 2006, 33, 1–27. [Google Scholar] [CrossRef]
- Pilari, S.; Huisinga, W. Lumping of physiologically-based pharmacokinetic models and a mechanistic derivation of classical compartmental models. J. Pharmacokinet. Pharmacodyn. 2010, 37, 365–405. [Google Scholar] [CrossRef] [PubMed]
- Jeong, Y.-S.; Yim, C.-S.; Ryu, H.-M.; Noh, C.-K.; Song, Y.-K.; Chung, S.-J. Estimation of the minimum permeability coefficient in rats for perfusion-limited tissue distribution in whole-body physiologically-based pharmacokinetics. Eur. J. Pharm. Biopharm. 2017, 115, 1–17. [Google Scholar] [CrossRef]
- Hemauer, S.J.; Patrikeeva, S.L.; Nanovskaya, T.N.; Hankins, G.D.; Ahmed, M.S. Role of human placental apical membrane transporters in the efflux of glyburide, rosiglitazone, and metformin. Am. J. Obstet. Gynecol. 2010, 202, 383.e1–383.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poulin, P.; Theil, F. Prediction of Pharmacokinetics Prior to In Vivo Studies. 1. Mechanism-Based Prediction of Volume of Distribution. J. Pharm. Sci. 2002, 91, 129–156. [Google Scholar] [CrossRef]
- Berezhkovskiy, L.M. Volume of Distribution at Steady State for a Linear Pharmacokinetic System with Peripheral Elimination. J. Pharm. Sci. 2004, 93, 1628–1640. [Google Scholar] [CrossRef] [PubMed]
- Amidon, G.L.; Lennernäs, H.; Shah, V.P.; Crison, J.R. A theoretical basis for a biopharmaceutic drug classification: The correlation of in vitro drug product dissolution and in vivo bioavailability. Pharm. Res. 1995, 12, 413–420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Øie, S.; Tozer, T.N. Effect of Altered Plasma Protein Binding on Apparent Volume of Distribution. J. Pharm. Sci. 1979, 68, 1203–1205. [Google Scholar] [CrossRef]
- Bailey, C.J.; Turner, R.C. Metformin. N. Engl. J. Med. 1996, 334, 574–579. [Google Scholar] [CrossRef]
- De Fronzo, R.A. Pharmacologic Therapy for Type 2 Diabetes Mellitus. Ann. Intern. Med. 1999, 131, 281–303. [Google Scholar] [CrossRef] [PubMed]
- Bailey, C.; Flatt, P.; Marks, V. Drugs inducing hypoglycemia. Pharmacol. Ther. 1989, 42, 361–384. [Google Scholar] [CrossRef]
- Jeppesen, J.; Zhou, M.-Y.; Chen, Y.-D.I.; Reaven, G.M. Effect of Metformin on Postprandial Lipemia in Patients with Fairly to Poorly Controlled NIDDM. Diabetes Care 1994, 17, 1093–1099. [Google Scholar] [CrossRef]
- De Fronzo, R.A.; Goodman, A.M. Efficacy of Metformin in Patients with Non-Insulin-Dependent Diabetes Mellitus. N. Engl. J. Med. 1995, 333, 541–549. [Google Scholar] [CrossRef] [Green Version]
- De Fronzo, R.A.; Barzilai, N.; Simonson, D.C. Mechanism of Metformin Action in Obese and Lean Noninsulin-Dependent Diabetic Subjects. J. Clin. Endocrinol. Metab. 1991, 73, 1294–1301. [Google Scholar] [CrossRef]
- Reaven, G.M.; Johnston, P.; Hollenbeck, C.B.; Skowronski, R.; Zhang, J.C.; Goldfine, I.D.; Chen, Y.D. Combined metformin-sulfonylurea treatment of patients with noninsulin-dependent diabetes in fair to poor glycemic control. J. Clin. Endocrinol. Metab. 1992, 74, 1020–1026. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Yesilkanal, A.E.; Wynne, J.P.; Frankenberger, C.; Liu, J.; Yan, J.; Elbaz, M.; Rabe, D.; Rustandy, F.D.; Tiwari, P.; et al. Effective breast cancer combination therapy targeting BACH1 and mitochondrial metabolism. Nature 2019, 568, 254–258. [Google Scholar] [CrossRef] [PubMed]
- Saini, N.; Yang, X. Metformin as an anti-cancer agent: Actions and mechanisms targeting cancer stem cells. Acta Biochim. Biophys. Sin. 2017, 50, 133–143. [Google Scholar] [CrossRef] [Green Version]
- Bridges, H.R.; Jones, A.J.Y.; Pollak, M.N.; Hirst, J. Effects of metformin and other biguanides on oxidative phosphorylation in mitochondria. Biochem. J. 2014, 462, 475–487. [Google Scholar] [CrossRef] [Green Version]
- Dykens, J.A.; Jamieson, J.; Marroquin, L.; Nadanaciva, S.; Billis, P.A.; Will, Y. Biguanide-induced mitochondrial dysfunction yields increased lactate production and cytotoxicity of aerobically-poised HepG2 cells and human hepatocytes in vitro. Toxicol. Appl. Pharmacol. 2008, 233, 203–210. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; An, H.; Liu, T.; Qin, C.; Sesaki, H.; Guo, S.; Radovick, S.; Hussain, M.; Maheshwari, A.; Wondisford, F.E.; et al. Metformin Improves Mitochondrial Respiratory Activity through Activation of AMPK. Cell Rep. 2019, 29, 1511–1523.e1515. [Google Scholar] [CrossRef] [PubMed]
- Malin, S.K.; Kashyap, S.R. Effects of metformin on weight loss: Potential mechanisms. Curr. Opin. Endocrinol. Diabetes Obes. 2014, 21, 323–329. [Google Scholar] [CrossRef] [PubMed]
- Barzilai, N.; Crandall, J.P.; Kritchevsky, S.B.; Espeland, M.A. Metformin as a Tool to Target Aging. Cell Metab. 2016, 23, 1060–1065. [Google Scholar] [CrossRef] [Green Version]
- Kulkarni, A.S.; Gubbi, S.; Barzilai, N. Benefits of Metformin in Attenuating the Hallmarks of Aging. Cell Metab. 2020, 32, 15–30. [Google Scholar] [CrossRef]
- Shurrab, N.T.; Arafa, E.-S.A. Metformin: A review of its therapeutic efficacy and adverse effects. Obes. Med. 2020, 17, 100186. [Google Scholar] [CrossRef]
- Choi, Y.H.; Lee, M.G. Effects of enzyme inducers and inhibitors on the pharmacokinetics of metformin in rats: Involvement of CYP2C11, 2D1 and 3A1/2 for the metabolism of metformin. Br. J. Pharmacol. 2006, 149, 424–430. [Google Scholar] [CrossRef] [Green Version]
- Kanter, G.S. Glomerular filtration and renal plasma flow during hyperthermia. Am. J. Physiol. Content 1960, 198, 1044–1048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boxenbaum, H.; Fertig, J.B. Scaling of antipyrine intrinsic clearance of unbound drug in 15 mammalian species. Eur. J. Drug Metab. Pharmacokinet. 1984, 9, 177–183. [Google Scholar] [CrossRef]
- Yates, F.E.; Kugler, P.N. Similarity Principles and Intrinsic Geometries: Contrasting Approaches to Interspecies Scaling. J. Pharm. Sci. 1986, 75, 1019–1027. [Google Scholar] [CrossRef]
- Lin, J.H. Species similarities and differences in pharmacokinetics. Drug Metab. Dispos. 1995, 23, 1008–1021. [Google Scholar]
- Basit, A.; Radi, Z.; Vaidya, V.S.; Karasu, M.; Prasad, B. Kidney Cortical Transporter Expression across Species Using Quantitative Proteomics. Drug Metab. Dispos. 2019, 47, 802–808. [Google Scholar] [CrossRef] [PubMed]
- Groves, C.E.; Suhre, W.B.; Cherrington, N.J.; Wright, S.H. Sex Differences in the mRNA, Protein, and Functional Expression of Organic Anion Transporter (Oat) 1, Oat3, and Organic Cation Transporter (Oct) 2 in Rabbit Renal Proximal Tubules. J. Pharmacol. Exp. Ther. 2005, 316, 743–752. [Google Scholar] [CrossRef] [Green Version]
- Suhre, W.M.; Ekins, S.; Chang, C.; Swaan, P.W.; Wright, S.H. Molecular Determinants of Substrate/Inhibitor Binding to the Human and Rabbit Renal Organic Cation Transporters hOCT2 and rbOCT2. Mol. Pharmacol. 2005, 67, 1067–1077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burt, H.; Neuhoff, S.; Almond, L.; Gaohua, L.; Harwood, M.; Jamei, M.; Rostami-Hodjegan, A.; Tucker, G.; Rowland-Yeo, K. Metformin and cimetidine: Physiologically based pharmacokinetic modelling to investigate transporter mediated drug–drug interactions. Eur. J. Pharm. Sci. 2016, 88, 70–82. [Google Scholar] [CrossRef] [PubMed]
- Nishiyama, K.; Toshimoto, K.; Lee, W.; Ishiguro, N.; Bister, B.; Sugiyama, Y. Physiologically-Based Pharmacokinetic Modeling Analysis for Quantitative Prediction of Renal Transporter–Mediated Interactions between Metformin and Cimetidine. CPT Pharmacomet. Syst. Pharmacol. 2019, 8, 396–406. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.; Wang, Q.; Sun, Y.; Shen, M.; Li, H.; Duan, Y. A New PAMPA Model Proposed on the Basis of a Synthetic Phospholipid Membrane. PLoS ONE 2015, 10, e0116502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veng-Pedersen, P. Theorems and implications of a model independent elimination/distribution function decomposition of linear and some nonlinear drug dispositions. I. Derivations and theoretical analysis. J. Pharmacokinet. Biopharm. 1984, 12, 627–648. [Google Scholar] [CrossRef]
- Gillespie, W.R.; Veng-Pedersen, P. Theorems and implications of a model-independent elimination/distribution function decomposition of linear and some nonlinear drug dispositions. II. Clearance concepts applied to the evaluation of distribution kinetics. J. Pharmacokinet. Biopharm. 1985, 13, 441–451. [Google Scholar] [CrossRef]
- Rodgers, T.; Leahy, D.; Rowland, M. Physiologically Based Pharmacokinetic Modeling 1: Predicting the Tissue Distribution of Moderate-to-Strong Bases. J. Pharm. Sci. 2005, 94, 1259–1276. [Google Scholar] [CrossRef]
- Assmus, F.; Houston, J.B.; Galetin, A. Incorporation of lysosomal sequestration in the mechanistic model for prediction of tissue distribution of basic drugs. Eur. J. Pharm. Sci. 2017, 109, 419–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trapp, S.; Rosania, G.R.; Horobin, R.W.; Kornhuber, J. Quantitative modeling of selective lysosomal targeting for drug design. Eur. Biophys. J. 2008, 37, 1317–1328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shu, Y.; Sheardown, S.A.; Brown, C.; Owen, R.P.; Zhang, S.; Castro, R.A.; Ianculescu, A.G.; Yue, L.; Lo, J.C.; Burchard, E.G.; et al. Effect of genetic variation in the organic cation transporter 1 (OCT1) on metformin action. J. Clin. Investig. 2007, 117, 1422–1431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, M.; Zhu, Q.; Zhu, A.; Gemski, C.; Ma, B.; Guan, E.; Li, A.P.; Xiao, G.; Xia, C.Q. Comparison of uptake transporter functions in hepatocytes in different species to determine the optimal model for evaluating drug transporter activities in humans. Xenobiotica 2018, 49, 852–862. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Prasad, B.; Salphati, L.; Chu, X.; Gupta, A.; Hop, C.E.; Evers, R.; Unadkat, J.D. Interspecies Variability in Expression of Hepatobiliary Transporters across Human, Dog, Monkey, and Rat as Determined by Quantitative Proteomics. Drug Metab. Dispos. 2014, 43, 367–374. [Google Scholar] [CrossRef] [Green Version]
- Meyer, M.J.; Tuerkova, A.; Römer, S.; Wenzel, C.; Seitz, T.; Gaedcke, J.; Oswald, S.; Brockmöller, J.; Zdrazil, B.; Tzvetkov, M.V. Differences in Metformin and Thiamine Uptake between Human and Mouse Organic Cation Transporter 1: Structural Determinants and Potential Consequences for Intrahepatic Concentrations. Drug Metab. Dispos. 2020, 48, 1380–1392. [Google Scholar] [CrossRef]
- Kimura, N.; Masuda, S.; Tanihara, Y.; Ueo, H.; Okuda, M.; Katsura, T.; Inui, K.-I. Metformin is a Superior Substrate for Renal Organic Cation Transporter OCT2 rather than Hepatic OCT1. Drug Metab. Pharmacokinet. 2005, 20, 379–386. [Google Scholar] [CrossRef] [Green Version]
- Hatton, G.B.; Yadav, V.; Basit, A.W.; Merchant, H.A. Animal Farm: Considerations in Animal Gastrointestinal Physiology and Relevance to Drug Delivery in Humans. J. Pharm. Sci. 2015, 104, 2747–2776. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.; Chien, H.-C.; Yee, S.W.; Giacomini, M.M.; Chen, E.C.; Piao, M.; Hao, J.; Twelves, J.; Lepist, E.-I.; Ray, A.S.; et al. Metformin Is a Substrate and Inhibitor of the Human Thiamine Transporter, THTR-2 (SLC19A3). Mol. Pharm. 2015, 12, 4301–4310. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.-S.; Jonker, J.W.; Kato, Y.; Kusuhara, H.; Schinkel, A.H.; Sugiyama, Y. Involvement of Organic Cation Transporter 1 in Hepatic and Intestinal Distribution of Metformin. J. Pharmacol. Exp. Ther. 2002, 302, 510–515. [Google Scholar] [CrossRef] [Green Version]
- Gertz, M.; Harrison, A.; Houston, J.B.; Galetin, A. Prediction of Human Intestinal First-Pass Metabolism of 25 CYP3A Substrates from In Vitro Clearance and Permeability Data. Drug Metab. Dispos. 2010, 38, 1147–1158. [Google Scholar] [CrossRef]
- Lawrence, X.Y.; Amidon, G.L. A compartmental absorption and transit model for estimating oral drug absorption. Int. J. Pharm. 1999, 186, 119–125. [Google Scholar]
- Musther, H.; Olivares-Morales, A.; Hatley, O.J.; Liu, B.; Hodjegan, A.R. Animal versus human oral drug bioavailability: Do they correlate? Eur. J. Pharm. Sci. 2014, 57, 280–291. [Google Scholar] [CrossRef] [PubMed]
- Xie, F.; Ke, A.B.; Bowers, G.D.; Zamek-Gliszczynski, M.J. Metformin’s intrinsic blood-to-plasma partition ratio (B/P): Reconciling the perceived high in vivo B/P> 10 with the in vitro equilibrium value of unity. J. Pharmacol. Exp. Ther. 2015, 354, 225–229. [Google Scholar] [CrossRef]
- Kajbaf, F.; Bennis, Y.; Hurtel-Lemaire, A.-S.; Andréjak, M.; Lalau, J.-D. Unexpectedly long half-life of metformin elimination in cases of metformin accumulation. Diabet. Med. 2015, 33, 105–110. [Google Scholar] [CrossRef] [PubMed]
- Lee, N.; Duan, H.; Hebert, M.F.; Liang, C.J.; Rice, K.M.; Wang, J. Taste of a Pill. J. Biol. Chem. 2014, 289, 27055–27064. [Google Scholar] [CrossRef] [Green Version]
- Toyama, K.; Yonezawa, A.; Masuda, S.; Osawa, R.; Hosokawa, M.; Fujimoto, S.; Inagaki, N.; Inui, K.; Katsura, T. Loss of multidrug and toxin extrusion 1 (MATE1) is associated with metformin-induced lactic acidosis. Br. J. Pharmacol. 2012, 166, 1183–1191. [Google Scholar] [CrossRef] [Green Version]
- Chaudhari, K.; Wang, J.; Xu, Y.; Winters, A.; Wang, L.; Dong, X.; Cheng, E.Y.; Liu, R.; Yang, S.-H. Determination of metformin bio-distribution by LC-MS/MS in mice treated with a clinically relevant paradigm. PLoS ONE 2020, 15, e0234571. [Google Scholar] [CrossRef] [PubMed]
- Łabuzek, K.; Suchy, D.; Gabryel, B.; Bielecka, A.; Liber, S.; Okopień, B. Quantification of metformin by the HPLC method in brain regions, cerebrospinal fluid and plasma of rats treated with lipopolysaccharide. Pharmacol. Rep. 2010, 62, 956–965. [Google Scholar] [CrossRef]
- Ma, Y.-R.; Shi, A.-X.; Qin, H.-Y.; Zhang, T.; Wu, Y.-F.; Zhang, G.-Q.; Wu, X.-A. Metoprolol decreases the plasma exposure of metformin via the induction of liver, kidney and muscle uptake in rats. Biopharm. Drug Dispos. 2016, 37, 511–521. [Google Scholar] [CrossRef]
- Gormsen, L.C.; Sundelin, E.I.; Jensen, J.B.; Vendelbo, M.H.; Jakobsen, S.; Munk, O.L.; Christensen, M.M.H.; Brøsen, K.; Frokiaer, J.; Jessen, N. In Vivo Imaging of Human 11C-Metformin in Peripheral Organs: Dosimetry, Biodistribution, and Kinetic Analyses. J. Nucl. Med. 2016, 57, 1920–1926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morse, B.L.; Fallon, J.K.; Kolur, A.; Hogan, A.T.; Smith, P.C.; Hillgren, K.M. Comparison of Hepatic Transporter Tissue Expression in Rodents and Interspecies Hepatic OCT1 Activity. AAPS J. 2021, 23, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Berezhkovskiy, L.M. The Connection Between the Steady State (Vss) and Terminal (Vβ) Volumes of Distribution in Linear Pharmacokinetics and The General Proof That Vβ ≥ Vss. J. Pharm. Sci. 2007, 96, 1638–1652. [Google Scholar] [CrossRef] [PubMed]
- Wolfensohn, S.; Lloyd, M. Handbook of Laboratory Animal Management and Welfare; John Wiley & Sons: Hoboken, NJ, USA, 2003. [Google Scholar]
- Davies, B.; Morris, T. Physiological Parameters in Laboratory Animals and Humans. Pharm. Res. 1993, 10, 1093–1095. [Google Scholar] [CrossRef] [PubMed]
- Von Hendy-Willson, V.E.; Pressler, B.M. An overview of glomerular filtration rate testing in dogs and cats. Vet. J. 2011, 188, 156–165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walsh, D.; Royal, H. Evaluation of a single injection of 99mTc-labeled diethylenetriaminepentaacetic acid for measuring glomerular filtration rate in horses. Am. J. Vet. Res. 1992, 53, 776–780. [Google Scholar]
- Kararli, T.T. Comparison of the gastrointestinal anatomy, physiology, and biochemistry of humans and commonly used laboratory animals. Biopharm. Drug Dispos. 1995, 16, 351–380. [Google Scholar] [CrossRef] [PubMed]
- Ferraris, R.P.; Lee, P.P.; Diamond, J.M. Origin of regional and species differences in intestinal glucose uptake. Am. J. Physiol. Gastrointest. Liver Physiol. 1989, 257, 689. [Google Scholar] [CrossRef]
- Merchant, H.A.; McConnell, E.L.; Liu, F.; Ramaswamy, C.; Kulkarni, R.P.; Basit, A.W.; Murdan, S. Assessment of gastrointestinal pH, fluid and lymphoid tissue in the guinea pig, rabbit and pig, and implications for their use in drug development. Eur. J. Pharm. Sci. 2011, 42, 3–10. [Google Scholar] [CrossRef] [Green Version]
- Bettini, G.; Muracchini, M.; Della Salda, L.; Preziosi, R.; Morini, M.; Guglielmini, C.; Sanguinetti, V.; Marcato, P. Hypertrophy of intestinal smooth muscle in cats. Res. Vet. Sci. 2003, 75, 43–53. [Google Scholar] [CrossRef]
- Clauss, M.; Frey, R.; Kiefer, B.; Lechner-Doll, M.; Loehlein, W.; Polster, C.; Streich, W.J. The maximum attainable body size of herbivorous mammals: Morphophysiological constraints on foregut, and adaptations of hindgut fermenters. Oecologia 2003, 136, 14–27. [Google Scholar] [CrossRef] [Green Version]
- Myagmarjalbuu, B.; Moon, M.J.; Heo, S.H.; Jeong, S.I.; Park, J.-S.; Jun, J.Y.; Jeong, Y.Y.; Kang, H.K. Establishment of a Protocol for Determining Gastrointestinal Transit Time in Mice Using Barium and Radiopaque Markers. Korean J. Radiol. 2013, 14, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Quini, C.C.; Américo, M.F.; Corá, L.A.; Calabresi, M.F.; Alvarez, M.; Oliveira, R.B.; A Miranda, J.R. Employment of a noninvasive magnetic method for evaluation of gastrointestinal transit in rats. J. Biol. Eng. 2012, 6, 6. [Google Scholar] [CrossRef] [PubMed]
- Davies, R.R.; Davies, J.A.R. Rabbit gastrointestinal physiology. Vet. Clin. N. Am. Exot. Anim. Pract. 2003, 6, 139–153. [Google Scholar] [CrossRef]
- Chandler, M.L.; Guilford, G.; Lawoko, C.R. Radiopaque Markers to Evaluate Gastric Emptying and Small Intestinal Transit Time in Healthy Cats. J. Vet. Intern. Med. 1997, 11, 361–364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steinmann, M.; Bezugley, R.J.; Bond, S.L.; Pomrantz, J.S.; Léguillette, R. A wireless endoscopy capsule suitable for imaging of the equine stomach and small intestine. J. Vet. Intern. Med. 2020, 34, 1622–1630. [Google Scholar] [CrossRef] [PubMed]
- D’Argenio, D.Z.; Schumitzky, A.; Wang, X. ADAPT 5 User’s Guide: Pharmacokinetic/Pharmacodynamic Systems Analysis Software; Biomedical Simulations Resource: Los Angeles, CA, USA, 2009. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reference | Species | Strain | Sex | Body Weight (kg) | Dosing Route | Dose (mg/kg) | Assay | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Higgins et al. (2012) [54] | Mouse | FVB | M | - | IV * | 5 | LC-MS/MS | 81.7 | 40.0 | 60 a | 1840 | |
| PO * | 10 | LC-MS/MS | 138 | |||||||||
| Nakamichi et al. (2013) [55] | Mouse | C57BL/6J | M | - | IV Infusion | 0.12 mg/kg/min | HPLC-UV | 60.7 | 60.3 | |||
| PO * | 50, 175 | HPLC-UV | 77.5, 59.8 ** | |||||||||
| Choi et al. (2006) [52] | Rat | Sprague-Dawley | M | 0.20–0.31 | IV * | 50, 100, 200 | HPLC-UV | 23.6–26.4 | 17.8–19.5 | 24.8 b | 586–693 | |
| PO | 50, 100, 200 | HPLC-UV | 76.0–82.0 | 37.4–39.6 | ||||||||
| Choi et al. (2010) [53] | Rat | Sprague-Dawley | M | 0.19–0.30 | IV | 100 | HPLC-UV | 14.7 | 11.6 | 383 | ||
| PO * | 100 | HPLC-UV | 34.1 | 30.9 | ||||||||
| Bouriche et al. (2020) [56] | Rabbit | New Zealand | F | - | IV * | 5 | HPLC-UV | 2.05 | 13.1 c | 321 | ||
| PO * | 5 | HPLC-UV | 5.67 | |||||||||
| Choi and Choi (2003) [57] | Rabbit | New Zealand | M | 2.0–2.2 | PO * | 15 | HPLC-UV | 6.01 | ||||
| Michels et al. (1999) [58] | Cat | Domestic shorthair | - | 4.3–6.5 | IV * | 25 | HPLC-UV | 2.5 | 2.17 | 13.8 d | 550 | |
| PO * | 25 | HPLC-UV | 5.21 | |||||||||
| Nelson et al. (2004) [59] | Cat | - | M | 4–5 | PO * | 50 mg | HPLC-UV | 9.05 | ||||
| Shen et al. (2016) [60] | Monkey | Cynomolgus | M | 5–7 | IV * | 3.9 | LC-MS/MS | 11.2 | 10.7 | 9.61e | 802 ** | |
| Heinig and Bucheli (2004) [61] | Monkey | Cynomolgus | - | - | PO * | 250 | LC-MS/MS | 93.5 ** | ||||
| Patel et al. (2017) [62] | Minipig | Hanford | M | 14.3 | IV * | 0.5 | LC-MS/MS | 9.7 | 9.36 f | 2260 ** | ||
| PO * | 5 | LC-MS/MS | 14.8 ** | |||||||||
| Johnston et al. (2017) [51] | Dog | Mixed | - | 25.7–29.2 | IV * | 24.8 | FIA-MS/MS | 24.1 | 17.9 g | 10100 | ||
| PO * | 19.1 | FIA-MS/MS | 77.7 | |||||||||
| Sirtori et al. (1978) [36] | Man | Healthy | 4 M 1 F | 64–81 | IV * | 926 mg | GC-MS | 6.13 | 4.65 | 7.81 b | 432 ** | |
| Pentikäinen et al. (1979) [40] | Man | Healthy | 1 M 2 F | 58–63 | IV * | 500 mg | LSC | 7.61 | 7.52 | 856 ** | ||
| 2 M 3 F | 56–80 | PO * | 500 mg | LSC | 16.3 | 7.06 | ||||||
| Tucker et al. (1981) [37] | Man | Healthy | 4 M | 64–83 | IV * | 250 mg | GC-EC | 10.1 | 7.83 | 511 ** | ||
| PO * | 500, 1500 mg | GC-EC | 18.9, 21.5 | 7.42, 7.16 | ||||||||
| Lee and Kwon (2004) [63] | Man | Healthy | 22 M | 55–89 | PO * | 500 mg | HPLC-UV | 16.2 | ||||
| Hustace et al. (2009) [64] | Horse | - | - | 531.8 | IV * | 6 g | HPLC-UV | 10.8 | 12.1 h | 2250 ** | ||
| PO * | 6 g | HPLC-UV | 152 |
| Definition | Estimate (CV%) | |
|---|---|---|
| Tissue-to-plasma partition coefficient | 1.29 (9.67) | |
| Total sum of fractional distribution parameters () | 0.0457 (8.12) | |
| Fractional distribution parameter for Tissue 1 | 0.0390 (8.91) | |
| Fractional distribution parameter for Tissue 2 | 0.00677 (7.59) c | |
| Fraction of total tissue volume for Tissue 1 | 0.172 (8.87) |
| Species (Body Weight, kg) | (mL/min/kg) | ||||
|---|---|---|---|---|---|
| Mouse (0.025) | 81.7 | 81.8 | 32.9 (6.05) | 65.8 (26.9) | 60 |
| Rat (0.25) | 23.6–26.4 | 20.8–21.3 | 23.5 (3.80) | 23.2 (4.00) | 24.8 |
| Rabbit (2) | 2.05 | 2.05 | 1.43 (14.5) | 1.30 (38.2) | 13.1 |
| Cat (5) | 2.5 | 2.55 | 2.90 (4.78) | 2.78 (7.11) | 13.8 |
| Monkey (7) | 11.2 | 11.0 | 12.5 (5.87) | 15.1 (7.71) | 9.61 |
| Minipig (14) | 9.7 | 9.18 | 6.08 (6.37) | 9.32 (9.72) | 9.36 |
| Dog (28) | 24.1 | 20.2 | 9.01 (4.15) | 19.3 (15.8) | 17.9 |
| Man (70) | 6.13–10.1 | 6.19–9.47 | 5.20 (3.77) | 7.74 (4.19) | 7.81 |
| Horse (530) | 10.8 | 8.25 | 5.51 (5.49) | 6.89 (11.0) | 12.1 |
| Tissue a | Kp | In Vitro PAMPA P | Tissue a | Kp | to In Vivo Profile | ||
|---|---|---|---|---|---|---|---|
| Kidney | 0.82 | 0.88 | 0.176 | Kidney | 0.82 | 0.0423 | 3.66 |
| Lung | 0.83 | 0.0648 | 0.199 | Heart | 0.82 | 0.00137 | 7.54 |
| Heart | 0.82 | 0.832 | 0.324 | Gut b | 0.81 | 0.0357 | 8.53 |
| Liver | 0.76 | 0.86 | 0.391 | Liver | 0.76 | 0.0393 | 8.56 |
| Adipose | 0.16 | 0.959 | 0.589 | Adipose | 0.16 | 0.0632 | 8.94 |
| Gut b | 0.81 | 0.975 | 0.636 | Lung | 0.83 | 0.0727 | 9.42 |
| Spleen | 0.81 | 0.588 | 0.893 | Muscle | 0.79 | 0.0179 | 25.1 |
| Bone c | 0.47 | 0.744 | 1.23 | Spleen | 0.81 | 0.0274 | 29.3 |
| Brain | 0.86 | 0.425 | 2.24 | Bone c | 0.47 | 0.0113 | 33.3 |
| Muscle | 0.79 | 1 | 4.84 | Brain | 0.86 | 0.1930 | 84.6 |
| Skin | 0.69 | 0.913 | 7.30 | Skin | 0.69 | 0.0486 | 137 |
| Estimate (CV%) | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Mouse | Rat | Rabbit | Cat | Monkey | Minipig | Dog | Man | Horse | |
| 0.921 (81.0) | 0.575 (10.2) | 0.479 (19.9) | 0.405 (20.6) | 0.615 (21.2) | 0.875 (20.5) | 4.31 (38.1) | 0.484 (13.3) | 0.175 (27.4) | |
| 1.44 (50.0) | 0.466 (9.62) | 2.15 (93.5) | 2.69 (18.6) | 3.27 (17.5) | 4.13 (20.8) | 7.53 (30.2) | 0.669 (11.9) | 0.884 (213) | |
| 0.379 (163) | 0.0694 (14.5) | 0.106 (36.2) | 0.0789 (38.5) | 0.158 (32.3) | 0.966 (34.9) | 0.765 (83.6) | 0.993 (44.4) | 0.106 (46.3) | |
| 0.239 (69.5) | 0.0157 (11.7) | 0.0184 (29.0) | 0.0321 (14.3) | 0.0482 (21.9) | 0.0472 (22.7) | 0.130 (30.8) | 0.171 (22.3) | 0.0455 (81.1) | |
| (mL/min) a | 1.65 (26.9) | 5.81 (4.00) | 2.60 (38.2) | 13.9 (7.11) | 106 (7.71) | 131 (9.72) | 540 (15.8) | 542 (4.19) | 3650 (11.0) |
| 0.995 (27.5) | 0.675 (7.28) | 0.334 (11.0) | 0.501 (8.63) | 0.157 (10.3) | 0.474 (13.6) | 0.203 (17.2) | 0.485 (5.54) | 0.0873 (11.7) | |
| (h−1) | 0.375 (15.4) | 0.246 (4.05) | 0.424 (16.0) | 0.402 (11.1) | 0.209 (9.71) | 0.390 (15.7) | 0.390 (13.0) | 0.253 (5.42) | 0.144 (11.7) |
| (L/kg) b | 1.21 (51.5) | 0.536 (7.48) | 1.49 (79.8) | 1.56 (16.9) | 1.89 (16.3) | 2.65 (18.8) | 6.00 (30.5) | 0.603 (7.36) | 0.574 (156) |
| Species | Tissue | . | Mean (Range) ** |
|---|---|---|---|
| No. of values | |||
| Mouse | Liver | 16 | 3.47 (1.72–7.10) |
| Brain | 5 | 0.213 (0.0354–0.257) | |
| Kidney | 12 | 8.74 (3.35–20.5) | |
| Muscle | 8 | 1.03 (0.359–2.06) | |
| Heart | 3 | 0.610 (0.519–0.712) | |
| Adipose | 1 | 0.471 | |
| Stomach | 4 | 6.38 (4.67–9.03) | |
| Small intestine | 6 | 11.3 (0.837–21.1) | |
| Colon | 4 | 7.72 (4.52–13.9) | |
| Salivary gland | 2 | 3.03 (2.60–3.45) | |
| Rat | Liver | 10 | 3.07 (0.368–6.83) |
| Brain | 5 | 0.8 (0.2–1.48) | |
| Kidney | 9 | 4.04 (0.128–5.92) | |
| Muscle | 2 | 0.597 (0.455–0.738) | |
| Spleen | 1 | 0.956 | |
| Gut | 1 | 4.63 | |
| No. of values | |||
| Rat | Blood | 2 | 1.18 (0.98–1.37) |
| Man | Blood | 2 | 1.03 (0.83–1.23) |
| (free fraction in the) | No. of values | ||
| Rat | Plasma | 3 | 0.873 (0.849–0.897) |
| Dog | Plasma | 3 | 0.904 (0.83–0.951) |
| Blood | 1 | 0.920 | |
| Man | Plasma | 6 | 0.938 (0.75–1.0) |
| Blood | 1 | 0.932 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jeong, Y.-S.; Jusko, W.J. Meta-Assessment of Metformin Absorption and Disposition Pharmacokinetics in Nine Species. Pharmaceuticals 2021, 14, 545. https://doi.org/10.3390/ph14060545
Jeong Y-S, Jusko WJ. Meta-Assessment of Metformin Absorption and Disposition Pharmacokinetics in Nine Species. Pharmaceuticals. 2021; 14(6):545. https://doi.org/10.3390/ph14060545
Chicago/Turabian StyleJeong, Yoo-Seong, and William J. Jusko. 2021. "Meta-Assessment of Metformin Absorption and Disposition Pharmacokinetics in Nine Species" Pharmaceuticals 14, no. 6: 545. https://doi.org/10.3390/ph14060545
APA StyleJeong, Y. -S., & Jusko, W. J. (2021). Meta-Assessment of Metformin Absorption and Disposition Pharmacokinetics in Nine Species. Pharmaceuticals, 14(6), 545. https://doi.org/10.3390/ph14060545