
Evaluation for Potential Drug–Drug Interaction of MT921 Using In Vitro Studies and Physiologically–Based Pharmacokinetic Models

 , , ,
, , ,  and
and
Abstract
:1. Introduction
2. Results
2.1. In Vitro Potential on Transporters
2.1.1. Substrate Specificity of MT921 for OATP1B1, OATP1B3, OAT3, NTCP, and ASBT
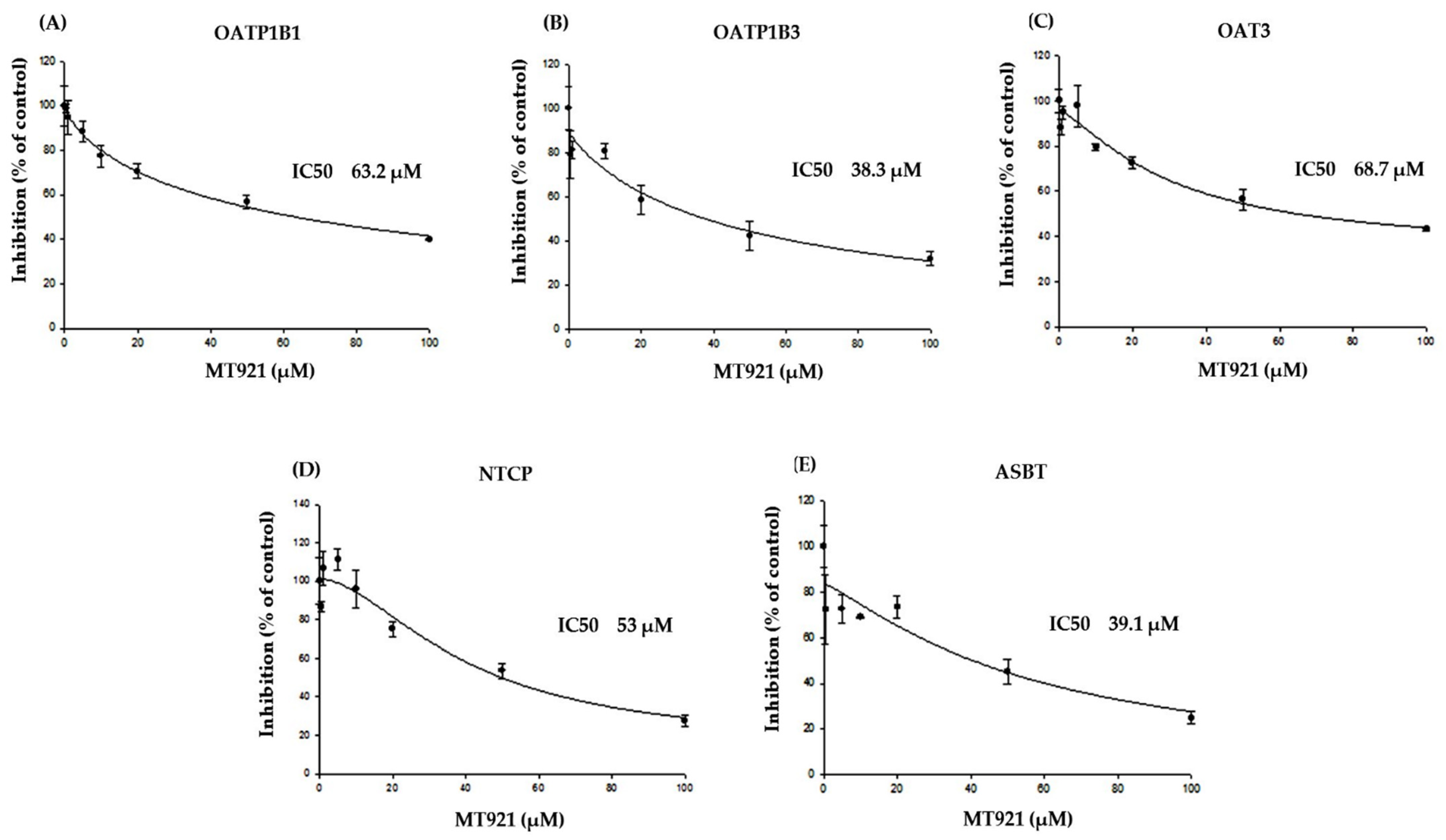
2.1.2. Inhibitory Effects of MT921 on The Transport Activities OATP1B1, OATP1B3, OAT3, NTCP, and ASBT
2.2. PBPK Modeling and Simulation
2.2.1. MT921 Model
2.2.2. Amlodipine Model
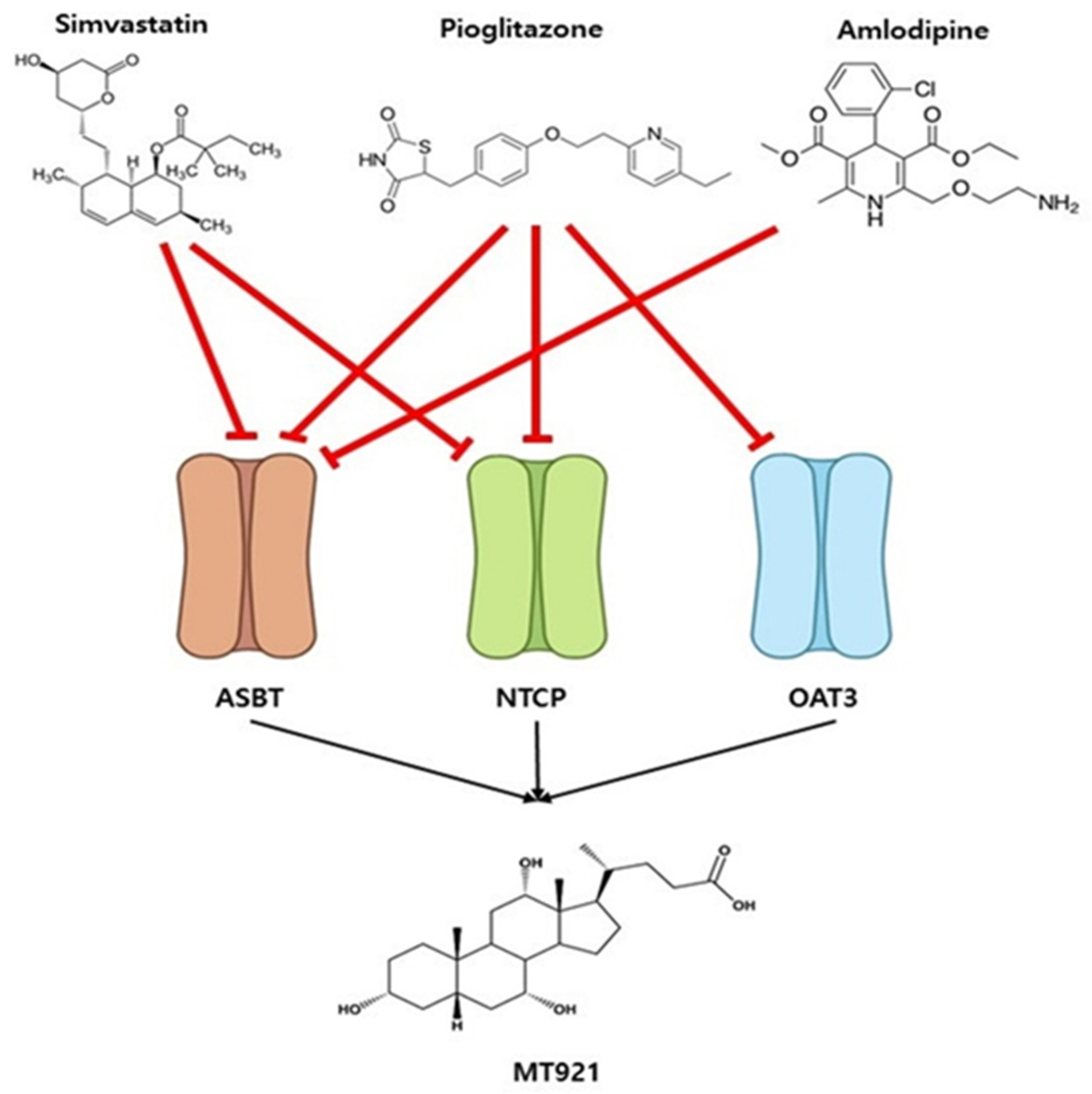
2.2.3. Prediction of Potential DDI between MT921 and Chronic Disease Drugs
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. In Vitro Effect on Transporters
4.2.1. Substrate Specificity of MT921 for OATP1B1, OATP1B3, OAT3, NTCP, and ASBT
4.2.2. Inhibitory Effects of MT921 on the Transport Activities of OATP1B1, OATP1B3, OAT3, NTCP, and ASBT
4.2.3. LC-MS/MS Analysis
4.2.4. Data Analysis
4.3. PBPK Modeling and Simulation
4.3.1. Software
4.3.2. PBPK Model Development
4.3.3. MT921 Model Development
4.3.4. Amlodipine Model Development
4.3.5. PBPK Model Evaluation
4.3.6. Sensitivity Analysis
4.3.7. Prediction of Potential DDI between MT921 and the Inhibitors of Uptake Transporters
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chiang, J.Y.L. Bile acids: Regulation of synthesis. J. Lipid Res. 2009, 50, 1955–1966. [Google Scholar] [CrossRef] [Green Version]
- Pavlović, N.; Goločorbin-Kon, S.; Ðanić, M.; Stanimirov, B.; Al-Salami, H.; Stankov, K.; Mikov, M. Bile Acids and Their Derivatives as Potenial Modifiers of Drug Release and Pharmacokinetic Profile. Front. Pharmacol. 2018, 9, 1–23. [Google Scholar] [CrossRef]
- Chiang, J.Y.L.; Ferrell, J.M. Up to date on cholesterol 7α-hydroxylase (CYP7A1) in bile acid synthesis. Liver Res. 2020, 4, 47–63. [Google Scholar] [CrossRef]
- Chiang, J.Y.L.; Li, T. Bile Acid Signaling in metabolic Disease and Drug Therapy. Pharmacol. Rev. 2014, 66, 948–983. [Google Scholar] [CrossRef] [Green Version]
- Dawson, P.A.; Lan, T.; Rao, A. Bile acid transporters. J. Lipid Res. 2009, 50, 2340–2357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alrefai, W.A.; Gill, R.K. Bile Acid Transporters: Structure, Function, Regulation and Pathophysiological Implications. Pharm. Res. 2007, 24, 1803–1823. [Google Scholar] [CrossRef]
- Thakkar, N.; Slizgi, J.R.; Brouwer, K.L.R. Effect of Liver Disease on Hepatic Transporter Expression and Function. J. Pharm. Sci. 2017, 106, 2282–2294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiang, J.Y.L. Bile Acid Metabolism and Signaling. Compr. Physiol. 2013, 3, 1191–1212. [Google Scholar] [CrossRef] [Green Version]
- Bang, J.S.; Lee, Y.J.; Jeong, J.H.; Sohn, U.D. Roles of Bile Acid as an Active Biological Substance. Kor. J. Clin. Pharm. 2011, 21, 49–56. [Google Scholar]
- Humphrey, S.; Beleznay, K.; Beleznay, J.D.A. Sodium Deoxycholate for Submental Contouring. Skin Ther. Lett. 2016, 21, 1–4. [Google Scholar]
- Fang, Z.-Z.; He, R.-R.; Cao, Y.-F.; Tanaka, N.; Jiang, C.; Krausz, K.W.; Qi, Y.; Dong, P.-P.; Ai, C.-Z.; Sun, X.-Y.; et al. A model of in vitro UDP-glucuronosyltransferase inhibition by bile acids predicts possible metabolic disorders. J. Lipid Res. 2013, 54, 3334–3344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ämlöv, J.; Sundström, J.; Schmitz, G.; Ingelsson, E.; Lind, L. Impact of BMI and the Metabolic Syndrome on the Risk of Diabetes in Middle-Aged Men. Diabetes Care 2011, 34, 61–65. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, N.T.; Magno, C.P.; Lane, K.T.; Hinojosa, M.W.; Lane, J.S. Association of Hypertension, Diabetes, Dyslipidemia, and Metabolic Syndrome with Obesity: Findings from the National Health and Nutrition Examination Survey, 1999 to 2004. Kor. J. Clin. Pharm. 2008, 207, 928–934. [Google Scholar] [CrossRef] [PubMed]
- Center for Drug Evaluation and Research (CDER). Guidance for Industry: In Vitro Drug Interaction Studies-Cytochrome P450 Enzyme- and Transporter-Mediated Drug Interactions; Food and Drug Administration: Silver Spring, MD, USA, 2020; Available online: http://www.fda.gov/media/134582/download (accessed on 10 January 2021).
- Yi, S.; Kim, T.-E.; Yoon, S.H.; Cho, J.-Y.; Shin, S.-G.; Jang, I.-J.; Yu, K.-S. Pharmacokinetic Interaction of Fimasartan, a New Angiotensin II Receptor Antagonist, with Amlodipine in Healthy Volunteers. J. Cardiovasc. Pharmacol. 2011, 57, 682–689. [Google Scholar] [CrossRef] [PubMed]
- Williams, D.M.; Cubeddu, L.X. Amlodipine Pharmacokinetics in Healthy Volunteers. J. Clin. Pharmacol. 1988, 28, 990–994. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Jia, J.; Liu, G.; Li, S.; Lu, C.; Liu, Y.; Yu, C. Pharmacokinetics and Bioequivalence Evaluation of Two Formulations of 10-Mg Amlodipine Besylate: An Open-Label, Single-Dose, Randomized, Two-Way Crossover Study in Healthy Chinese Male Volunteers. Clin. Ther. 2009, 31, 777–783. [Google Scholar] [CrossRef]
- Faulkner, J.K.; McGibney, D.; Chasseaud, L.F.; Perry, J.L.; Taylor, I.W. The Pharmacokinetics of Amlodipine in Healthy Volunteers after Single Intravenous and Oral Doses and after 14 Repeated Oral Doses given Once Daily. Br. J. Clin. Pharmacol. 1986, 22, 21–25. [Google Scholar] [CrossRef] [Green Version]
- Jeon, J.Y.; Kim, J.R.; Chung, J.Y.; Cho, J.-Y.; Yu, K.-S.; Jang, I.-J.; Shin, S.G. Pharmacokinetic and Pharmacodynamic Comparative Study of Amlodipine Adipate and Amlodipine Besylate. J. Korean Soc. Clin. Pharmacol. Ther. 2006, 14, 106–115. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.-R.; Kim, S.; Huh, W.; Ko, J.-W. No Pharmacokinetic Interactions between Candesartan and Amlodipine Following Multiple Oral Administrations in Healthy Subjects. Drug Des. Devel. Ther. 2018, 12, 2475–2483. [Google Scholar] [CrossRef] [Green Version]
- Abad-Santos, F.; Novalbos, J.; Gálvez-Múgica, M.-A.; Gallego-Sandín, S.; Almeida, S.; Vallée, F.; García, A.G. Assessment of Sex Differences in Pharmacokinetics and Pharmacodynamics of Amlodipine in a Bioequivalence Study. Pharmacol. Res. 2005, 51, 445–452. [Google Scholar] [CrossRef]
- Choi, S.; Jeon, S.; Yim, D.-S.; Han, S. Contribution of Trough Concentration Data in the Evaluation of Multiple-Dose Pharmacokinetics for Drugs with Delayed Distributional Equilibrium and Long Half-Life. Drug Des. Devel Ther. 2020, 14, 811–821. [Google Scholar] [CrossRef]
- Ma, Y.; Qin, F.; Sun, X.; Lu, X.; Li, F. Determination and Pharmacokinetic Study of Amlodipine in Human Plasma by Ultra Performance Liquid Chromatography-Electrospray Ionization Mass Spectrometry. J. Pharm. Biomed Anal. 2007, 43, 1540–1545. [Google Scholar] [CrossRef] [PubMed]
- Josefsson, M.; Zackrisson, A.L.; Ahlner, J. Effect of Grapefruit Juice on the Pharmacokinetics of Amlodipine in Healthy Volunteers. Eur. J. Clin. Pharmacol. 1996, 51, 189–193. [Google Scholar] [CrossRef] [PubMed]
- Han, J.M.; Yee, J.; Chung, J.E.; Lee, K.E.; Park, K.; Gwak, H.S. Effects of Cytochrome P450 Oxidoreductase Genotypes on the Pharmacokinetics of Amlodipine in Healthy Korean Subjects. Mol. Genet Genomic. Med. 2020, 8, e1201. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Wang, Y.; Lin, S.; Fang, L.; Lou, S.; Zhao, D.; Zhu, J.; Yang, Q.; Wang, Y. Evaluation of Pharmacokinetics and Safety with Bioequivalence of Amlodipine in Healthy Chinese Volunteers: Bioequivalence Study Findings. J. Clin. Lab. Anal. 2020, 34, e23228. [Google Scholar] [CrossRef] [PubMed]
- Kirigaya, Y.; Shiramoto, M.; Ishizuka, T.; Uchimaru, H.; Irie, S.; Kato, M.; Shimizu, T.; Nakatsu, T.; Nishikawa, Y.; Ishizuka, H. Pharmacokinetic Interactions of Esaxerenone with Amlodipine and Digoxin in Healthy Japanese Subjects. BMC Pharmacol. Toxicol. 2020, 21, 55. [Google Scholar] [CrossRef] [PubMed]
- Sailer, R.; Arnold, P.; Erenmemişoğlu, A.; Martin, W.; Tamur, U.; Kanzik, I.; Hincal, A.A. Pharmacokinetics and Bioequivalence Study of a Generic Amlodipine Tablet Formulation in Healthy Male Volunteers. Arzneimittelforschung 2007, 57, 462–466. [Google Scholar] [CrossRef] [PubMed]
- Park, J.-Y.; Kim, K.-A.; Lee, G.-S.; Park, P.-W.; Kim, S.-L.; Lee, Y.-S.; Lee, Y.-W.; Shin, E.-K. Randomized, Open-Label, Two-Period Crossover Comparison of the Pharmacokinetic and Pharmacodynamic Properties of Two Amlodipine Formulations in Healthy Adult Male Korean Subjects. Clin. Ther. 2004, 26, 715–723. [Google Scholar] [CrossRef]
- Baier, V.; Cordes, H.; Thiel, C.; Castell, J.V.; Neumann, U.P.; Blank, L.M.; Kuepfer, L. A Physiology-Based Model of Human Bile Acid Metabolism for Predicting Bile Acid Tissue Levels After Drug Administration in Healthy Subjects and BRIC Type 2 Patients. Front. Physiol. 2019, 10. [Google Scholar] [CrossRef] [Green Version]
- Mukherjee, D.; Zha, J.; Menon, R.M.; Shebley, M. Guiding Dose Adjustment of Amlodipine after Co-Administration with Ritonavir Containing Regimens Using a Physiologically-Based Pharmacokinetic/Pharmacodynamic Model. J. Pharm. Pharm. 2018, 45, 443–456. [Google Scholar] [CrossRef] [Green Version]
- Rhee, S.; Lee, H.A.; Lee, S.; Kim, E.; Jeon, I.; Song, I.-S.; Yu, K.-S. Physiologically Based Pharmacokinetic Modeling of Fimasartan, Amlodipine, and Hydrochlorothiazide for the Investigation of Drug–Drug Interaction Potentials. Pharm. Res. 2018, 35, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Engelking, L.R.; Barnes, S.; Dasher, C.A.; Naftel, D.C.; Hirschowitz, B.I. Radiolabelled Bile Acid Clearance in Control Subjects and Patients with Liver Disease. Clin. Sci. 1979, 57, 499–508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roda, A.; Minutello, A.; Angellotti, M.; Fini, A. Bile Acid Structure-Activity Relationship: Evaluation of Bile Acid Lipophilicity Using 1-Octanol/Water Partition Coefficient and Reverse Phase HPLC. J. Lipid Res. 1990, 31, 1433–1443. [Google Scholar] [CrossRef]
- Salvioli, G.; Lugli, R.; Pradelli, J.M.; Gigliotti, G. Bile Acid Binding in Plasma: The Importance of Lipoproteins. FEBS Lett. 1985, 187, 272–276. [Google Scholar] [CrossRef] [Green Version]
- Open Systems Pharmacology Suite Community. Open Systems Pharmacology Suite Manual. Available online: https://docs.open-systems-pharmacology.org/shared-tools-and-example-workflows/sensitivity-analysis (accessed on 26 May 2021).
- Zhu, Y.; Wang, F.; Li, Q.; Zhu, M.; Du, A.; Tang, W.; Chen, W. Amlodipine Metabolism in Human Liver Microsomes and Roles of CYP3A4/5 in the Dihydropyridine Dehydrogenation. Drug Metab. Dispos. 2014, 42, 245–249. [Google Scholar] [CrossRef] [Green Version]
- Zheng, X. Inhibition and Substrate Requirements of Human Apical Sodium-Dependent Bile Acid Transporter (ASBT) and Its Potential as a Prodrug Target. Ph.D. Thesis, University of Maryland, Baltimore, ML, USA, 2010. [Google Scholar]
- Dong, Z.; Ekins, S.; Polli, J.E. Structure-Activity Relationship for FDA Approved Drugs as Inhibitors of the Human Sodium Taurocholate Cotransporting Polypeptide (NTCP). Mol. Pharm. 2013, 10, 1008–1019. [Google Scholar] [CrossRef]
- Dong, Z.; Ekins, S.; Polli, J.E. Quantitative NTCP Pharmacophore and Lack of Association between DILI and NTCP Inhibition. Eur. J. Pharm. Sci. 2015, 66, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Zou, L.; Matsson, P.; Stecula, A.; Ngo, H.X.; Zur, A.A.; Giacomini, K.M. Drug Metabolites Potently Inhibit Renal Organic Anion Transporters, OAT1 and OAT3. J. Pharm. Sci. 2021, 110, 347–353. [Google Scholar] [CrossRef]
- Türk, D.; Hanke, N.; Lehr, T. A Physiologically-Based Pharmacokinetic Model of Trimethoprim for MATE1, OCT1, OCT2, and CYP2C8 Drug-Drug-Gene Interaction Predictions. Pharmaceutics 2020, 12, 1074. [Google Scholar] [CrossRef]
- Zhou, Y.-T.; Yu, L.-S.; Zeng, S.; Huang, Y.-W.; Xu, H.-M.; Zhou, Q. Pharmacokinetic Drug-Drug Interactions between 1,4-Dihydropyridine Calcium Channel Blockers and Statins: Factors Determining Interaction Strength and Relevant Clinical Risk Management. Ther. Clin. Risk Manag. 2014, 10, 17–26. [Google Scholar] [CrossRef] [Green Version]
- Xin, H.-W.; Schwab, M.; Klotz, U. Transport studies with 5-aminosalicylate. Eur. J. Clin. Pharmacol. 2006, 62, 871–875. [Google Scholar] [CrossRef] [PubMed]
- Hirano, M.; Maeda, K.; Shitara, Y.; Sugiyama, Y. Contribution of OATP2 (OATP1B1) and OATP8 (OATP1B3) to the hepatic uptake of pitavastatin in humans. J. Pharmacol. Exp. Ther. 2004, 311, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Ueo, H.; Motohashi, H.; Katsura, T.; Inui, K.-i. Human organic anion transporter hOAT3 is a potent transporter of cephalosporin antibiotics, in comparison with hOAT1. Biochem. Pharmacol. 2005, 70, 1104–1113. [Google Scholar] [CrossRef]
- Maeda, K.; Kambara, M.; Tian, Y.; Hofmann, A.F.; Sugiyama, Y. Uptake of ursodeoxycholate and its conjugates by human hepatocytes: Role of Na+-taurocholate cotransporting polypeptide (NTCP), organic anion transporting polypeptide (OATP) 1B1 (OATP-C), and OATP1B3 (OATP8). Mol. Pharm. 2006, 3, 70–77. [Google Scholar] [CrossRef]
- Ho, R.H.; Leake, B.F.; Urquhart, B.L.; Gregor, J.C.; Dawson, P.A.; Kim, R.B. Functional characterization of genetic variants in the apical sodium-dependent bile acid transporter (ASBT.; SLC10A2). J. Gastroenterol. Hepatol. 2011, 26, 1740–1748. [Google Scholar] [CrossRef] [Green Version]
- Craddock, A.L.; Love, M.W.; Daniel, R.W.; Kirby, L.C.; Walters, H.C.; Wong, M.H. Expression and transport properties of the human ileal and renal sodium-dependent bile acid transporter. Am. J. Physiol. 1998, 274, G157–G169. [Google Scholar] [CrossRef] [PubMed]
- Scherer, M.; Gnewuch, C.; Schmitz, G.; Liebisch, G. Rapid quantification of bile acids and their conjugates in serum by liquid chromatography-tandem mass spectrometry. J. Chromatogr. B. 2009, 877, 3920–3925. [Google Scholar] [CrossRef]
- Koo, T.-S.; Kwon, H.-J.; Kim, M.-H.; Kim, D.-D.; Shim, C.-K.; Chung, S.-J.; Chong, S. Functional impairment of rat taurine transporter by activation of nitrogen oxide through superoxide. Drug. Metab. Pharmacokinet. 2012, 27, 286–293. [Google Scholar] [CrossRef] [Green Version]
- Kwon, M.; Choi, Y.A.; Choi, M.-K.; Song, I.-S. Organic cation transporter-mediated drug-drug interaction potential between berberine and metformin. Arch. Pharm. Res. 2015, 38, 849–856. [Google Scholar] [CrossRef]
- Rohatgi, A. WebPlotDigitizer; Web Based Tool to Extract Data from Plot, Images, and Mapss Version 4.4. (Pacifica, California, USA). 2020. Available online: http://automeris.io/WebPlotDigitizer (accessed on 5 May 2021).
- Meyer, M.; Schneckener, S.; Ludewing, B.; Kuepfer, L.; Lippert, J. Using expression data for Quantification of Active Processes in Physiologically Based Pharmacokinetic Modeling. Drug Metab. Dispos. 2012, 40, 892–901. [Google Scholar] [CrossRef]
- Schmitt, W. General Approach for the Calculation of Tissue to Plasma Partition Coefficients. Toxicol. In Vitro 2008, 22, 457–467. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, G. Anatomical and Physiological Characteristics for Asian Reference Man-Male and Female of Different Age: Tanaka Model; NIRS-M-115; Division of Radioecology, National Institute of Radiological Sciences: Chiba, Japan, 1996. [Google Scholar]
- Valentin, J. Basic Anatomical and Physiological Data for Use in Radiological Protection: Reference Values: ICRP Publication 89. ICRP 2002, 32, 1–277. [Google Scholar] [CrossRef]
- Rodgers, T.; Leahy, D.; Rowland, M. Physiologically Based Pharmacokinetic Modeling 1: Predicting the Tissue Distribution of Moderate-to-Strong Bases. J. Pharm. Sci. 2005, 94, 1259–1276. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.J.; Tanna, S.; Sahota, T. In Vivo Study of a Polymeric Glucose-Sensitive Insulin Delivery System Using a Rat Model. J. Pharm. Sci. 2010, 99, 4215–4227. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Ekins, S.; Raufman, J.-P.; Polli, J.E. Computational Models for Drug Inhibition of the Human Apical Sodium-Dependent Bile Acid Transporter. Mol. Pharm. 2009, 6, 1591–1603. [Google Scholar] [CrossRef] [Green Version]
- Hanke, N.; Teifel, M.; Moj, D.; Wojtyniak, J.-G.; Britz, H.; Aicher, B.; Sindermann, H.; Ammer, N.; Lehr, T. A Physiologically Based Pharmacokinetic (PBPK) Parent-Metabolite Model of the Chemotherapeutic Zoptarelin Doxorubicin-Integration of in Vitro Results, Phase I and Phase II Data and Model Application for Drug-Drug Interaction Potential Analysis. Cancer Chemother Pharmacol. 2018, 81, 291–304. [Google Scholar] [CrossRef]
- Türk, D.; Hanke, N.; Wolf, S.; Frechen, S.; Eissing, T.; Wendl, T.; Schwab, M.; Lehr, T. Physiologically Based Pharmacokinetic Models for Prediction of Complex CYP2C8 and OATP1B1 (SLCO1B1) Drug-Drug-Gene Interactions: A Modeling Network of Gemfibrozil, Repaglinide, Pioglitazone, Rifampicin, Clarithromycin and Itraconazole. Clin. Pharm. 2019, 58, 1595–1607. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Concentration (µM) | Transporter | Uptake (pmol/mg protein/min) | Uptake Ratio (Transporter vs. Mock) |
|---|---|---|---|
| 10 | Mock | 7.61 ± 0.91 | 1.00 |
| HEK 293-OATP1B1 | 12.68 ± 0.64 | 1.67 | |
| 100 | Mock | 72.15 ± 3.91 | 1.00 |
| HEK 293-OATP1B1 | 72.98 ± 4.46 | 1.01 | |
| 10 | Mock | 7.61 ± 0.91 | 1.00 |
| HEK 293-OATP1B3 | 23.24 ± 1.08 | 3.05 | |
| 100 | Mock | 72.15 ± 3.91 | 1.00 |
| HEK 293-OATP1B3 | 126.56 ± 5.44 | 1.75 | |
| 10 | Mock | 7.61 ± 0.91 | 1.00 |
| HEK 293-OAT3 | 15.46 ± 2.09 | 2.03 | |
| 100 | Mock | 72.15 ± 3.91 | 1.00 |
| HEK 293-OAT3 | 63.53 ± 5.35 | 0.88 | |
| 10 | Mock | 4.85 ± 0.55 | 1.00 |
| HEK 293-NTCP | 56.74 ± 6.66 | 11.71 | |
| 100 | Mock | 57.16 ± 0.67 | 1.00 |
| HEK 293-NTCP | 215.08 ± 8.65 | 3.76 | |
| 10 | Mock | 4.38 ± 0.42 | 1.00 |
| HEK 293-ASBT | 78.33 ± 5.66 | 17.89 | |
| 100 | Mock | 50.22 ± 5.65 | 1.00 |
| HEK 293-ASBT | 253.49 ± 13.66 | 5.05 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ryu, H.-j.; Moon, H.-k.; Lee, J.; Yang, G.-h.; Yang, S.-y.; Yun, H.-y.; Chae, J.-w.; Kang, W.-h. Evaluation for Potential Drug–Drug Interaction of MT921 Using In Vitro Studies and Physiologically–Based Pharmacokinetic Models. Pharmaceuticals 2021, 14, 654. https://doi.org/10.3390/ph14070654
Ryu H-j, Moon H-k, Lee J, Yang G-h, Yang S-y, Yun H-y, Chae J-w, Kang W-h. Evaluation for Potential Drug–Drug Interaction of MT921 Using In Vitro Studies and Physiologically–Based Pharmacokinetic Models. Pharmaceuticals. 2021; 14(7):654. https://doi.org/10.3390/ph14070654
Chicago/Turabian StyleRyu, Hyo-jeong, Hyun-ki Moon, Junho Lee, Gi-hyeok Yang, Sung-yoon Yang, Hwi-yeol Yun, Jung-woo Chae, and Won-ho Kang. 2021. "Evaluation for Potential Drug–Drug Interaction of MT921 Using In Vitro Studies and Physiologically–Based Pharmacokinetic Models" Pharmaceuticals 14, no. 7: 654. https://doi.org/10.3390/ph14070654
APA StyleRyu, H. -j., Moon, H. -k., Lee, J., Yang, G. -h., Yang, S. -y., Yun, H. -y., Chae, J. -w., & Kang, W. -h. (2021). Evaluation for Potential Drug–Drug Interaction of MT921 Using In Vitro Studies and Physiologically–Based Pharmacokinetic Models. Pharmaceuticals, 14(7), 654. https://doi.org/10.3390/ph14070654