
Response Predictive Markers and Synergistic Agents for Drug Repositioning of Statins in Ovarian Cancer

 ,
,  ,
,
Abstract
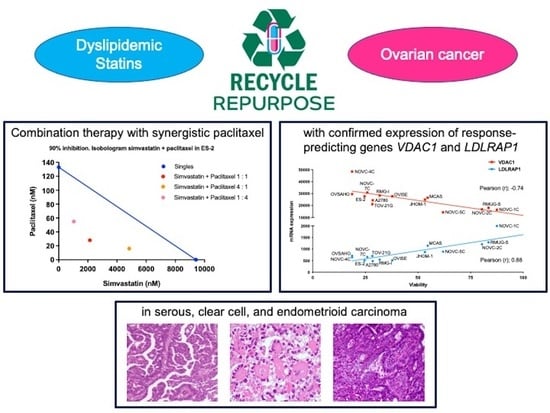
:
1. Introduction
2. Results
2.1. Mode of Action of Statins Other Than Protein Prenylation
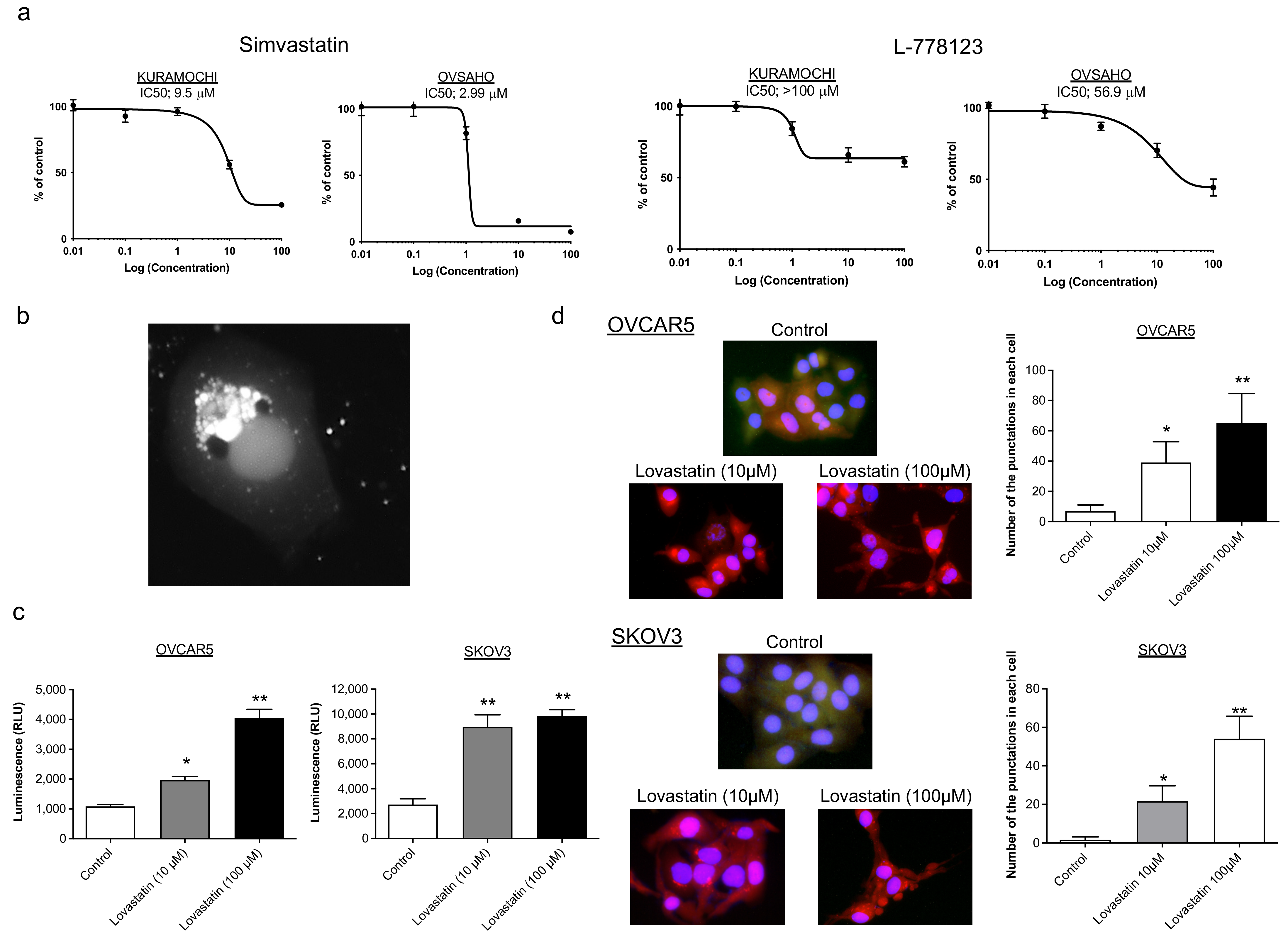
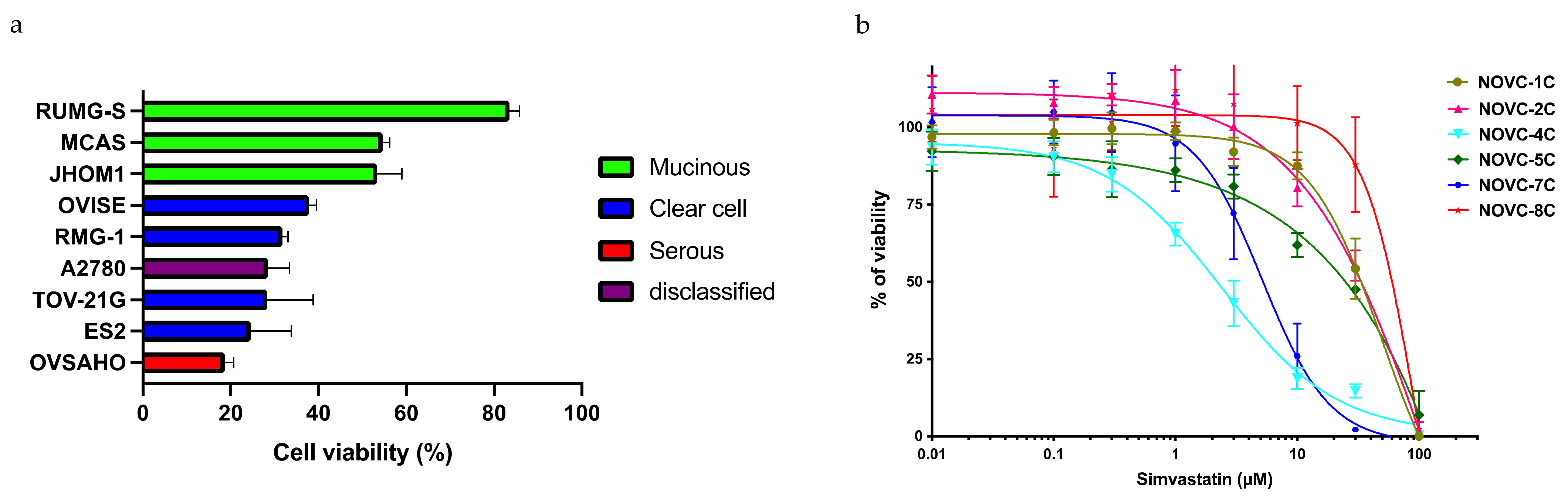
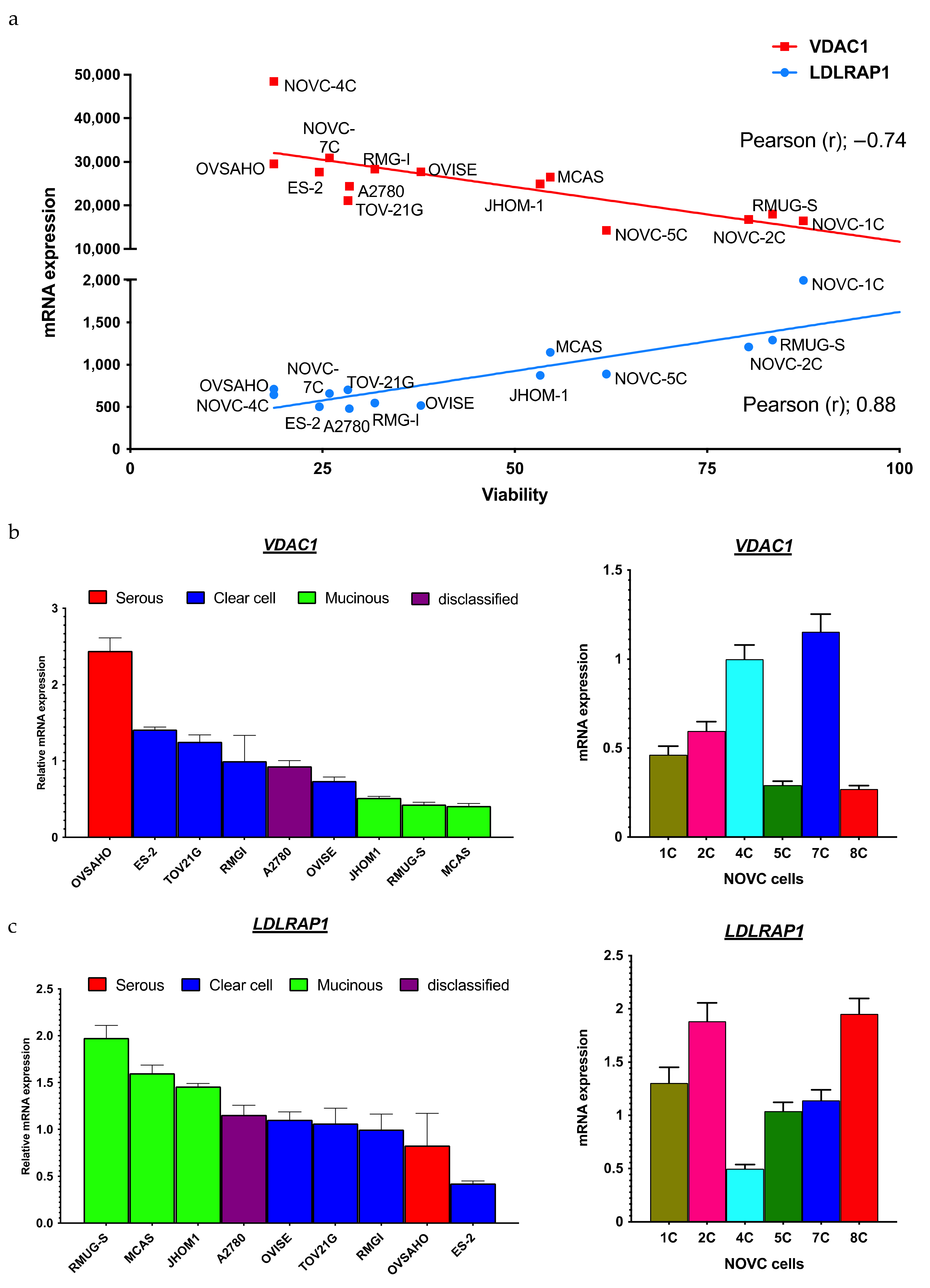
2.2. Identification of Biomarkers That Predict the Response to Statins
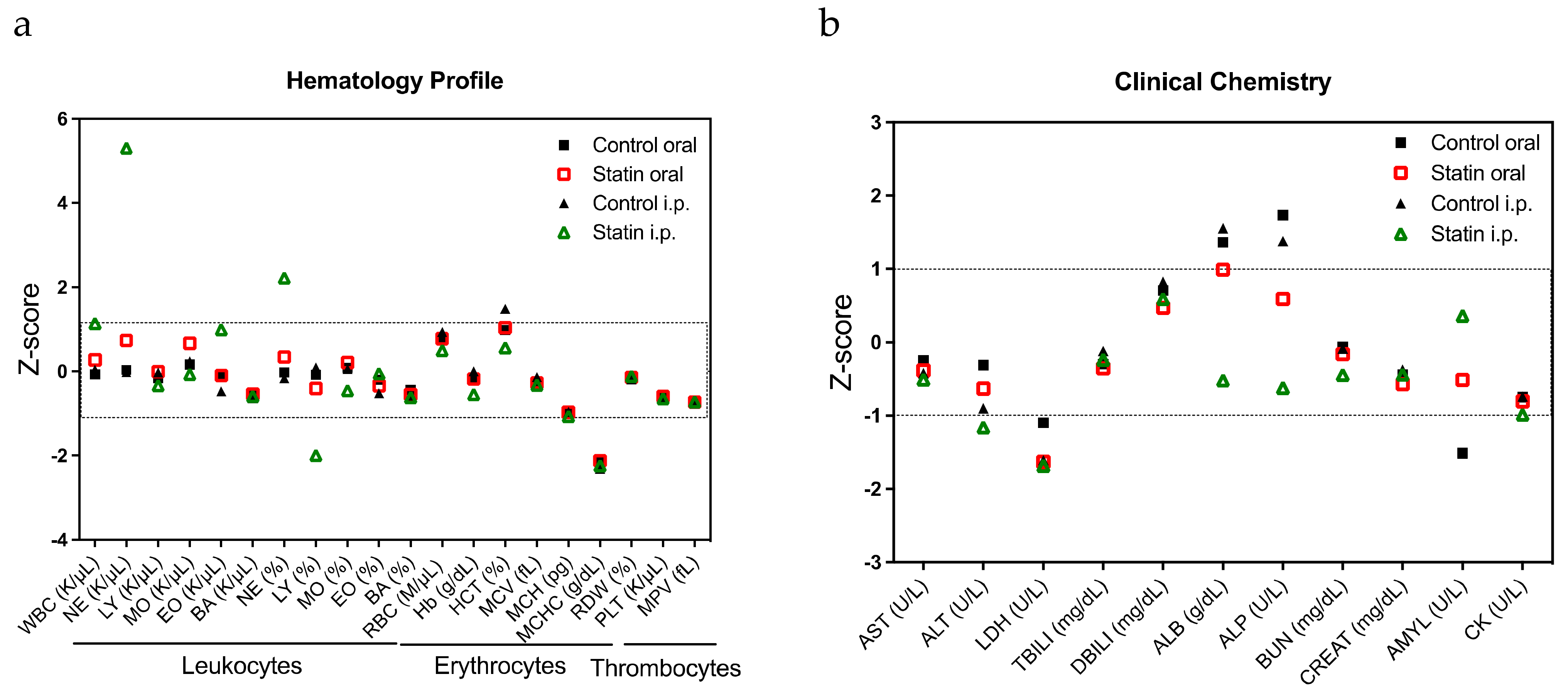
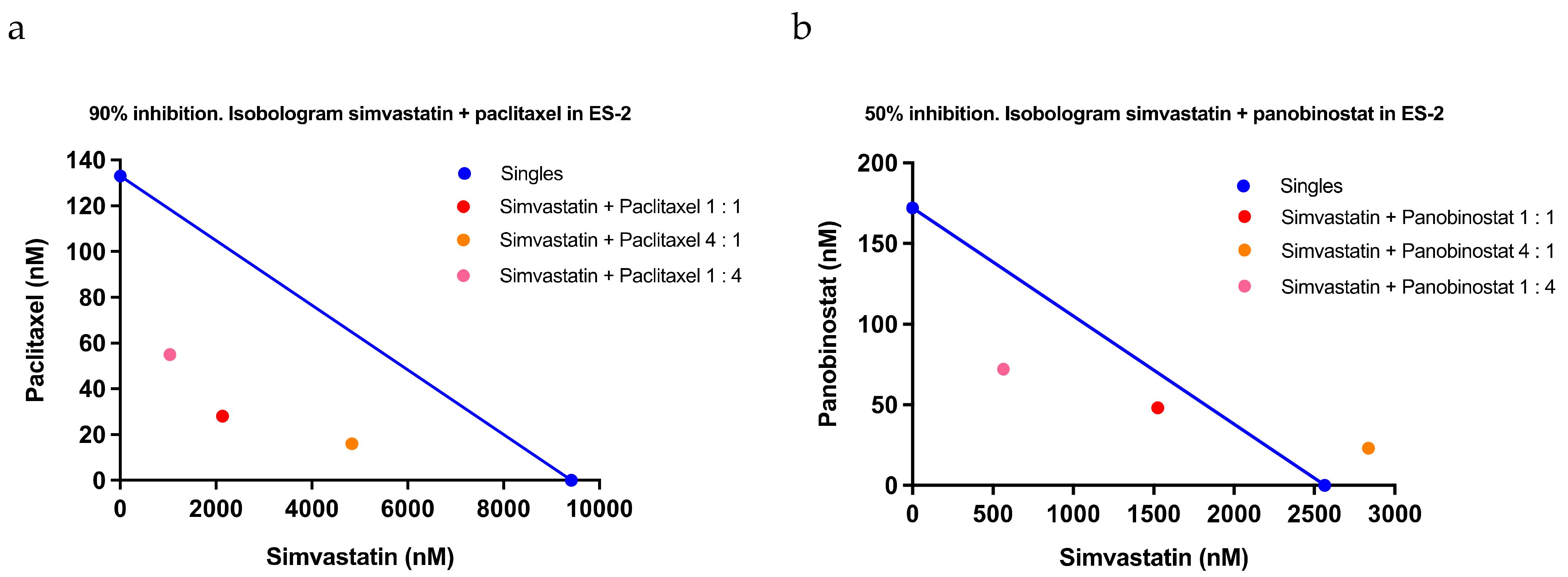
2.3. Confirmation of the Response to Statins in Clinical Samples, Their Safety in Mice, and Evaluation of Drug Combination Effects
3. Discussion
4. Materials and Methods
4.1. Drugs
4.2. Cell Lines and Cell Cultures
4.3. Cell Viability Assays
4.4. Microarray Analysis of KURAMOCHI and OVSAHO Cells Incubated with Simvastatin or L-778123
4.5. Analysis of Cell Apoptosis and Autophagy
4.6. Correlation Analysis between mRNA Expression and Cell Viability Using Pearson’s Product-Moment Correlation Coefficient
4.7. Quantitative RT-PCR
4.8. Histoculture Drug Response Assays
4.9. Combination Index Analysis
4.10. Hemogram and Biochemical Analysis
4.11. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pignata, S.; Pisano, C.; Di Napoli, M.; Cecere, S.C.; Tambaro, R.; Attademo, L. Treatment of recurrent epithelial ovarian cancer. Cancer 2019, 125 (Suppl. S24), 4609–4615. [Google Scholar] [CrossRef] [PubMed]
- Hay, M.; Thomas, D.W.; Craighead, J.L.; Economides, C.; Rosenthal, J. Clinical development success rates for investigational drugs. Nat. Biotechnol. 2014, 32, 40–51. [Google Scholar] [CrossRef] [PubMed]
- Xue, H.; Li, J.; Xie, H.; Wang, Y. Review of Drug Repositioning Approaches and Resources. Int. J. Biol. Sci. 2018, 14, 1232–1244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masuda, T.; Tsuruda, Y.; Matsumoto, Y.; Uchida, H.; Nakayama, K.I.; Mimori, K. Drug repositioning in cancer: The current situation in Japan. Cancer Sci. 2020, 111, 1039–1046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sleire, L.; Førde, H.E.; Netland, I.A.; Leiss, L.; Skeie, B.S.; Enger, P. Drug repurposing in cancer. Pharmacol. Res. 2017, 124, 74–91. [Google Scholar] [CrossRef]
- Pushpakom, S.; Iorio, F.; Eyers, P.A.; Escott, K.J.; Hopper, S.; Wells, A.; Doig, A.; Guilliams, T.; Latimer, J.; McNamee, C.; et al. Drug repurposing: Progress, challenges and recommendations. Nat. Rev. Drug Discov. 2019, 18, 41–58. [Google Scholar] [CrossRef]
- Kobayashi, Y.; Banno, K.; Kunitomi, H.; Tominaga, E.; Aoki, D. Current state and outlook for drug repositioning anticipated in the field of ovarian cancer. J. Gynecol. Oncol. 2019, 30, e10. [Google Scholar] [CrossRef]
- Bellosta, S.; Corsini, A. Statin drug interactions and related adverse reactions: An update. Expert Opin. Drug Saf. 2018, 17, 25–37. [Google Scholar] [CrossRef]
- Mo, H.; Jeter, R.; Bachmann, A.; Yount, S.T.; Shen, C.L.; Yeganehjoo, H. The Potential of Isoprenoids in Adjuvant Cancer Therapy to Reduce Adverse Effects of Statins. Front. Pharmacol. 2018, 9, 1515. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, S.F.; Nordestgaard, B.G.; Bojesen, S.E. Statin use and reduced cancer-related mortality. N. Engl. J. Med. 2012, 367, 1792–1802. [Google Scholar] [CrossRef] [Green Version]
- Cardwell, C.R.; Hicks, B.M.; Hughes, C.; Murray, L.J. Statin use after colorectal cancer diagnosis and survival: A population-based cohort study. J. Clin. Oncol. 2014, 32, 3177–3183. [Google Scholar] [CrossRef] [PubMed]
- Yu, O.; Eberg, M.; Benayoun, S.; Aprikian, A.; Batist, G.; Suissa, S.; Azoulay, L. Use of statins and the risk of death in patients with prostate cancer. J. Clin. Oncol. 2014, 32, 5–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, T.; Khalaf, N.; Ramsey, D.; El-Serag, H.B. Statin use is associated with a decreased risk of Barrett’s esophagus. Gastroenterology 2014, 147, 314–323. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, Y.; Kashima, H.; Wu, R.C.; Jung, J.G.; Kuan, J.C.; Gu, J.; Xuan, J.; Sokoll, L.; Visvanathan, K.; Shih Ie, M.; et al. Mevalonate Pathway Antagonist Suppresses Formation of Serous Tubal Intraepithelial Carcinoma and Ovarian Carcinoma in Mouse Models. Clin. Cancer Res. 2015, 21, 4652–4662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobayashi, Y.; Kashima, H.; Rahmanto, Y.S.; Banno, K.; Yu, Y.; Matoba, Y.; Watanabe, K.; Iijima, M.; Takeda, T.; Kunitomi, H.; et al. Drug repositioning of mevalonate pathway inhibitors as antitumor agents for ovarian cancer. Oncotarget 2017, 8, 72147–72156. [Google Scholar] [CrossRef] [Green Version]
- Armando, R.G.; Mengual Gómez, D.L.; Gomez, D.E. New drugs are not enough-drug repositioning in oncology: An update. Int. J. Oncol. 2020, 56, 651–684. [Google Scholar] [CrossRef] [Green Version]
- Ricci, G.; Ciccone, M.M.; Giordano, P.; Cortese, F. Statins: Pharmacokinetics, Pharmacodynamics and Cost-Effectiveness Analysis. Curr. Vasc. Pharmacol. 2019, 17, 213–221. [Google Scholar] [CrossRef]
- Maligres, P.E.; Waters, M.S.; Weissman, S.A.; McWilliams, J.C.; Lewis, S.; Cowen, J.; Reamer, R.A.; Volante, R.P.; Reider, P.J.; Askin, D. Preparation of a clinically investigated ras farnesyl transferase inhibitor. J. Heterocycl. Chem. 2003, 40, 229–241. [Google Scholar] [CrossRef]
- Anglesio, M.S.; Wiegand, K.C.; Melnyk, N.; Chow, C.; Salamanca, C.; Prentice, L.M.; Senz, J.; Yang, W.; Spillman, M.A.; Cochrane, D.R.; et al. Type-specific cell line models for type-specific ovarian cancer research. PLoS ONE 2013, 8, e72162. [Google Scholar] [CrossRef]
- Domcke, S.; Sinha, R.; Levine, D.A.; Sander, C.; Schultz, N. Evaluating cell lines as tumour models by comparison of genomic profiles. Nat. Commun. 2013, 4, 2126. [Google Scholar] [CrossRef]
- Shoshan-Barmatz, V.; Shteinfer-Kuzmine, A.; Verma, A. VDAC1 at the Intersection of Cell Metabolism, Apoptosis, and Diseases. Biomolecules 2020, 10, 1485. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, Y.; Banno, K.; Kunitomi, H.; Takahashi, T.; Takeda, T.; Nakamura, K.; Tsuji, K.; Tominaga, E.; Aoki, D. Warburg effect in Gynecologic cancers. J. Obstet. Gynaecol. Res. 2019, 45, 542–548. [Google Scholar] [CrossRef] [PubMed]
- Garcia, C.K.; Wilund, K.; Arca, M.; Zuliani, G.; Fellin, R.; Maioli, M.; Calandra, S.; Bertolini, S.; Cossu, F.; Grishin, N.; et al. Autosomal recessive hypercholesterolemia caused by mutations in a putative LDL receptor adaptor protein. Science 2001, 292, 1394–1398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Onwuka, J.U.; Okekunle, A.P.; Olutola, O.M.; Akpa, O.M.; Feng, R. Lipid profile and risk of ovarian tumours: A meta-analysis. BMC Cancer 2020, 20, 200. [Google Scholar] [CrossRef]
- Yamaguchi, K.; Kitamura, S.; Furutake, Y.; Murakami, R.; Yamanoi, K.; Taki, M.; Ukita, M.; Hamanishi, J.; Mandai, M. Acquired Evolution of Mitochondrial Metabolism Regulated by HNF1B in Ovarian Clear Cell Carcinoma. Cancers 2021, 13, 2413. [Google Scholar] [CrossRef] [PubMed]
- Irvin, S.; Clarke, M.A.; Trabert, B.; Wentzensen, N. Systematic review and meta-analysis of studies assessing the relationship between statin use and risk of ovarian cancer. Cancer Causes Control 2020, 31, 869–879. [Google Scholar] [CrossRef]
- Majidi, A.; Na, R.; Dixon-Suen, S.; Jordan, S.J.; Webb, P.M. Common medications and survival in women with ovarian cancer: A systematic review and meta-analysis. Gynecol. Oncol. 2020, 157, 678–685. [Google Scholar] [CrossRef]
- Wang, Y.; Ren, F.; Song, Z.; Chen, P.; Liu, S.; Ouyang, L. Statin use and the risk of ovarian and endometrial cancers: A meta-analysis. BMC Cancer 2019, 19, 730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yarmolinsky, J.; Bull, C.J.; Vincent, E.E.; Robinson, J.; Walther, A.; Smith, G.D.; Lewis, S.J.; Relton, C.L.; Martin, R.M. Association Between Genetically Proxied Inhibition of HMG-CoA Reductase and Epithelial Ovarian Cancer. JAMA 2020, 323, 646–655. [Google Scholar] [CrossRef]
- Mannella, C.A.; Bonner, W.D., Jr. Biochemical characteristics of the outer membranes of plant mitochondria. Biochim. Biophys. Acta 1975, 413, 213–225. [Google Scholar] [CrossRef]
- Sampson, M.J.; Lovell, R.S.; Craigen, W.J. The murine voltage-dependent anion channel gene family. Conserved structure and function. J. Biol. Chem. 1997, 272, 18966–18973. [Google Scholar] [CrossRef] [Green Version]
- Cesar Mde, C.; Wilson, J.E. All three isoforms of the voltage-dependent anion channel (VDAC1, VDAC2, and VDAC3) are present in mitochondria from bovine, rabbit, and rat brain. Arch. Biochem. Biophys. 2004, 422, 191–196. [Google Scholar] [CrossRef]
- Reymann, S.; Flörke, H.; Heiden, M.; Jakob, C.; Stadtmüller, U.; Steinacker, P.; Lalk, V.E.; Pardowitz, I.; Thinnes, F.P. Further evidence for multitopological localization of mammalian porin (VDAC) in the plasmalemma forming part of a chloride channel complex affected in cystic fibrosis and encephalomyopathy. Biochem. Mol. Med. 1995, 54, 75–87. [Google Scholar] [CrossRef]
- Xu, X.; Decker, W.; Sampson, M.J.; Craigen, W.J.; Colombini, M. Mouse VDAC isoforms expressed in yeast: Channel properties and their roles in mitochondrial outer membrane permeability. J. Membr. Biol. 1999, 170, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Rostovtseva, T.K.; Kazemi, N.; Weinrich, M.; Bezrukov, S.M. Voltage gating of VDAC is regulated by nonlamellar lipids of mitochondrial membranes. J. Biol. Chem. 2006, 281, 37496–37506. [Google Scholar] [CrossRef] [Green Version]
- Mazure, N.M. VDAC in cancer. Biochim. Biophys. Acta Bioenergy 2017, 1858, 665–673. [Google Scholar] [CrossRef]
- Pastorino, J.G.; Shulga, N.; Hoek, J.B. Mitochondrial binding of hexokinase II inhibits Bax-induced cytochrome c release and apoptosis. J. Biol. Chem. 2002, 277, 7610–7618. [Google Scholar] [CrossRef] [Green Version]
- Shoshan-Barmatz, V.; Ben-Hail, D. VDAC, a multi-functional mitochondrial protein as a pharmacological target. Mitochondrion 2012, 12, 24–34. [Google Scholar] [CrossRef] [PubMed]
- Lipper, C.H.; Stofleth, J.T.; Bai, F.; Sohn, Y.S.; Roy, S.; Mittler, R.; Nechushtai, R.; Onuchic, J.N.; Jennings, P.A. Redox-dependent gating of VDAC by mitoNEET. Proc. Natl. Acad. Sci. USA 2019, 116, 19924–19929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedlander, M.L.; Russell, K.; Millis, S.; Gatalica, Z.; Bender, R.; Voss, A. Molecular Profiling of Clear Cell Ovarian Cancers: Identifying Potential Treatment Targets for Clinical Trials. Int. J. Gynecol. Cancer 2016, 26, 648–654. [Google Scholar] [CrossRef] [Green Version]
- Itamochi, H.; Oishi, T.; Oumi, N.; Takeuchi, S.; Yoshihara, K.; Mikami, M.; Yaegashi, N.; Terao, Y.; Takehara, K.; Ushijima, K.; et al. Whole-genome sequencing revealed novel prognostic biomarkers and promising targets for therapy of ovarian clear cell carcinoma. Br. J. Cancer 2017, 117, 717–724. [Google Scholar] [CrossRef]
- Caumanns, J.J.; Berns, K.; Wisman, G.B.A.; Fehrmann, R.S.N.; Tomar, T.; Klip, H.; Meersma, G.J.; Hijmans, E.M.; Gennissen, A.M.C.; Duiker, E.W.; et al. Integrative Kinome Profiling Identifies mTORC1/2 Inhibition as Treatment Strategy in Ovarian Clear Cell Carcinoma. Clin. Cancer Res. 2018, 24, 3928–3940. [Google Scholar] [CrossRef] [Green Version]
- Westhoff, M.A.; Faham, N.; Marx, D.; Nonnenmacher, L.; Jennewein, C.; Enzenmüller, S.; Gonzalez, P.; Fulda, S.; Debatin, K.M. Sequential dosing in chemosensitization: Targeting the PI3K/Akt/mTOR pathway in neuroblastoma. PLoS ONE 2013, 8, e83128. [Google Scholar] [CrossRef]
- Li, L.; Yao, Y.C.; Gu, X.Q.; Che, D.; Ma, C.Q.; Dai, Z.Y.; Li, C.; Zhou, T.; Cai, W.B.; Yang, Z.H.; et al. Plasminogen kringle 5 induces endothelial cell apoptosis by triggering a voltage-dependent anion channel 1 (VDAC1) positive feedback loop. J. Biol. Chem. 2014, 289, 32628–32638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berberich, A.J.; Hegele, R.A. The complex molecular genetics of familial hypercholesterolaemia. Nat. Rev. Cardiol. 2019, 16, 9–20. [Google Scholar] [CrossRef]
- Pisciotta, L.; Oliva, C.P.; Pes, G.M.; Scala, D.L.; Bellocchio, A.; Fresa, R.; Cantafora, A.; Arca, M.; Calandra, S.; Bertolini, S. Autosomal recessive hypercholesterolemia (ARH) and homozygous familial hypercholesterolemia (FH): A phenotypic comparison. Atherosclerosis 2006, 188, 398–405. [Google Scholar] [CrossRef]
- Rostovtseva, T.K.; Sheldon, K.L.; Hassanzadeh, E.; Monge, C.; Saks, V.; Bezrukov, S.M.; Sackett, D.L. Tubulin binding blocks mitochondrial voltage-dependent anion channel and regulates respiration. Proc. Natl. Acad. Sci. USA 2008, 105, 18746–18751. [Google Scholar] [CrossRef] [Green Version]
- Reina, S.; De Pinto, V. Anti-Cancer Compounds Targeted to VDAC: Potential and Perspectives. Curr. Med. Chem. 2017, 24, 4447–4469. [Google Scholar] [CrossRef] [PubMed]
- Fang, D.; Maldonado, E.N. VDAC Regulation: A Mitochondrial Target to Stop Cell Proliferation. Adv. Cancer Res. 2018, 138, 41–69. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Seto, E. HDACs and HDAC Inhibitors in Cancer Development and Therapy. Cold Spring Harb. Perspect. Med. 2016, 6, a026831. [Google Scholar] [CrossRef] [Green Version]
- Ogiwara, H.; Takahashi, K.; Sasaki, M.; Kuroda, T.; Yoshida, H.; Watanabe, R.; Maruyama, A.; Makinoshima, H.; Chiwaki, F.; Sasaki, H.; et al. Targeting the Vulnerability of Glutathione Metabolism in ARID1A-Deficient Cancers. Cancer Cell 2019, 35, 177–190.e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chou, T.C. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010, 70, 440–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Pearson’s Correlation Coefficient (r) | p-Value | Spearman’s Correlation Coefficient (r) | p-Value |
|---|---|---|---|---|
| Top 10 | ||||
| NRDC | −0.90 | 7.38 × 10−7 | −0.78 | 2.49 × 10−4 |
| PPID | −0.79 | 1.67 × 10−7 | −0.82 | 6.33 × 10−5 |
| XRN2 | −0.78 | 2.30 × 10−4 | −0.76 | 4.25 × 10−4 |
| VDAC1 | −0.74 | 6.44 × 10−4 | −0.75 | 5.73 × 10−4 |
| HSP90AB1 | −0.74 | 7.19 × 10−4 | −0.77 | 3.10 × 10−4 |
| DOCK7 | −0.74 | 7.45 × 10−4 | −0.67 | 3.28 × 10−3 |
| VARS1 | −0.73 | 9.71 × 10−4 | −0.82 | 5.24 × 10−5 |
| HSDL1 | −0.72 | 9.92 × 10−4 | −0.74 | 7.16 × 10−4 |
| ING2 | −0.72 | 1.02 × 10−3 | −0.62 | 8.36 × 10−3 |
| AGK | −0.72 | 1.04 × 10−3 | −0.79 | 1.83 × 10−4 |
| Bottom 10 | ||||
| LDLRAP1 | 0.88 | 3.99 × 10−6 | 0.82 | 6.04 × 10−5 |
| EPN3 | 0.87 | 4.52 × 10−6 | 0.73 | 7.85 × 10−4 |
| P4HTM | 0.82 | 5.09 × 10−5 | 0.68 | 2.57 × 10−3 |
| VPS37C | 0.80 | 1.23 × 10−4 | 0.66 | 3.96 × 10−3 |
| PHF2 | 0.80 | 1.24 × 10−4 | 0.77 | 3.21 × 10−4 |
| JADE2 | 0.80 | 1.33 × 10−4 | 0.71 | 1.37 × 10−3 |
| CXorf56 | 0.80 | 1.34 × 10−4 | 0.67 | 2.98 × 10−3 |
| PPL | 0.79 | 1.52 × 10−4 | 0.83 | 3.91 × 10−5 |
| OVOL1 | 0.79 | 1.60 × 10−4 | 0.75 | 5.73 × 10−4 |
| IFNLR1 | 0.78 | 2.49 × 10−4 | 0.75 | 5.02 × 10−4 |
| SLC1A4 | 0.77 | 2.67 × 10−4 | 0.77 | 2.99 × 10−4 |
| No | Histological Type | Age (Years) | Stage | Simvastatin (%) | Paclitaxel (%) | Carboplatin |
|---|---|---|---|---|---|---|
| 1 | High-grade serous | 65 | IIB | 77.6 | 82.7 | 61.0 |
| 2 | 45 | IIIA2 | 65.1 | 72.4 | 60.3 | |
| 3 | 46 | IVB | 53.5 | 65.0 | 15.5 | |
| 4 | 80 | IVA | 44.6 | 86.3 | 58.9 | |
| 5 | 71 | IIIC | 38.1 | 41.5 | 33.2 | |
| 6 | 40 | IIIB | 24.9 | 69.2 | 52.7 | |
| 7 | 71 | IIIC | 12.6 | 54.1 | 11.2 | |
| 8 | 48 | IIIA | 10.3 | 81.1 | 18.4 | |
| 9 | 74 | IIIC | 6.0 | 51.4 | 28.6 | |
| 10 | Clear cell | 54 | IA | 67.6 | 82.2 | 19.1 |
| 11 | 74 | IC2 | 67.5 | 76.3 | 29.4 | |
| 12 | 49 | IC3 | 61.4 | 77.6 | 32.7 | |
| 13 | 51 | IC1 | 61.0 | 34.2 | 56.2 | |
| 14 | 59 | IIIC | 42.5 | 50.5 | 59.1 | |
| 15 | 46 | IVB | 18.0 | 41.7 | 33.3 | |
| 16 | Endometrioid | 33 | IC1 | 77.7 | 83.2 | 62.9 |
| 17 | 79 | IC1 | 72.9 | 78.4 | 45.2 | |
| 18 | 63 | IC1 | 72.5 | 85.2 | 53.3 | |
| 19 | 59 | IIB | 15.7 | 51.0 | 29.4 | |
| 20 | Mucinous | 52 | IC2 | 22.7 | 85.3 | 38.8 |
| Combination Experiment | CI50 | CI75 | CI90 | Average | Standard Deviation |
|---|---|---|---|---|---|
| AZD8055 | |||||
| Simvastatin + AZD8055 1:1 | 0.78 | 0.77 | 0.98 | 0.84 | 0.12 |
| Simvastatin + AZD8055 4:1 | 1.36 | 1.12 | 0.91 | 1.13 | 0.23 |
| Simvastatin + AZD8055 1:4 | 0.72 | 0.73 | >2.00 | >1.15 | NA |
| Average | 0.95 | 0.87 | >1.30 | ||
| Standard deviation | 0.35 | 0.22 | NA | ||
| Copanlisib | |||||
| Simvastatin + Copanlisib 1:1 | 1.04 | 0.77 | >2.00 | >0.27 | NA |
| Simvastatin + Copanlisib 4:1 | 1.04 | 1.17 | 1.52 | 1.24 | 0.24 |
| Simvastatin + Copanlisib 1:4 | 0.90 | 0.60 | >2.00 | >1.17 | NA |
| Average | 0.99 | 0.85 | >1.84 | ||
| Standard deviation | 0.08 | 0.29 | NA | ||
| Dabrafenib | |||||
| Simvastatin + Dabrafenib 1:1 | 1.04 | 0.67 | 0.74 | 0.82 | 0.20 |
| Simvastatin + Dabrafenib 4:1 | 0.71 | 0.48 | >2.00 | >1.06 | NA |
| Simvastatin + Dabrafenib 1:4 | 0.66 | >2.00 | >2.00 | >1.55 | NA |
| Average | 0.80 | > 1.05 | >1.58 | ||
| Standard deviation | 0.21 | NA | NA | ||
| Doxorubicin | |||||
| Simvastatin + Doxorubicin 1:1 | 1.05 | 1.14 | 1.11 | 1.10 | 0.04 |
| Simvastatin + Doxorubicin 4:1 | 1.73 | 1.47 | 1.08 | 1.43 | 0.33 |
| Simvastatin + Doxorubicin 1:4 | 1.04 | 1.01 | 0.96 | 1.00 | 0.04 |
| Average | 1.27 | 1.20 | 1.05 | ||
| Standard deviation | 0.40 | 0.24 | 0.08 | ||
| Etoposide | |||||
| Simvastatin + Etoposide 1:1 | 1.44 | 1.11 | 0.78 | 1.11 | 0.33 |
| Simvastatin + Etoposide 4:1 | 1.16 | 1.14 | 0.98 | 1.09 | 0.10 |
| Simvastatin + Etoposide 1:4 | 0.98 | 0.95 | 0.91 | 0.95 | 0.04 |
| Average | 1.19 | 1.07 | 0.89 | ||
| Standard deviation | 0.23 | 0.10 | 0.10 | ||
| Irinotecan | |||||
| Simvastatin + Irinotecan 1:1 | 1.48 | 1.11 | 0.79 | 1.13 | 0.34 |
| Simvastatin + Irinotecan 4:1 | 1.13 | 0.96 | 0.79 | 0.96 | 0.17 |
| Simvastatin + Irinotecan 1:4 | 1.20 | 1.06 | 0.95 | 1.07 | 0.13 |
| Average | 1.27 | 1.04 | 0.84 | ||
| Standard deviation | 0.18 | 0.08 | 0.09 | ||
| Niraparib | |||||
| Simvastatin + Niraparib 1:1 | 1.30 | 1.18 | 0.86 | 1.11 | 0.23 |
| Simvastatin + Niraparib 4:1 | 0.93 | 0.81 | 0.70 | 0.81 | 0.11 |
| Simvastatin + Niraparib 1:4 | 1.07 | 1.06 | 0.83 | 0.99 | 0.13 |
| Average | 1.10 | 1.02 | 0.80 | ||
| Standard deviation | 0.19 | 0.19 | 0.08 | ||
| Paclitaxel | |||||
| Simvastatin + Paclitaxel 1:1 | 0.62 | 1.13 | 0.52 | 0.76 | 0.33 |
| Simvastatin + Paclitaxel 4:1 | 1.01 | 1.35 | 0.14 | 0.83 | 0.62 |
| Simvastatin + Paclitaxel 1:4 | 0.73 | 0.67 | 0.37 | 0.59 | 0.19 |
| Average | 0.79 | 1.05 | 0.34 | ||
| Standard deviation | 0.20 | 0.34 | 0.19 | ||
| Panobinostat | |||||
| Simvastatin + Panobinostat 1:1 | 0.88 | 0.56 | 0.30 | 0.58 | 0.29 |
| Simvastatin + Panobinostat 4:1 | 1.24 | 0.82 | 0.34 | 0.80 | 0.45 |
| Simvastatin + Panobinostat 1:4 | 0.64 | 0.32 | >2.00 | >0.98 | NA |
| Average | 0.92 | 0.56 | >0.88 | ||
| Standard deviation | 0.30 | 0.25 | NA | ||
| Trametinib | |||||
| Simvastatin + Trametinib 1:1 | 1.34 | 1.31 | 1.27 | 1.31 | 0.03 |
| Simvastatin + Trametinib 4:1 | 0.97 | 0.84 | 0.70 | 0.84 | 0.14 |
| Simvastatin + Trametinib 1:4 | 1.02 | 1.15 | 1.36 | 1.17 | 0.17 |
| Average | 1.11 | 1.10 | 1.11 | ||
| Standard deviation | 0.20 | 0.24 | 0.36 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kobayashi, Y.; Takeda, T.; Kunitomi, H.; Chiwaki, F.; Komatsu, M.; Nagai, S.; Nogami, Y.; Tsuji, K.; Masuda, K.; Ogiwara, H.; et al. Response Predictive Markers and Synergistic Agents for Drug Repositioning of Statins in Ovarian Cancer. Pharmaceuticals 2022, 15, 124. https://doi.org/10.3390/ph15020124
Kobayashi Y, Takeda T, Kunitomi H, Chiwaki F, Komatsu M, Nagai S, Nogami Y, Tsuji K, Masuda K, Ogiwara H, et al. Response Predictive Markers and Synergistic Agents for Drug Repositioning of Statins in Ovarian Cancer. Pharmaceuticals. 2022; 15(2):124. https://doi.org/10.3390/ph15020124
Chicago/Turabian StyleKobayashi, Yusuke, Takashi Takeda, Haruko Kunitomi, Fumiko Chiwaki, Masayuki Komatsu, Shimpei Nagai, Yuya Nogami, Kosuke Tsuji, Kenta Masuda, Hideaki Ogiwara, and et al. 2022. "Response Predictive Markers and Synergistic Agents for Drug Repositioning of Statins in Ovarian Cancer" Pharmaceuticals 15, no. 2: 124. https://doi.org/10.3390/ph15020124
APA StyleKobayashi, Y., Takeda, T., Kunitomi, H., Chiwaki, F., Komatsu, M., Nagai, S., Nogami, Y., Tsuji, K., Masuda, K., Ogiwara, H., Sasaki, H., Banno, K., & Aoki, D. (2022). Response Predictive Markers and Synergistic Agents for Drug Repositioning of Statins in Ovarian Cancer. Pharmaceuticals, 15(2), 124. https://doi.org/10.3390/ph15020124