
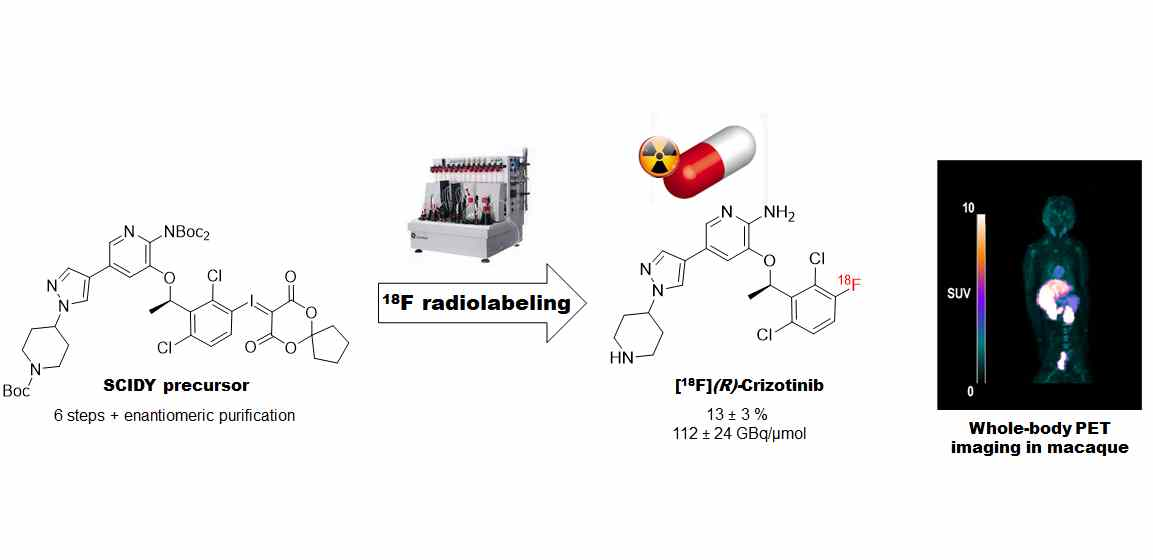
Isotopic Radiolabeling of Crizotinib with Fluorine-18 for In Vivo Pet Imaging

 , , , , , and
, , , , , and
Abstract
:
1. Introduction
2. Results and Discussion
2.1. Chemistry
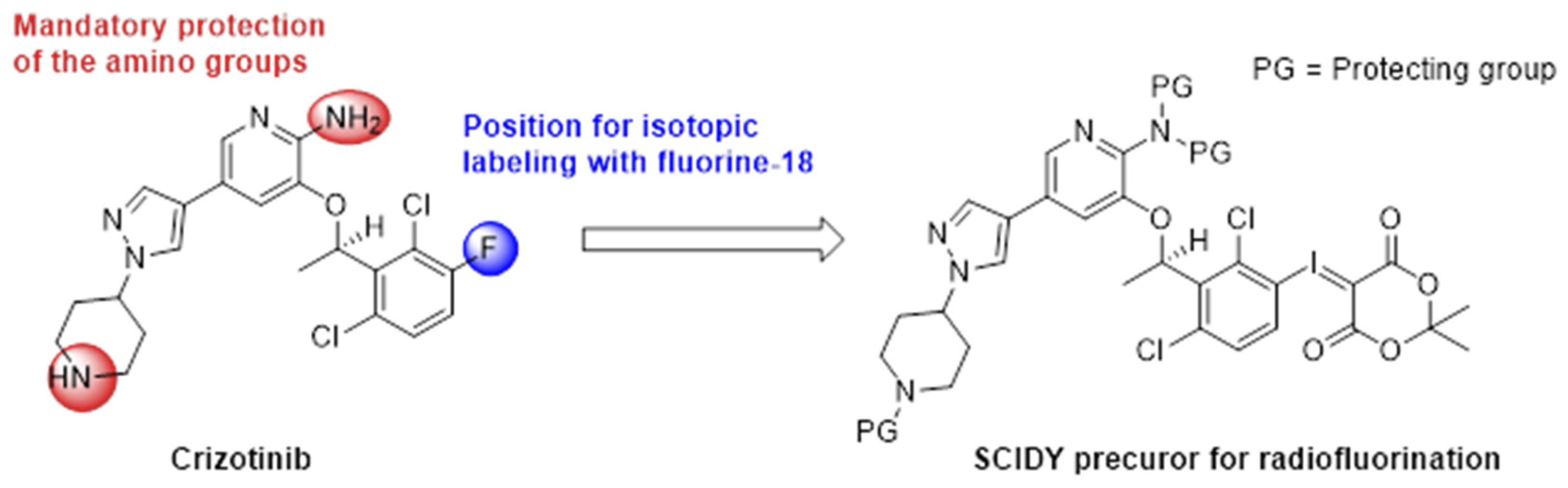
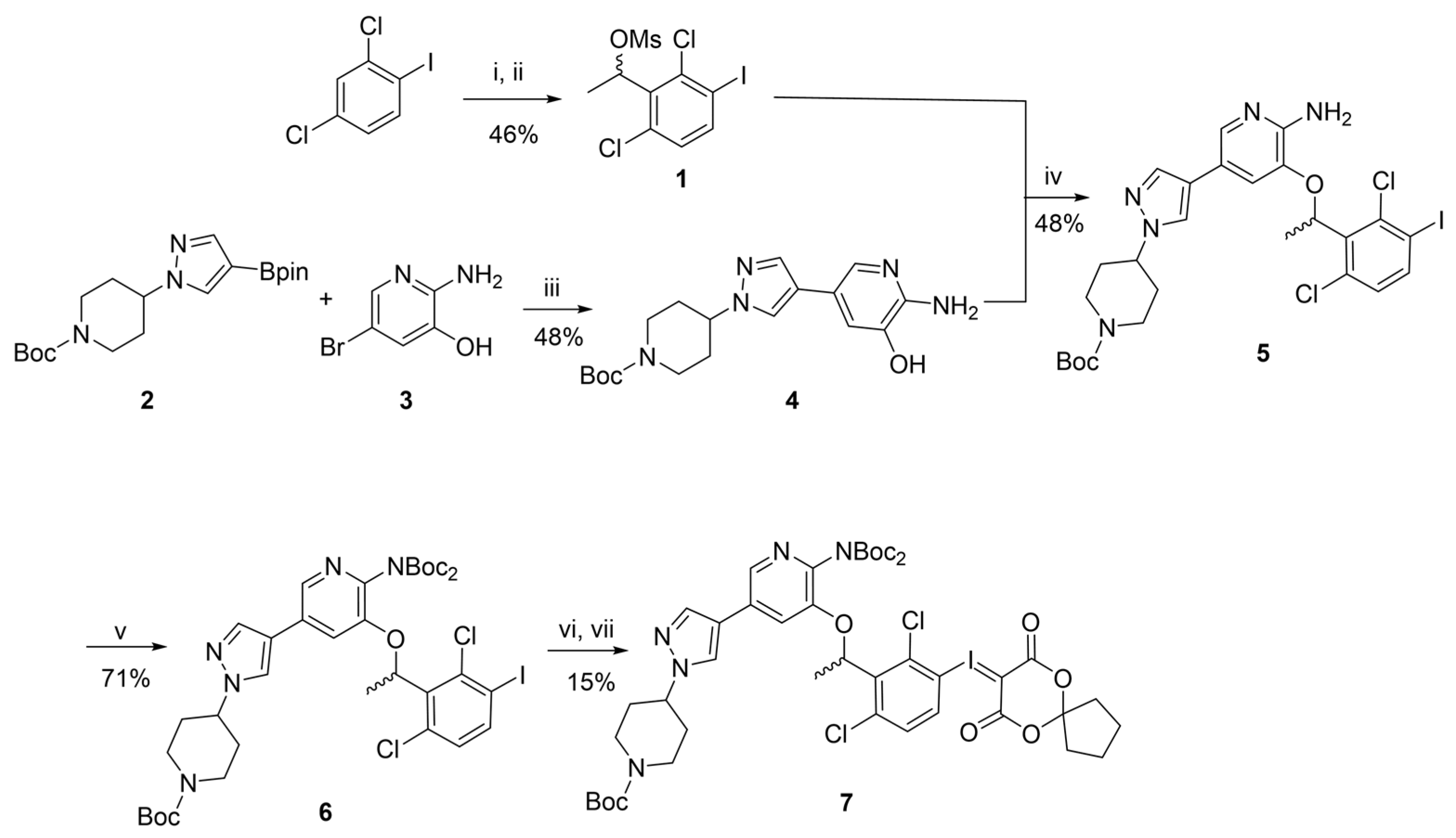
2.1.1. Synthesis of the Racemic Precursor
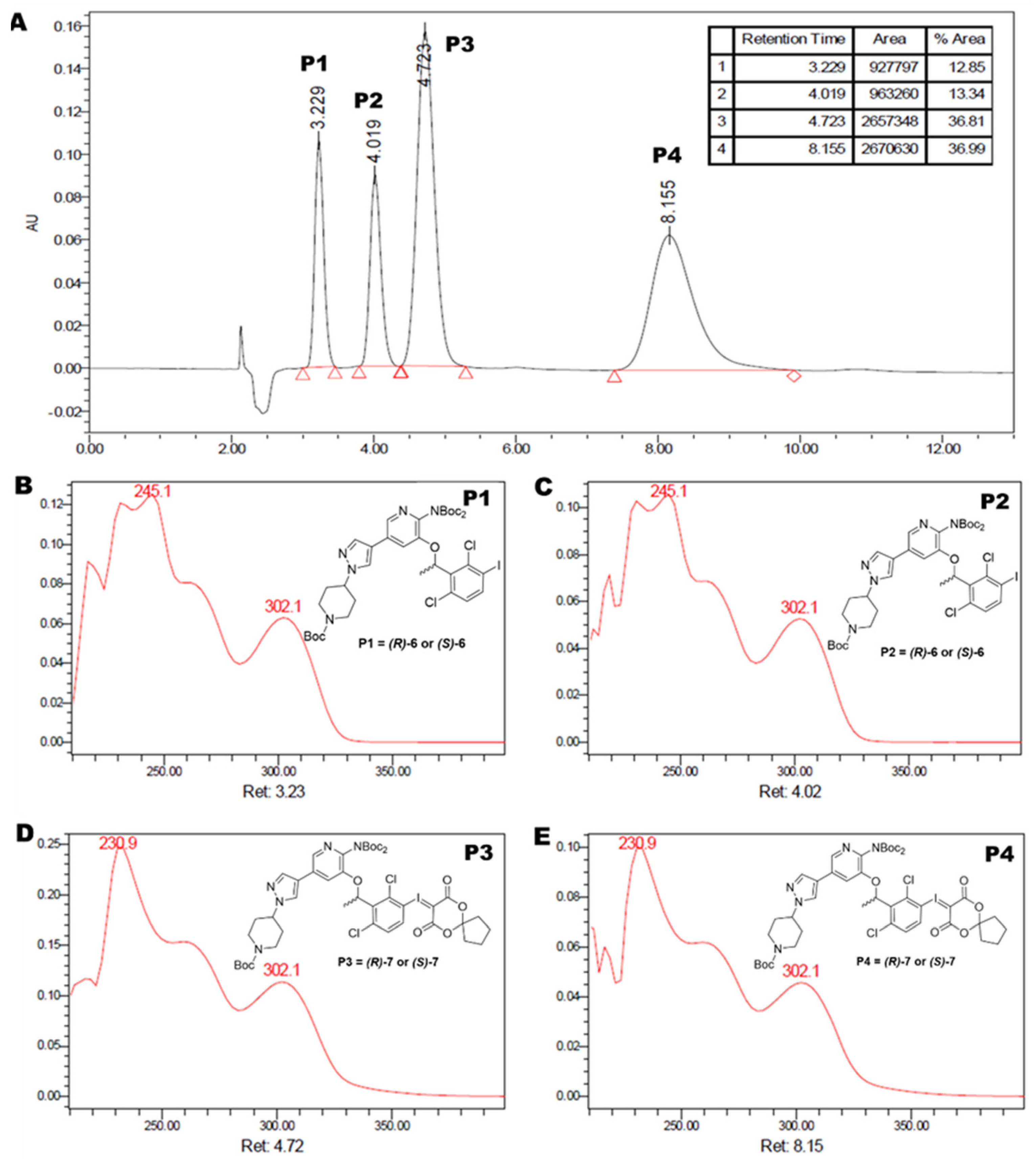
2.1.2. Enantiomeric Purification of the Precursor
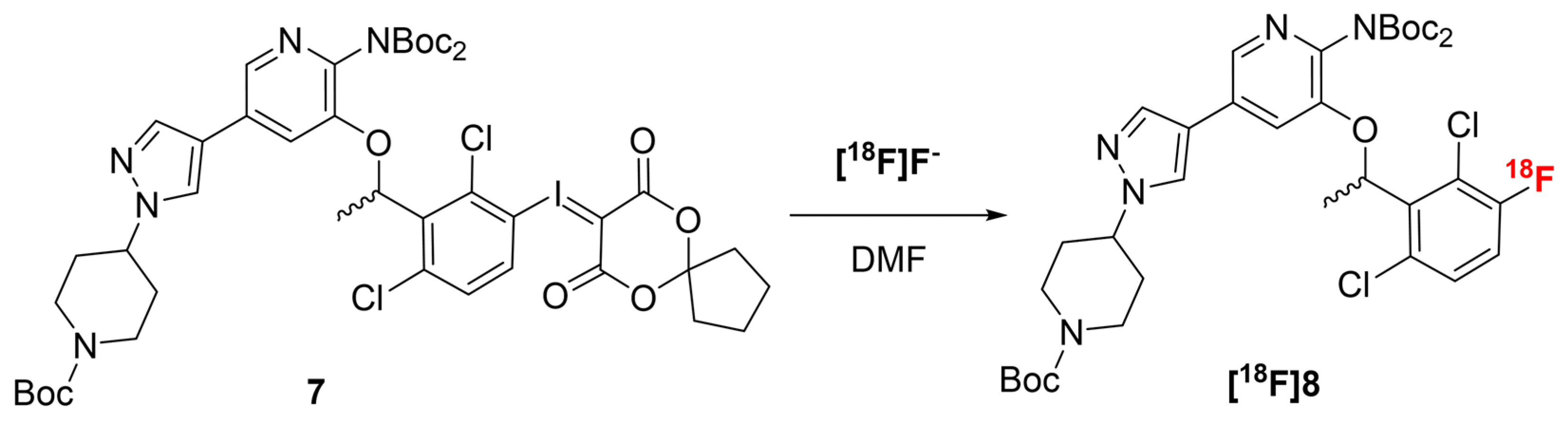
2.2. Radiochemistry
2.2.1. Radiofluorination Optimization
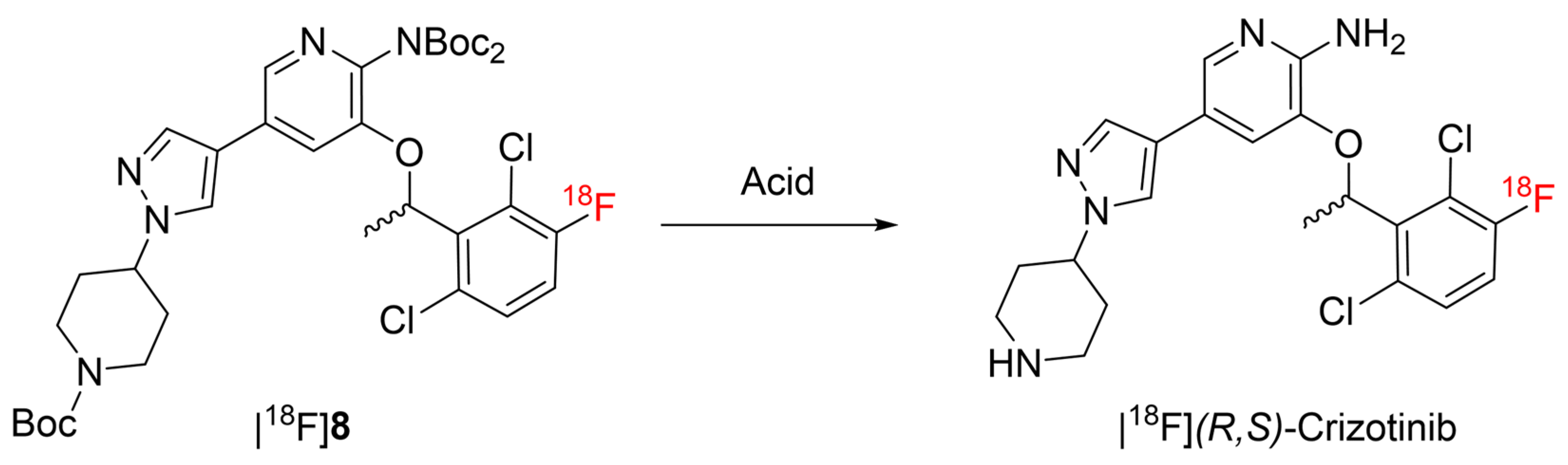
2.2.2. Deprotection Optimization
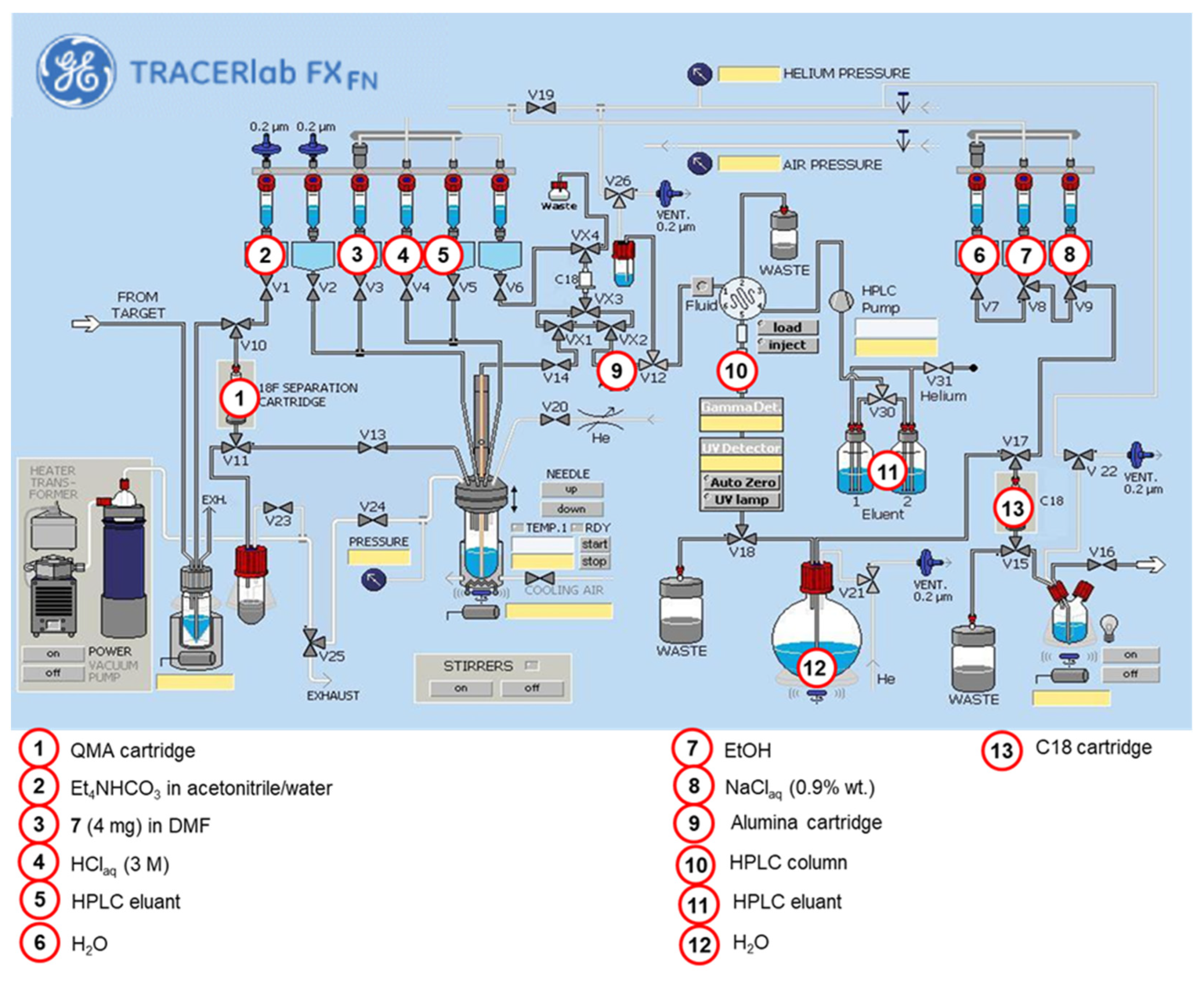
2.2.3. Automated Radiosynthesis of [18F](R,S)-crizotinib
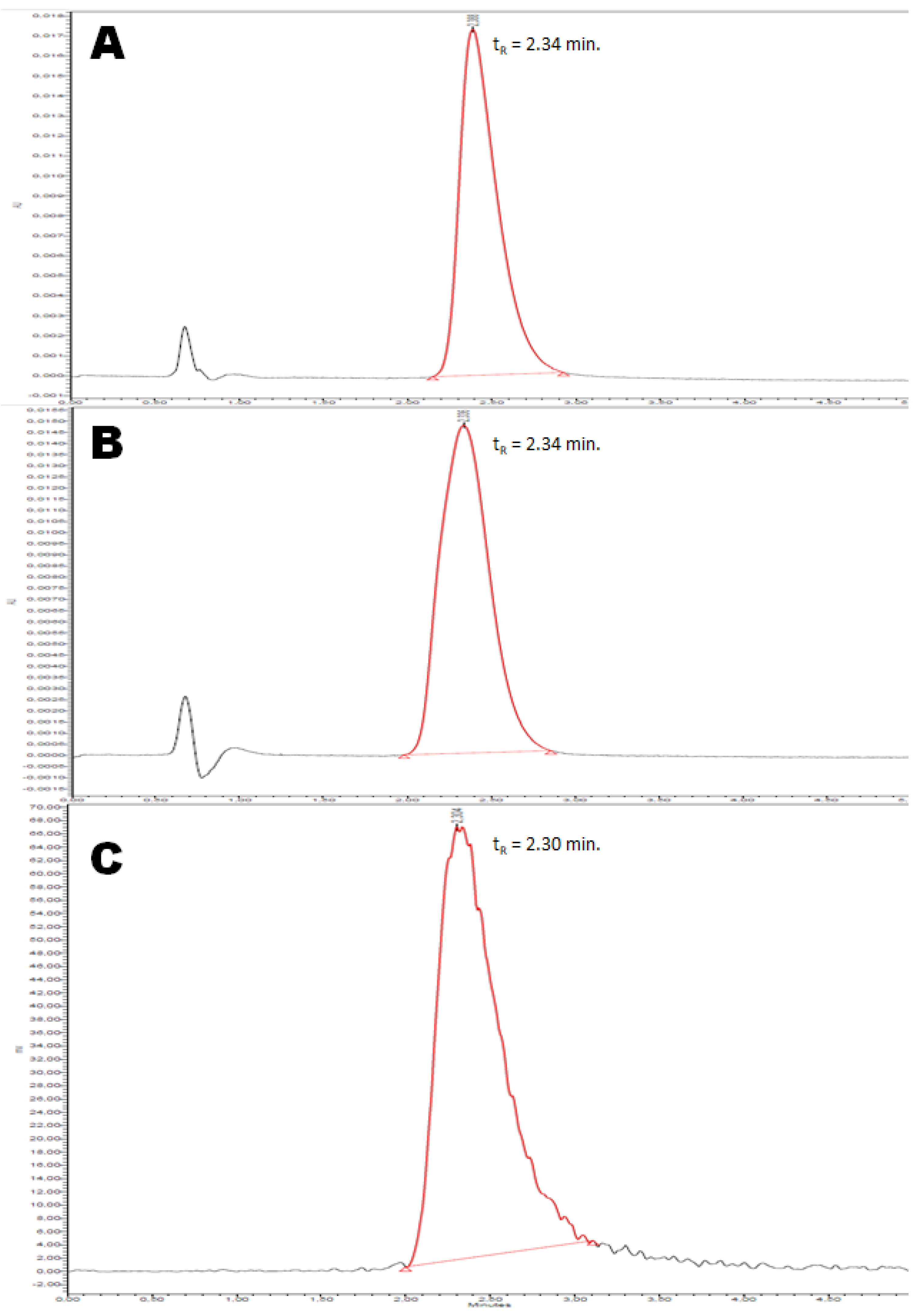
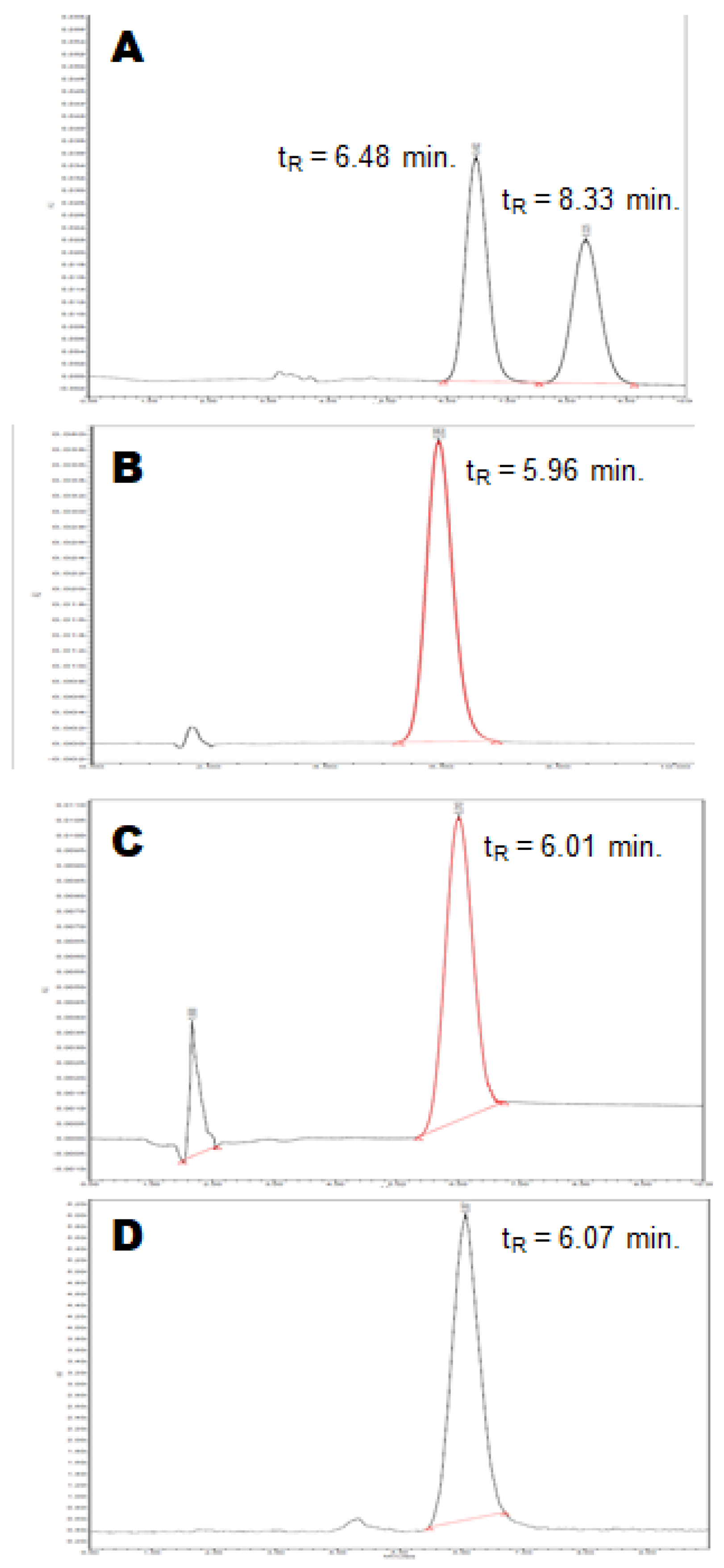
2.2.4. Quality Control
2.2.5. Radiosynthesis of Enantiomerically Pure [18F](R)-crizotinib
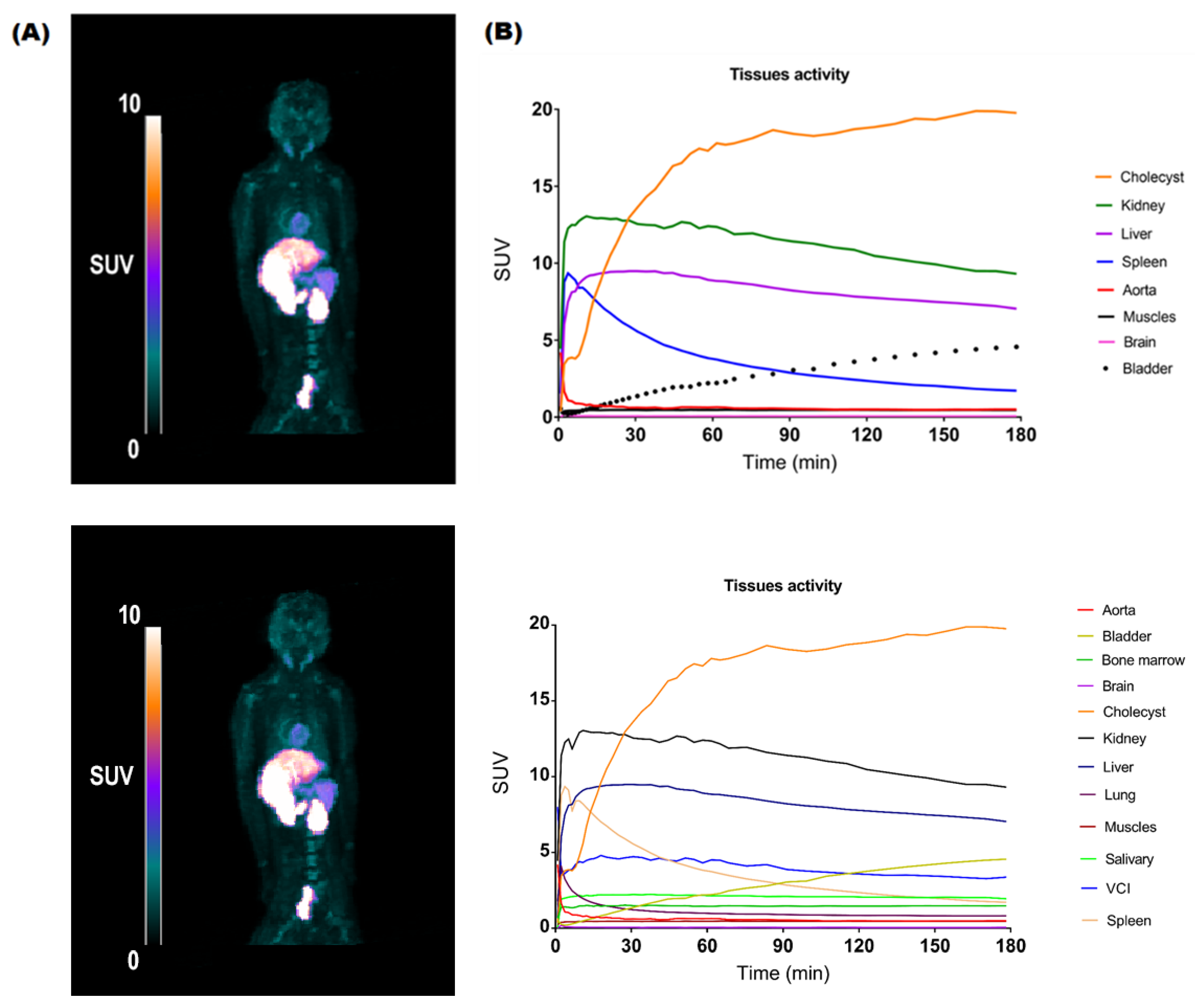
2.3. PET Imaging
3. Materials and Methods
3.1. Chemistry
3.1.1. Chemicals
3.1.2. Enantiomeric Purification of 7
3.2. Radiochemistry
3.2.1. General Procedures
3.2.2. Optimization of the Radiofuorination of 7
3.2.3. Optimization of the Deprotection of [18F]8 with TFA
3.2.4. Optimization of the Deprotection of [18F]8 with HCl
3.2.5. Automated Radiosynthesis of [18F](R,S)-Crizotinib
3.2.6. Quality Control of [18F](R,S)-Crizotinib
3.2.7. Automated Radiosynthesis of [18F](R)-Crizotinib
3.2.8. Quality Control of [18F](R)-Crizotinib
3.3. PET Imaging
3.3.1. Animal
3.3.2. PET Imaging
3.3.3. PET Data Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shaw, A.T.; Yasothan, U.; Kirkpatrick, P. Crizotinib. Nat. Rev. Drug Discov. 2011, 10, 897–898. [Google Scholar] [CrossRef]
- Kwak, E.L.; Bang, Y.-J.; Camidge, D.R.; Shaw, A.T.; Solomon, B.; Maki, R.G.; Ou, S.-H.I.; Dezube, B.J.; Jänne, P.A.; Costa, D.B.; et al. Anaplastic Lymphoma Kinase Inhibition in Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2010, 363, 1693–1703. [Google Scholar] [CrossRef] [Green Version]
- Lung Cancer—Non-Small Cell—Statistics. Cancer.Net. Available online: https://www.cancer.net/cancer-types/lung-cancer-non-small-cell/statistics (accessed on 19 April 2022).
- Soda, M.; Choi, Y.L.; Enomoto, M.; Takada, S.; Yamashita, Y.; Ishikawa, S.; Fujiwara, S.; Watanabe, H.; Kurashina, K.; Hatanaka, H.; et al. Identification of the Transforming EML4-ALK Fusion Gene in Non-Small-Cell Lung Cancer. Nature 2007, 448, 561–566. [Google Scholar] [CrossRef]
- Preusser, M.; Berghoff, A.S.; Koller, R.; Zielinski, C.C.; Hainfellner, J.A.; Liebmann-Reindl, S.; Popitsch, N.; Geier, C.B.; Streubel, B.; Birner, P. Spectrum of Gene Mutations Detected by next Generation Exome Sequencing in Brain Metastases of Lung Adenocarcinoma. Eur. J. Cancer 2015, 51, 1803–1811. [Google Scholar] [CrossRef]
- Shi, W.; Dicker, A.P. CNS Metastases in Patients with Non–Small-Cell Lung Cancer and ALK Gene Rearrangement. J. Clin. Oncol. 2015, 34, 107–109. [Google Scholar] [CrossRef]
- Gainor, J.F.; Ou, S.-H.I.; Logan, J.; Borges, L.F.; Shaw, A.T. The Central Nervous System as a Sanctuary Site in ALK-Positive Non–Small-Cell Lung Cancer. J. Thorac. Oncol. 2013, 8, 1570–1573. [Google Scholar] [CrossRef] [Green Version]
- Katayama, R.; Sakashita, T.; Yanagitani, N.; Ninomiya, H.; Horiike, A.; Friboulet, L.; Gainor, J.F.; Motoi, N.; Dobashi, A.; Sakata, S.; et al. P-Glycoprotein Mediates Ceritinib Resistance in Anaplastic Lymphoma Kinase-Rearranged Non-Small Cell Lung Cancer. EBioMedicine 2015, 3, 54–66. [Google Scholar] [CrossRef] [Green Version]
- Tang, S.C.; Nguyen, L.N.; Sparidans, R.W.; Wagenaar, E.; Beijnen, J.H.; Schinkel, A.H. Increased Oral Availability and Brain Accumulation of the ALK Inhibitor Crizotinib by Coadministration of the P-Glycoprotein (ABCB1) and Breast Cancer Resistance Protein (ABCG2) Inhibitor Elacridar. Int. J. Cancer 2014, 134, 1484–1494. [Google Scholar] [CrossRef]
- Deo, A.K.; Theil, F.-P.; Nicolas, J.-M. Confounding Parameters in Preclinical Assessment of Blood–Brain Barrier Permeation: An Overview with Emphasis on Species Differences and Effect of Disease States. Mol. Pharm. 2013, 10, 1581–1595. [Google Scholar] [CrossRef]
- Huttunen, K.M.; Terasaki, T.; Urtti, A.; Montaser, A.B.; Uchida, Y. Pharmacoproteomics of Brain Barrier Transporters and Substrate Design for the Brain Targeted Drug Delivery. Pharm. Res. 2022, 39, 1363–1392. [Google Scholar] [CrossRef]
- Ito, K.; Uchida, Y.; Ohtsuki, S.; Aizawa, S.; Kawakami, H.; Katsukura, Y.; Kamiie, J.; Terasaki, T. Quantitative Membrane Protein Expression at the Blood–Brain Barrier of Adult and Younger Cynomolgus Monkeys. J. Pharm. Sci. 2011, 100, 3939–3950. [Google Scholar] [CrossRef] [PubMed]
- Pandit, R.; Chen, L.; Götz, J. The Blood-Brain Barrier: Physiology and Strategies for Drug Delivery. Adv. Drug Deliv. Rev. 2020, 165–166, 1–14. [Google Scholar] [CrossRef]
- Pottier, G.; Marie, S.; Goutal, S.; Auvity, S.; Peyronneau, M.-A.; Stute, S.; Boisgard, R.; Dollé, F.; Buvat, I.; Caillé, F.; et al. Imaging the Impact of the P-Glycoprotein (ABCB1) Function on the Brain Kinetics of Metoclopramide. J. Nucl. Med. 2016, 57, 309–314. [Google Scholar] [CrossRef] [Green Version]
- Tournier, N.; Goutal, S.; Auvity, S.; Traxl, A.; Mairinger, S.; Wanek, T.; Helal, O.-B.; Buvat, I.; Soussan, M.; Caillé, F.; et al. Strategies to Inhibit ABCB1- and ABCG2-Mediated Efflux Transport of Erlotinib at the Blood–Brain Barrier: A PET Study on Nonhuman Primates. J. Nucl. Med. 2017, 58, 117–122. [Google Scholar] [CrossRef] [Green Version]
- Tournier, N.; Saba, W.; Cisternino, S.; Peyronneau, M.-A.; Damont, A.; Goutal, S.; Dubois, A.; Dollé, F.; Scherrmann, J.-M.; Valette, H.; et al. Effects of Selected OATP and/or ABC Transporter Inhibitors on the Brain and Whole-Body Distribution of Glyburide. AAPS J. 2013, 15, 1082–1090. [Google Scholar] [CrossRef] [Green Version]
- Tournier, N.; Goutal, S.; Mairinger, S.; Hernández-Lozano, I.; Filip, T.; Sauberer, M.; Caillé, F.; Breuil, L.; Stanek, J.; Freeman, A.F.; et al. Complete Inhibition of ABCB1 and ABCG2 at the Blood–brain Barrier by Co-Infusion of Erlotinib and Tariquidar to Improve Brain Delivery of the Model ABCB1/ABCG2 Substrate [11C]erlotinib. J. Cereb. Blood Flow Metab. 2021, 41, 1634–1646. [Google Scholar] [CrossRef]
- Lin, Q.; Zhang, Y.; Fu, Z.; Hu, B.; Si, Z.; Zhao, Y.; Shi, H.; Cheng, D. Synthesis and Evaluation of 18F Labeled Crizotinib Derivative [18F]FPC as a Novel PET Probe for Imaging c-MET-Positive NSCLC Tumor. Bioorg. Med. Chem. 2020, 28, 115577. [Google Scholar] [CrossRef]
- Radaram, B.; Piwnica-Worms, D.; Alauddin, M. Development of Novel Probes for PET Imaging of Lung Cancer Overexpressing Anaplastic Lymphoma Kinase: Synthesis and Radiolabeling of Crizotinib and Alectinib Analogues. J. Nucl. Med. 2018, 59 (Suppl. S1), 183. [Google Scholar]
- Tredwell, M.; Preshlock, S.M.; Taylor, N.J.; Gruber, S.; Huiban, M.; Passchier, J.; Mercier, J.; Génicot, C.; Gouverneur, V. A General Copper-Mediated Nucleophilic 18F Fluorination of Arenes. Angew. Chem. 2014, 126, 7885–7889. [Google Scholar] [CrossRef]
- Makaravage, K.J.; Brooks, A.F.; Mossine, A.V.; Sanford, M.S.; Scott, P.J.H. Copper-Mediated Radiofluorination of Arylstannanes with [18F]KF. Org. Lett. 2016, 18, 5440–5443. [Google Scholar] [CrossRef]
- Lee, E.; Kamlet, A.S.; Powers, D.C.; Neumann, C.N.; Boursalian, G.B.; Furuya, T.; Choi, D.C.; Hooker, J.M.; Ritter, T. A Fluoride-Derived Electrophilic Late-Stage Fluorination Reagent for PET Imaging. Science 2011, 334, 639–642. [Google Scholar] [CrossRef]
- Neumann, C.N.; Hooker, J.M.; Ritter, T. Concerted Nucleophilic Aromatic Substitution with 19F−and 18F−. Nature 2016, 534, 369–373. [Google Scholar] [CrossRef] [Green Version]
- Beyzavi, M.H.; Mandal, D.; Strebl, M.G.; Neumann, C.N.; D’Amato, E.M.; Chen, J.; Hooker, J.M.; Ritter, T. 18F-Deoxyfluorination of Phenols via Ru π-Complexes. ACS Cent. Sci. 2017, 3, 944–948. [Google Scholar] [CrossRef] [Green Version]
- Kohlhepp, S.V.; Gulder, T. Hypervalent iodine(III) Fluorinations of Alkenes and Diazo Compounds: New Opportunities in Fluorination Chemistry. Chem. Soc. Rev. 2016, 45, 6270–6288. [Google Scholar] [CrossRef]
- Rotstein, B.H.; Stephenson, N.A.; Vasdev, N.; Liang, S.H. Spirocyclic Hypervalent iodine(III)-Mediated Radiofluorination of Non-Activated and Hindered Aromatics. Nat. Commun. 2014, 5, 4365. [Google Scholar] [CrossRef] [Green Version]
- Rotstein, B.H.; Wang, L.; Liu, R.Y.; Patteson, J.; Kwan, E.E.; Vasdev, N.; Liang, S.H. Mechanistic Studies and Radiofluorination of Structurally Diverse Pharmaceuticals with Spirocyclic iodonium(III) Ylides. Chem. Sci. 2016, 7, 4407–4417. [Google Scholar] [CrossRef] [Green Version]
- Liang, S.H.; Wang, L.; Stephenson, N.A.; Rotstein, B.H.; Vasdev, N. Facile 18F Labeling of Non-Activated Arenes via a Spirocyclic iodonium(III) Ylide Method and Its Application in the Synthesis of the mGluR5 PET Radiopharmaceutical [18F]FPEB. Nat. Protoc. 2019, 14, 1530–1545. [Google Scholar] [CrossRef]
- Qian, J.-Q.; Yan, P.-C.; Che, D.-Q.; Zhou, Q.-L.; Li, Y.-Q. A Novel Approach for the Synthesis of Crizotinib through the Key Chiral Alcohol Intermediate by Asymmetric Hydrogenation Using Highly Active Ir-Spiro-PAP Catalyst. Tetrahedron Lett. 2014, 55, 1528–1531. [Google Scholar] [CrossRef]
- Liu, N.; Wang, Y.; Huang, G.; Ji, C.; Fan, W.; Li, H.; Cheng, Y.; Tian, H. Design, Synthesis and Biological Evaluation of 1H-pyrrolo[2,3-B]pyridine and 1H-pyrazolo[3,4-B]pyridine Derivatives as c-Met Inhibitors. Bioorganic Chem. 2016, 65, 146–158. [Google Scholar] [CrossRef]
- Bravo, A.; Fontana, F.; Fronza, G.; Minisci, F.; Serri, A. Oxidation of Alkyl and Aryl Iodides, Phenylacetaldehyde and Alkenes by Dimethyldioxirane. Reaction Products and Mechanism. Tetrahedron Lett. 1995, 36, 6945–6948. [Google Scholar] [CrossRef]
- Dai, X.; Guo, G.; Zou, P.; Cui, R.; Chen, W.; Chen, X.; Yin, C.; He, W.; Vinothkumar, R.; Yang, F.; et al. (S)-Crizotinib Induces Apoptosis in Human Non-Small Cell Lung Cancer Cells by Activating ROS Independent of MTH1. J. Exp. Clin. Cancer Res. 2017, 36, 120. [Google Scholar] [CrossRef]
- Grushin, V.V. Carboranylhalonium Ions: From Striking Reactivity to a Unified Mechanistic Analysis of Polar Reactions of Diarylhalonium Compounds. Acc. Chem. Res. 1992, 25, 529–536. [Google Scholar] [CrossRef]
- Lee, B.C.; Kim, J.S.; Kim, B.S.; Son, J.Y.; Hong, S.K.; Park, H.S.; Moon, B.S.; Jung, J.H.; Jeong, J.M.; Kim, S.E. Aromatic Radiofluorination and Biological Evaluation of 2-Aryl-6-[18F]fluorobenzothiazoles as a Potential Positron Emission Tomography Imaging Probe for β-Amyloid Plaques. Bioorg. Med. Chem. 2011, 19, 2980–2990. [Google Scholar] [CrossRef]
- Carroll, M.A.; Nairne, J.; Smith, G.; Widdowson, D.A. Radical Scavengers: A Practical Solution to the Reproducibility Issue in the Fluoridation of Diaryliodonium Salts. J. Fluor. Chem. 2007, 128, 127–132. [Google Scholar] [CrossRef]
- Moon, B.S.; Kil, H.S.; Park, J.H.; Kim, J.S.; Park, J.; Chi, D.Y.; Lee, B.C.; Kim, S.E. Facile Aromatic Radiofluorination of [18F]flumazenil from Diaryliodonium Salts with Evaluation of Their Stability and Selectivity. Org. Biomol. Chem. 2011, 9, 8346–8355. [Google Scholar] [CrossRef]
- Wang, J.; Liang, Y.-L.; Qu, L. Boiling water-catalyzed neutral and selective N-Boc deprotection. Chem. Commun. 2009, 34, 5144–5146. [Google Scholar] [CrossRef]
- Johnson, T.R.; Tan, W.; Goulet, L.; Smith, E.B.; Yamazaki, S.; Walker, G.S.; O’Gorman, M.T.; Bedarida, G.; Zou, H.Y.; Christensen, J.G.; et al. Metabolism, Excretion and Pharmacokinetics of [14C]crizotinib Following Oral Administration to Healthy Subjects. Xenobiotica 2015, 45, 45–59. [Google Scholar] [CrossRef]
- Bauer, M.; Karch, R.; Wulkersdorfer, B.; Philippe, C.; Nics, L.; Klebermass, E.-M.; Weber, M.; Poschner, S.; Haslacher, H.; Jäger, W.; et al. A Proof-of-Concept Study to Inhibit ABCG2- and ABCB1-Mediated Efflux Transport at the Human Blood–Brain Barrier. J. Nucl. Med. 2019, 60, 486–491. [Google Scholar] [CrossRef] [Green Version]
- De Koning, P.D.; McAndrew, D.; Moore, R.; Moses, I.B.; Boyles, D.C.; Kissick, K.; Stanchina, C.L.; Cuthbertson, T.; Kamatani, A.; Rahman, L.; et al. Fit-for-Purpose Development of the Enabling Route to Crizotinib (PF-02341066). Org. Process Res. Dev. 2011, 15, 1018–1026. [Google Scholar] [CrossRef]
- Auvity, S.; Saba, W.; Goutal, S.; Leroy, C.; Buvat, I.; Cayla, J.; Caillé, F.; Bottlaender, M.; Cisternino, S.; Tournier, N. Acute Morphine Exposure Increases the Brain Distribution of [18F]DPA-714, a PET Biomarker of Glial Activation in Nonhuman Primates. Int. J. Neuropsychopharmacol. 2017, 20, 67–71. [Google Scholar] [CrossRef] [Green Version]
- Yushkevich, P.A.; Piven, J.; Hazlett, H.C.; Smith, R.G.; Ho, S.; Gee, J.C.; Gerig, G. User-Guided 3D Active Contour Segmentation of Anatomical Structures: Significantly Improved Efficiency and Reliability. NeuroImage 2006, 31, 1116–1128. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry 1 | Base | Conditions 2 | Conversion 3 |
|---|---|---|---|
| 1 | K2CO3 + K222 | DMF, 120 °C, 10 min. | 18% |
| 2 | Et4NHCO3 | DMF, 120 °C, 10 min. | 51% |
| 3 | Et4NHCO3 | DMF, 160 °C, 10 min. | 64% |
| 4 | Et4NHCO3 | DMF, 160 °C, 20 min. | 64% |
| 5 | Et4NHCO3 + TEMPO (1 mg) | DMF, 160 °C, 10 min. | 48% |
| Entry 1 | Acid | Conditions | Conversion 2 |
|---|---|---|---|
| 1 | TFA | Dioxane/H2O (1/1 v/v) 120 °C, 10 min. | 47% |
| 2 | HCl 3M | DMF 120 °C, 10 min. | 72% |
| 3 | HCl 3M | DMF 160 °C, 10 min. | >99% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sardana, M.; Breuil, L.; Goutal, S.; Goislard, M.; Kondrashov, M.; Marchal, E.; Besson, F.L.; Dugave, C.; Wrigley, G.; Jonson, A.C.; et al. Isotopic Radiolabeling of Crizotinib with Fluorine-18 for In Vivo Pet Imaging. Pharmaceuticals 2022, 15, 1568. https://doi.org/10.3390/ph15121568
Sardana M, Breuil L, Goutal S, Goislard M, Kondrashov M, Marchal E, Besson FL, Dugave C, Wrigley G, Jonson AC, et al. Isotopic Radiolabeling of Crizotinib with Fluorine-18 for In Vivo Pet Imaging. Pharmaceuticals. 2022; 15(12):1568. https://doi.org/10.3390/ph15121568
Chicago/Turabian StyleSardana, Malvika, Louise Breuil, Sébastien Goutal, Maud Goislard, Mikhail Kondrashov, Etienne Marchal, Florent L. Besson, Christophe Dugave, Gail Wrigley, Anna C. Jonson, and et al. 2022. "Isotopic Radiolabeling of Crizotinib with Fluorine-18 for In Vivo Pet Imaging" Pharmaceuticals 15, no. 12: 1568. https://doi.org/10.3390/ph15121568
APA StyleSardana, M., Breuil, L., Goutal, S., Goislard, M., Kondrashov, M., Marchal, E., Besson, F. L., Dugave, C., Wrigley, G., Jonson, A. C., Kuhnast, B., Schou, M., Tournier, N., Elmore, C. S., & Caillé, F. (2022). Isotopic Radiolabeling of Crizotinib with Fluorine-18 for In Vivo Pet Imaging. Pharmaceuticals, 15(12), 1568. https://doi.org/10.3390/ph15121568