
Enhancing the Bioavailability and Efficacy of Vismodegib for the Control of Skin Cancer: In Vitro and In Vivo Studies


Abstract
:
1. Introduction
2. Results
2.1. HPLC Quantification of Vismodegib
2.2. Equilibrium Solubility Studies of Vismodegib
2.3. Preparation and Vitro Characterization of VLI and VLL Formulations
2.3.1. Entrapment Efficiency and DLS Characterization
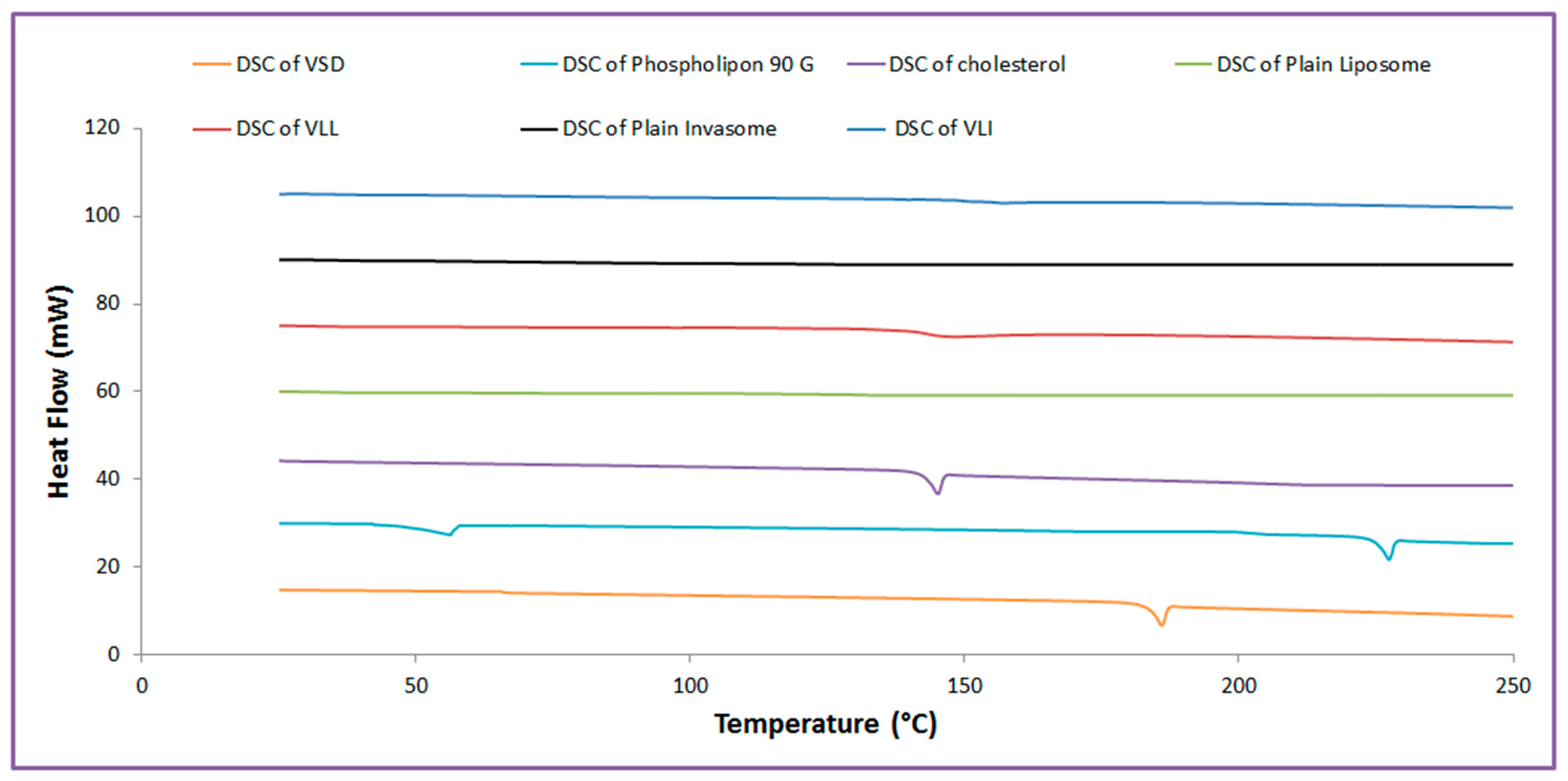
2.3.2. Thermal Analysis Examination
2.3.3. Transmission Electron Microscopy (TEM) Measurement
2.4. Preparation and In Vitro Characterization of Gel Formulations
2.4.1. Preparation and Characterization of VLI and VLL Gel Formulations
2.4.2. Ex Vivo Permeability and Disposition Studies
2.4.3. In Vitro Drug Release Kinetic Studies
2.5. In Vivo Anti-Tumor Characterization of VLI Gel Formulation
2.5.1. Anti-Tumor Activity of VLI Gel Formulation
2.5.2. Toxicity of VLI Gel
2.5.3. In Vivo Permeation and Bioavailability Studies
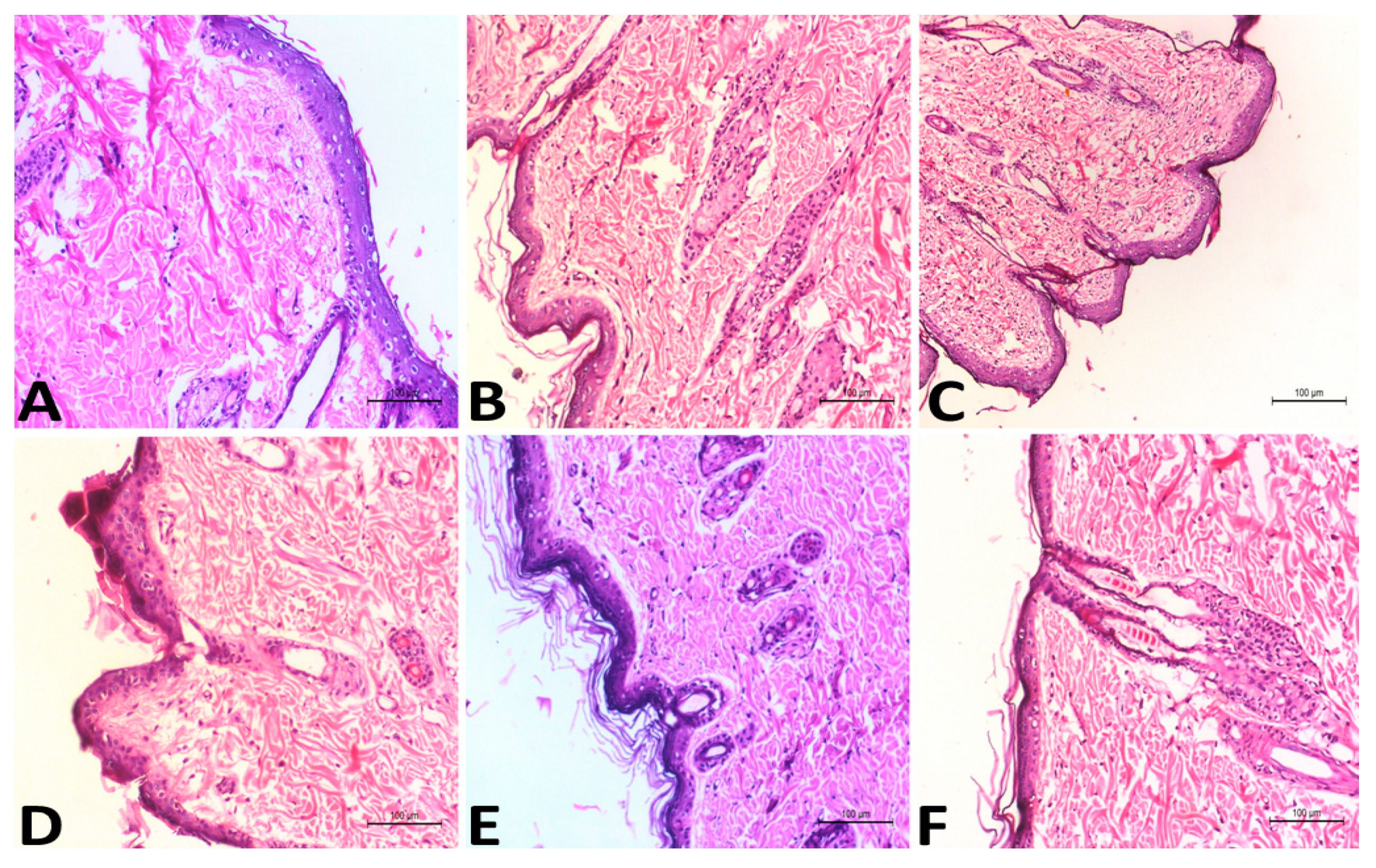
2.5.4. In Vivo Skin Disposition Studies
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. HPLC Quantification of Vismodegib
4.3. Equilibrium Solubility Study
4.4. Preparation of Vismodegib-Loaded Invasomes and Vismodegib-Loaded Liposomes
4.5. In Vitro Evaluation of VLI and VLL Formulations
4.5.1. Entrapment Efficiency Measurement
4.5.2. Vesicle Size and Zeta Potential Measurement
4.5.3. Thermal Analysis Examination
4.5.4. Transmission Electron Microscopy (TEM) Measurement
4.6. Preparation and In Vitro Characterization of Gel Formulations
4.6.1. Incorporation of VLI and VLL Formulations into Carbopol Gel Base
4.6.2. Viscosity Coefficient Determination
4.6.3. Ex Vivo Permeability and Skin Disposition Studies
4.6.4. In Vitro Release Studies
4.6.5. Kinetic Analysis of Release Data
4.7. In Vivo Anti-Tumor Characterization of VLI Gel Formulation
4.7.1. Animal Study
- Group 1 Negative control group
- Group 2 Positive control group
- Group 3 Free VSD suspension
- Group 4 Free VSD gel
- Group 5 VLL gel formulation
- Group 6 VLI gel formulation
4.7.2. Anti-Tumor Activity and Toxicity of VLI Gel Formulation
4.7.3. In Vivo Permeation and Bioavailability Studies
4.7.4. In Vivo Skin Disposition Studies
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gamal, F.A.; Kharshoum, R.M.; Sayed, O.M.; Ela, F.I.; Salem, H.F. Control of basal cell carcinoma via positively charged ethosomes of Vismodegib: In vitro and in vivo studies. J. Drug Deliv. Sci. Technol. 2020, 56, 101556. [Google Scholar] [CrossRef]
- Leiter, U.; Eigentler, T.; Garbe, C. Epidemiology of Skin Cancer. In Sunlight, Vitamin D and Skin Cancer; Springer: Berlin/Heidelberg, Germany, 2014; pp. 120–140. [Google Scholar]
- Leiter, U.; Keim, U.; Garbe, C. Epidemiology of Skin Cancer: Update 2019. In Sunlight, Vitamin D and Skin Cancer; Springer: Berlin/Heidelberg, Germany, 2020; pp. 123–139. [Google Scholar]
- Sayed, O.M.; Gamal, F.A.; Kharshoum, R.M.; Ela, F.I.; Salem, H.F. Treatment of basal cell carcinoma via binary Ethosomes of Vismodegib: In vitro and in vivo studies. AAPS PharmSciTech 2020, 21, 51. [Google Scholar]
- Abdulbaqi, I.M.; Darwis, Y.; Khan, N.; Abou Assi, R.; Khan, A.A. Ethosomal nanocarriers: The impact of constituents and formulation techniques on ethosomal properties, in vivo studies, and clinical trials. Int. J. Nanomed. 2016, 11, 2279. [Google Scholar] [CrossRef] [Green Version]
- Brown, M.B.; Martin, G.P.; Jones, S.A.; Akomeah, F.K. Dermal and transdermal drug delivery systems: Current and future prospects. Drug Deliv. 2006, 13, 175–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahmood, S.; Mandal, U.K.; Chatterjee, B. Transdermal delivery of raloxifene HCl via ethosomal system: Formulation, advanced characterizations and pharmacokinetic evaluation. Int. J. Pharm. 2018, 542, 36–46. [Google Scholar] [CrossRef] [PubMed]
- Goyal, R.; Macri, L.K.; Kaplan, H.M.; Kohn, J. Nanoparticles and nanofibers for topical drug delivery. J. Control. Release 2016, 240, 77–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahman, M.; Akhter, S.; Ahmad, J.; Ahmad, M.Z.; Beg, S.; Ahmad, F.J. Nanomedicine-based drug targeting for psoriasis: Potentials and emerging trends in nanoscale pharmacotherapy. Expert Opin. Drug Deliv. 2015, 12, 635–652. [Google Scholar] [CrossRef] [PubMed]
- Seeni, R.Z.; Krishnamurthy, S.; Chan, J.M. Nanoparticles for Improved Topical Drug Delivery for Skin Diseases. In Perspectives in Micro-and Nanotechnology for Biomedical Applications; World Scientific: Singapore, 2016; pp. 275–294. [Google Scholar]
- Gamal, A.; Saeed, H.; El-Ela, F.I.; Salem, H.F. Improving the Antitumor Activity and Bioavailability of Sonidegib for the Treatment of Skin Cancer. Pharmaceutics 2021, 13, 1560. [Google Scholar] [CrossRef]
- Touitou, E.; Dayan, N.; Bergelson, L.; Godin, B.; Eliaz, M. Ethosomes—Novel vesicular carriers for enhanced delivery: Characterization and skin penetration properties. J. Control. Release 2000, 65, 403–418. [Google Scholar] [CrossRef]
- Salem, H.F.; Gamal, A.; Saeed, H.; Tulbah, A.S. The Impact of Improving Dermal Permeation on the Efficacy and Targeting of Liposome Nanoparticles as a Potential Treatment for Breast Cancer. Pharmaceutics 2021, 13, 1633. [Google Scholar] [CrossRef]
- Dragicevic, N.; Verma, D.D.; Fahr, A. Invasomes: Vesicles for Enhanced Skin Delivery of Drugs. In Percutaneous Penetration Enhancers Chemical Methods in Penetration Enhancement; Springer: Berlin/Heidelberg, Germany, 2016; pp. 77–92. [Google Scholar]
- Ahmed, O.A.; Badr-Eldin, S.M. Development of an optimized avanafil-loaded invasomal transdermal film: Ex vivo skin permeation and in vivo evaluation. Int. J. Pharm. 2019, 570, 118657. [Google Scholar] [CrossRef]
- Kamran, M.; Ahad, A.; Aqil, M.; Imam, S.S.; Sultana, Y.; Ali, A. Design, formulation and optimization of novel soft nano-carriers for transdermal olmesartan medoxomil delivery: In vitro characterization and in vivo pharmacokinetic assessment. Int. J. Pharm. 2016, 505, 147–158. [Google Scholar] [CrossRef]
- El-Feky, G.S.; Mona, M.; Mahmoud, A.A. Flexible nano-sized lipid vesicles for the transdermal delivery of colchicine; in vitro/in vivo investigation. J. Drug Deliv. Sci. Technol. 2019, 49, 24–34. [Google Scholar] [CrossRef]
- Shah, S.M.; Ashtikar, M.; Jain, A.S.; Makhiji, D.T.; Nikam, Y.; Gude, R.P.; Steiniger, F.; Jagtap, A.A.; Nagarsenker, M.S.; Fahr, A. LeciPlex, invasomes, and liposomes: A skin penetration study. Int. J. Pharm. 2015, 490, 391–403. [Google Scholar] [CrossRef]
- Dragicevic-Curic, N.; Scheglmann, D.; Albrecht, V.; Fahr, A. Temoporfin-loaded invasomes: Development, characterization and in vitro skin penetration studies. J. Control. Release 2008, 127, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Dragicevic-Curic, N.; Scheglmann, D.; Albrecht, V.; Fahr, A. Development of different temoporfin-loaded invasomes—novel nanocarriers of temoporfin: Characterization, stability and in vitro skin penetration studies. Colloids Surf. B Biointerfaces 2009, 70, 198–206. [Google Scholar] [CrossRef]
- Qadri, G.R.; Ahad, A.; Aqil, M.; Imam, S.S.; Ali, A. Invasomes of isradipine for enhanced transdermal delivery against hypertension: Formulation, characterization, and in vivo pharmacodynamic study. Artif. Cells Nanomed. Biotechnol. 2017, 45, 139–145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bangham, A.D.; Hill, M.W.; Miller, N. Preparation and Use of Liposomes as Models of Biological Membranes. In Methods in Membrane Biology; Springer: Berlin/Heidelberg, Germany, 1974; pp. 1–68. [Google Scholar]
- Franklin, R.K.; Marcus, S.A.; Talaat, A.M.; Kukanich, B.K.; Sullivan, R.; Krugner, L.A.; Heath, T.D. A novel loading method for doxycycline liposomes for intracellular drug delivery: Characterization of in vitro and in vivo release kinetics and efficacy in a J774A.1 cell line model of mycobacterium smegmatis infection. Drug Metab. Dispos. 2015, 43, 1236–1245. [Google Scholar] [CrossRef] [Green Version]
- Ahad, A.; Aqil, M.; Kohli, K.; Sultana, Y.; Mujeeb, M.; Ali, A. Interactions between novel terpenes and main components of rat and human skin: Mechanistic view for transdermal delivery of propranolol hydrochloride. Curr. Drug Deliv. 2011, 8, 213–224. [Google Scholar] [CrossRef] [PubMed]
- Saffari, M.; Shirazi, F.H.; Moghimi, H.R. Terpene-loaded liposomes and isopropyl myristate as chemical permeation enhancers toward liposomal gene delivery in lung cancer cells; a comparative study. Iran. J. Pharm. Res. IJPR 2016, 15, 261. [Google Scholar] [PubMed]
- Fathalla, D.; Youssef, E.M.; Soliman, G.M. Liposomal and ethosomal gels for the topical delivery of anthralin: Preparation, comparative evaluation and clinical assessment in psoriatic patients. Pharmaceutics 2020, 12, 446. [Google Scholar] [CrossRef]
- Jain, S.; Patel, N.; Madan, P.; Lin, S. Formulation and rheological evaluation of ethosome-loaded carbopol hydrogel for transdermal application. Drug Dev. Ind. Pharm. 2016, 42, 1315–1324. [Google Scholar] [CrossRef]
- Poorahmary Kermany, B. Carbopol Hydrogels for Topical Administration: Treatment of Wounds. Master’s Thesis, Universitetet i Tromsø, Tromsø, Norway, 2010. [Google Scholar]
- Babaie, S.; Bakhshayesh, A.R.; Ha, J.W.; Hamishehkar, H.; Kim, K.H. Invasome: A novel nanocarrier for transdermal drug delivery. Nanomaterials 2020, 10, 341. [Google Scholar] [CrossRef] [Green Version]
- El-Kattan, A.F.; Asbill, C.S.; Kim, N.; Michniak, B.B. The effects of terpene enhancers on the percutaneous permeation of drugs with different lipophilicities. Int. J. Pharm. 2001, 215, 229–240. [Google Scholar] [CrossRef]
- Ahad, A.; Aqil, M.; Kohli, K.; Sultana, Y.; Mujeeb, M.; Ali, A. Role of novel terpenes in transcutaneous permeation of valsartan: Effectiveness and mechanism of action. Drug Dev. Ind. Pharm. 2011, 37, 583–596. [Google Scholar] [CrossRef]
- Ahad, A.; Al-Mohizea, A.M.; Al-Saleh, A.A.; Al-Jenoob, F.I.; Raish, M.; Yassin, A.E.; Alam, M.A. Formulation and characterization of novel soft nanovesicles for enhanced transdermal delivery of eprosartan mesylate. Saudi Pharm. J. 2017, 25, 1040–1046. [Google Scholar] [CrossRef]
- LoRusso, P.M.; Jimeno, A.; Dy, G.; Adjei, A.; Berlin, J.; Leichman, L.; Low, J.A.; Colburn, D.; Chang, I.; Cheeti, S.; et al. Pharmacokinetic dose-scheduling study of hedgehog pathway inhibitor vismodegib (GDC-0449) in patients with locally advanced or metastatic solid tumors. Clin. Cancer Res. 2011, 17, 5774–5782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Proctor, A.E.; Thompson, L.A.; O’Bryant, C.L. Vismodegib: An inhibitor of the Hedgehog signaling pathway in the treatment of basal cell carcinoma. Ann. Pharmacother. 2014, 48, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Calienni, M.N.; Vega, D.M.; Temprana, C.F.; Izquierdo, M.C.; Ybarra, D.E.; Bernabeu, E.; Moretton, M.; Alvira, F.C.; Chiappetta, D.; Alonso, S.D.; et al. The Topical Nanodelivery of Vismodegib Enhances Its Skin Penetration and Performance In Vitro While Reducing Its Toxicity In Vivo. Pharmaceutics 2021, 13, 186. [Google Scholar] [CrossRef] [PubMed]
- Pulusu, V.; Kommarajula, P. Development and validation of a new chromatographic method for the estimation of Vismodegib by RP-HPLC. J. Chromatogr. Sep. Tech. 2019, 10, 2. [Google Scholar]
- Salem, H.F.; Kharshoum, R.M.; Gamal, F.A.; Abo El-Ela, F.I.; Abdellatif, K.R.A. Treatment of breast cancer with engineered novel pH-sensitive triaryl-(Z)-olefin niosomes containing hydrogel: An in vitro and in vivo study. J. Liposome Res. 2020, 30, 126–135. [Google Scholar] [CrossRef] [PubMed]
- Salem, H.F.; Kharshoum, R.M.; Gamal, F.A.; Abo El-Ela, F.I.; Abdellatif, K.R.A. Evaluation and optimization of pH-responsive niosomes as a carrier for efficient treatment of breast cancer. Drug Deliv. Transl. Res. 2018, 8, 633–644. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.-N.; Zhang, Y.T.; Wang, Q.; Xu, L.; Feng, N.P. Enhanced in vitro and in vivo skin deposition of apigenin delivered using ethosomes. Int. J. Pharm. 2014, 460, 280–288. [Google Scholar] [CrossRef] [PubMed]
- Gamal, A.; Kharshoum, R.M.; Sayed, O.M.; Ela, F.I.; Salem, H.F. Proniosomal microcarriers: Impact of constituents on the physicochemical properties of proniosomes as a new approach to enhance inhalation efficiency of dry powder inhalers. AAPS PharmSciTech 2020, 21, 156. [Google Scholar] [CrossRef]
- Zhang, Y.; Huo, M.; Zhou, J.; Zou, A.; Li, W.; Yao, C.; Xie, S. DDSolver: An add-in program for modeling and comparison of drug dissolution profiles. AAPS J. 2010, 12, 263–271. [Google Scholar] [CrossRef] [Green Version]
- Zuo, J.; Gao, Y.; Bou, N.; Lobenberg, R. Evaluation of the DDSolver software applications. BioMed Res. Int. 2014, 2014, 204925. [Google Scholar] [CrossRef] [Green Version]
- Choudhury, H.; Gorain, B.; Karmakar, S.; Pal, T.K. Development and validation of RP-HPLC method: Scope of application in the determination of oil solubility of paclitaxel. J. Chromatogr. Sci. 2014, 52, 68–74. [Google Scholar] [CrossRef] [Green Version]
- Dias, M.F.; De, F.B.; Teixeira, J.; Guerra, M.C.; Fialho, S.L.; Cunha, A.S. In vivo evaluation of antitumoral and antiangiogenic effect of imiquimod-loaded polymeric nanoparticles. Biomed. Pharmacother. 2018, 103, 1107–1114. [Google Scholar] [CrossRef]
- Aziz, R.L.A.; Abdel-wahab, A.; El-Ela, F.I.; Hassan, N.E.; El-Nahass, E.; Ibrahim, M.A.; Khalil, A.A. Dose-dependent ameliorative effects of quercetin and l-Carnitine against atrazine-induced reproductive toxicity in adult male Albino rats. Biomed. Pharmacother. 2018, 102, 855–864. [Google Scholar] [CrossRef]
- Trontelj, J.; Vovk, T.; Bogataj, M.; Mrhar, A. HPLC analysis of raloxifene hydrochloride and its application to drug quality control studies. Pharmacol. Res. 2005, 52, 334–339. [Google Scholar] [CrossRef]
- Praça, F.S.G.; Medina, W.S.; Eloy, J.O.; Petrilli, R.; Campos, P.M.; Ascenso, A.; Bentley, M.V. Evaluation of critical parameters for in vitro skin permeation and penetration studies using animal skin models. Eur. J. Pharm. Sci. 2018, 111, 121–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Formulation Code | Phospholipid %w/w | Cholesterol %w/w | Cineole %v/v | Ethanol %v/v | %EE (%) (Mean ± SD) | Particle Size (nm) (Mean ± SD) | Zeta Potential (mV) (Mean ± SD) | PDI (Mean ± SD) |
|---|---|---|---|---|---|---|---|---|
| VLI | 2 | 0.15 | 1 | 3 | 87.73 ± 3.82 | 188.27 ± 3.25 | −20.5 ± 1.61 | 0.12 ± 0.02 |
| VLL | 2 | 0.15 | 0 | 0 | 72.52 ± 4.72 | 258.67 ± 9.00 | −7.72 ± 1.53 | 0.26 ± 0.02 |
| Formulation Code | Release (%) (Mean ± SD) | Steady-State Flux (µg/cm2/h) (Mean ± SD) | Skin Disposition (µg/cm2) (Mean ± SD) |
|---|---|---|---|
| Free VSD suspension | 99.60 ± 0.96 | 1.35 ± 0.03 | 147.23 ± 3.18 |
| VLL | 58.55 ± 1.04 | 2.73 ± 0.09 | 106.86 ± 2.98 |
| VLI | 71.96 ± 0.86 | 9.83 ± 0.11 | 31.09 ± 2.68 |
| Free VSD gel | 96.60 ± 1.24 | 1.17 ± 0.07 | 153.97 ± 3.95 |
| VLL gel | 49.76 ± 0.88 | 2.43 ± 0.06 | 119.56 ± 4.40 |
| VLI gel | 62.12 ± 1.27 | 9.14 ± 0.09 | 51.27 ± 4.84 |
| Pharmacokinetic Parameters | VLI Gel | VLL Gel | Oral Free VSD Suspension |
|---|---|---|---|
| Cpmax (μg/mL) | 7.11 ± 0.82 # | 8.02 ± 0.66 | 10.76 ± 0.57 |
| Tmax (h) | 8 | 8 | 4 |
| AUC(0–24) (μg.h/mL) | 102.70 ± 5.69 *,# | 80.82 ± 5.03 | 58.18 ± 4.03 |
| AUC(0–inf) (μg.h/mL) | 211.34 ± 9.70 *,# | 91.83 ± 7.13 | 58.86 ± 3.83 |
| AUMC(0–inf) (μg.h/mL) | 7002.80 ± 135.41 *,# | 1119.99 ± 112.32 | 361.34 ± 30.19 |
| MRT (h) | 33.13 ± 2.13 *,# | 12.19 ± 2.33 | 6.13 ± 2.17 |
| t0.5 (h) | 19.98 ± 1.85 *,# | 6.43 ± 1.05 | 3.83 ± 0.85 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Salem, H.F.; Gamal, A.; Saeed, H.; Kamal, M.; Tulbah, A.S. Enhancing the Bioavailability and Efficacy of Vismodegib for the Control of Skin Cancer: In Vitro and In Vivo Studies. Pharmaceuticals 2022, 15, 126. https://doi.org/10.3390/ph15020126
Salem HF, Gamal A, Saeed H, Kamal M, Tulbah AS. Enhancing the Bioavailability and Efficacy of Vismodegib for the Control of Skin Cancer: In Vitro and In Vivo Studies. Pharmaceuticals. 2022; 15(2):126. https://doi.org/10.3390/ph15020126
Chicago/Turabian StyleSalem, Heba F., Amr Gamal, Haitham Saeed, Marwa Kamal, and Alaa S. Tulbah. 2022. "Enhancing the Bioavailability and Efficacy of Vismodegib for the Control of Skin Cancer: In Vitro and In Vivo Studies" Pharmaceuticals 15, no. 2: 126. https://doi.org/10.3390/ph15020126
APA StyleSalem, H. F., Gamal, A., Saeed, H., Kamal, M., & Tulbah, A. S. (2022). Enhancing the Bioavailability and Efficacy of Vismodegib for the Control of Skin Cancer: In Vitro and In Vivo Studies. Pharmaceuticals, 15(2), 126. https://doi.org/10.3390/ph15020126