
Galactosylated Prodrugs: A Strategy to Improve the Profile of Nonsteroidal Anti-Inflammatory Drugs

 ,
,  , , , ,
, , , , 
 and
and
Abstract
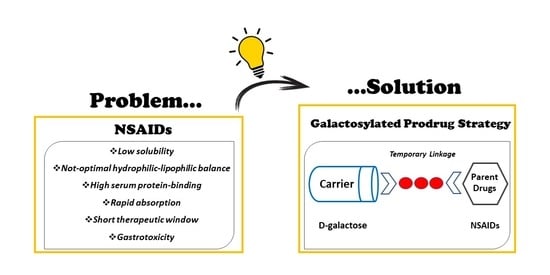
:
1. Introduction
2. Results
2.1. Physicochemical Characterization
2.1.1. Lipophilicity
2.1.2. Solubility Assay
2.1.3. Stability in SGF and PBS
2.1.4. Stability in Human Serum
2.1.5. Serum Protein Binding
2.2. Ex Vivo Absorption Evaluation
2.3. In Vivo Studies
2.3.1. Effect of Parent Drugs and Galactosylated Prodrugs on Paw Edema in Mice
2.3.2. Effect of Parent Drugs and Galactosylated Prodrugs on the Acetic-Acid-Induced Writhing Test in Mice
2.3.3. Effect of Parent Drugs and Galactosylated Prodrugs on Ulcerogenic Activity
3. Discussion
4. Materials and Methods
4.1. Chemicals and Materials
4.1.1. Lipophilicity
4.1.2. Solubility Assay
4.1.3. Stability in SGF and PBS
4.1.4. Stability in Human Serum
4.1.5. Serum Protein Binding
- vol.bound: calculated by dividing the weight of the bound fraction (difference between the weights of the empty sample reservoir and that after ultrafiltration) by its density (0.991 g/mL assessed by weighing five replicates of a known volume of bound fraction).
- vol.unbound: calculated by dividing the weight of the unbound fraction (difference between the weight of the ultrafiltrate cup before and after ultrafiltration) by its density (0.999 g/mL assessed by weighing five replicates of a known volume of unbound fraction).
- concbound: calculated using the RP-HPLC method.
- concunbound: calculated using the RP-HPLC method (calibration with standard additions).
4.1.6. HPLC Analysis
4.2. Permeation Measurements across Excised Rat Small Intestine
4.3. Animals
4.3.1. Carrageenan-Induced Hyperalgesia
4.3.2. Acetic Acid Writhing Test
4.3.3. Ulcerogenicity Studies
4.4. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rautio, J.; Kumpulainen, H.; Heimbach, T.; Oliyai, R.; Oh, D.; Järvinen, T.; Savolainen, J. Prodrugs: Design and clinical applications. Nat. Rev. Drug Discov. 2008, 7, 255–270. [Google Scholar] [CrossRef] [PubMed]
- Huttunen, K.; Raunio, H.; Rautio, J. Prodrugs: Design and clinical applications. Pharmacol. Rev. 2011, 63, 750–771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stella, V.J.; Nti-Addae, K.W. Prodrug strategies to overcome poor water solubility. Adv. Drug Deliv. Rev. 2007, 59, 677–694. [Google Scholar] [CrossRef] [PubMed]
- Abu-jaish, A.; Jumaa, S.; Karaman, R. Prodrug Overview. In Prodrugs Design: A New Era; Karaman, R., Ed.; Nova Publisher: Hauppauge, NY, USA, 2014; pp. 77–102. [Google Scholar]
- Chung, M.C.; Silva, A.T.D.A.; Castro, L.F.; Güido, R.V.C.; Nassute, J.C.; Ferreira, E.I. Latenciação e formas avançadas de transporte de fármacos. Rev. Bras. Ciênc. Farm. 2005, 41, 155–180. [Google Scholar] [CrossRef] [Green Version]
- Redasani, V.K.; Bari, S.B. Prodrug Design: Perspectives, Approaches and Applications in Medicinal Chemistry, 1st ed.; Academic Press: London, UK, 2015. [Google Scholar]
- Zovko, M.; Zorc, B.; Novak, P.; Tepeš, P.; Cetina-Cižmek, B.; Horvat, M. Macromolecular prodrugs: XI. Synthesis and characterization of polymer–estradiol conjugate. Int. J. Pharm. 2004, 285, 35–41. [Google Scholar] [PubMed]
- Fathia, M.I.; Mubark, E.O. Review: Carbohydrates chemistry. Asian J. Sci. Technol. 2017, 7, 5038–5043. [Google Scholar]
- Mishra, S.; Upadhaya, K.; Mishra, K.B.; Shukla, A.K.; Tripathi, R.P.; Tiwari, V.K. Carbohydrate-based therapeutics: A frontier in drug discovery and development. Stud. Nat. Prod. Chem. 2016, 49, 307–361. [Google Scholar]
- Bertozzi, C.R.; Kiessling, L.L. Chemical glycobiology. Science 2001, 291, 2357–2364. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Delbianco, M.; Anggara, K.; Michnowicz, T.; Pardo-Vargas, A.; Bharate, P.; Sen, S.; Pristl, M.; Rauschenbach, S.; Schlickum, U.; et al. Publisher Correction: Imaging single glycans. Nature 2020, 582, 375–378. [Google Scholar] [CrossRef]
- Wang, J.; Zhang, Y.; Lu, Q.; Xing, D.; Zhang, R. Exploring Carbohydrates for Therapeutics: A Review on Future Directions. Front. Pharmacol. 2021, 12, 756724. [Google Scholar] [CrossRef]
- Jornada, D.H.; dos Santos Fernandes, G.F.; Chiba, D.E.; de Melo, T.R.; dos Santos, J.L.; Chung, M.C. The Prodrug Approach: A Successful Tool for Improving Drug Solubility. Molecules 2016, 21, 42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schuh, L.; Müller, P.; Torvisco, A.; Stueger, H.; Wrodnigg, T.M.; Haas, M. Synthesis of D-Galactose-Substituted Acylsilanes and Acylgermanes. Model Compounds for Visible Light Photoinitiators with Intriguing High Solubility. Organometallics 2021, 40, 1185–1189. [Google Scholar] [CrossRef] [PubMed]
- Di Guida, F.; Pirozzi, C.; Magliocca, S.; Santoro, A.; Lama, A.; Russo, R.; Nieddu, M.; Burrai, L.; Boatto, G.; Mollica, M.P.; et al. Galactosylated Pro−Drug of Ursodeoxycholic Acid: Design, Synthesis, Characterization, and Pharmacological Effects in a Rat Model of Estrogen-Induced Cholestasis. Mol. Pharm. 2018, 15, 21–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perrone, M.R.; Vitale, P.; Ferorelli, S.; Boccarelli, A.; Coluccia, M.; Pannunzio, A.; Campanella, F.; Di Mauro, G.; Bonaccorso, C.; Fortuna, C.G.; et al. Effect of mofezolac-galactose distance in conjugates targeting cyclooxygenase (COX)-1 and CNS GLUT-1 carrier. Eur. J. Med. Chem. 2017, 141, 404–416. [Google Scholar] [CrossRef] [PubMed]
- Lima, A.O.D.L.; Salamanca-Fernández, E.; Del Rey, E.J.A.; Hoces, A.M.; Vera, M.G.; Tamayo, C.B. Safety considerations during prescription of non-steroidal anti-inflammatory drugs (NSAIDs), through a review of systematic reviews. An. Sist. Sanit. Navar. 2021, 44, 261–273. [Google Scholar] [CrossRef]
- Bindu, S.; Mazumder, S.; Bandyopadhyay, U. Non-steroidal anti-inflammatory drugs (NSAIDs) and organ damage: A current perspective. Biochem. Pharmacol. 2020, 180, 114147. [Google Scholar] [CrossRef]
- Tsutsumi, S.; Gotoh, T.; Tomisato, W.; Mima, S.; Hoshino, T.; Hwang, H.J.; Takenaka, H.; Tsuchiya, T.; Mori, M.; Mizushima, T. Endoplasmic reticulum stress response is involved in nonsteroidal anti-inflammatory drug-induced apoptosis. Cell Death Differ. 2004, 11, 1009–1016. [Google Scholar] [CrossRef] [Green Version]
- James, D.S. The multisystem adverse effects of NSAID therapy. J. Am. Osteopath. Assoc. 1999, 99 (Suppl. 11), S1–S7. [Google Scholar] [CrossRef] [Green Version]
- Hooper, W.P.; Gerber, H. Monte Carlo simulations of laser-generated sea surface aureole. Appl. Opt. 1988, 27, 5111–5118. [Google Scholar] [CrossRef]
- Arfe, A.; Scotti, L.; Varas-Lorenzo, C.; Nicotra, F.; Zambon, A.; Kollhorst, B.; Schink, T.; Garbe, E.; Herings, R.; Straatman, H.; et al. Non-steroidal anti-inflammatory drugs and risk of heart failure in four European countries: Nested case-control study. BMJ 2016, 354, i4857. [Google Scholar] [CrossRef] [Green Version]
- Dreischulte, T.; Morales, D.R.; Bell, S.; Guthrie, B. Combined use of nonsteroidal anti-inflammatory drugs with diuretics and/or renin-angiotensin system inhibitors in the community increases the risk of acute kidney injury. Kidney Int. 2015, 88, 396–403. [Google Scholar] [CrossRef] [PubMed]
- Angi, A.; Lakatos, L. Adverse effects of non-steroidal anti-inflammatory drugs in the lower gastrointestinal tract. Orv. Hetil. 2009, 150, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Sodano, F.; Avallone, B.; Tizzano, M.; Fogliano, C.; Rolando, B.; Gazzano, E.; Riganti, C.; Magliocca, S.; Cuozzo, M.; Albrizio, S.; et al. Ketogal Safety Profile in Human Primary Colonic Epithelial Cells and in Mice. Pharmaceuticals 2021, 14, 1149. [Google Scholar] [CrossRef] [PubMed]
- Sodano, F.; Lazzarato, L.; Rolando, B.; Spyrakis, F.; De Caro, C.; Magliocca, S.; Marabello, D.; Chegaev, K.; Gazzano, E.; Riganti, C.; et al. Paracetamol−Galactose Conjugate: A Novel Prodrug for an Old Analgesic Drug. Mol. Pharm. 2019, 16, 4181–4189. [Google Scholar] [CrossRef] [PubMed]
- Magliocca, S.; De Caro, C.; Lazzarato, L.; Russo, R.; Rolando, B.; Chegaev, K.; Marini, E.; Nieddu, M.; Burrai, L.; Boatto, G.; et al. Aceclofenac−Galactose Conjugate: Design, Synthesis, Characterization, and Pharmacological and Toxicological Evaluations. Mol. Pharm. 2018, 15, 3101–3110. [Google Scholar] [CrossRef] [PubMed]
- Magliocca, S.; Sodano, F.; Nieddu, M.; Burrai, L.; Boatto, G.; Rimoli, M.G. New Galactosylated NSAIDS prodrugs in a green context: Synthesis and stability. IJPSR 2017, 8, 1575–1581. [Google Scholar]
- Ruan, L.P.; Chen, S.; Yu, B.Y.; Zhu, D.N.; Cordell, G.A.; Qiu, S.X. Prediction of human absorption of natural compounds by the non-everted rat intestinal sac model. Eur. J. Med. Chem. 2006, 41, 605–610. [Google Scholar] [CrossRef]
- Luo, Z.; Liu, Y.; Zhao, B.; Tang, M.; Dong, H.; Zhang, L.; Lv, B.; Wei, L. Ex vivo and in situ approaches used to study intestinal absorption. J. Pharmacol. Toxicol. Methods 2013, 68, 208–216. [Google Scholar] [CrossRef]
- Kapoor, V.K.; Kaur, A. Drug-glycosidation and drug development. Mini Rev. Med. Chem. 2013, 13, 584–596. [Google Scholar] [CrossRef]
- Guerrero, A.; Guiho, R.; Herranz, N.; Uren, A.; Withers, D.J.; Martínez-Barbera, J.P.; Tietze, L.F.; Gil, J. Galactose-modified duocarmycin prodrugs as senolytics. Aging Cell. 2020, 19, e13133. [Google Scholar] [CrossRef] [Green Version]
- Verbeeck, R.K.; Blackburn, J.L.; Loewen, G.R. Clinical Pharmacokinetics of Non-steroidal Anti-inflammatory Drugs. Clin. Pharm. 1983, 8, 297–331. [Google Scholar] [CrossRef] [PubMed]
- Ashraf, Z.; Alamgeer, M.K.; Hassan, M.; Abdullah, S.; Waheed, M.; Ahsan, H.; Kim, S.J. Flurbiprofen–antioxidant mutual prodrugs as safer nonsteroidal anti-inflammatory drugs: Synthesis, pharmacological investigation, and computational molecular modeling. Drug Des. Devel. Ther. 2016, 10, 2401–2419. [Google Scholar] [PubMed] [Green Version]
- Dhakane, V.D.; Thakare, V.N.; Dongare, S.B.; Bhale, P.S.; Mule, Y.B.; Bandgar, B.P.; Chavan, H.V. Preparation and Pharmacological Evaluation of Novel Orally Active Ester Prodrugs of Ketoprofen with Non-Ulcerogenic Property. Chem. Biol. Drug Des. 2016, 87, 878–884. [Google Scholar] [CrossRef] [PubMed]
- Maciel, H.P.; Cardoso, L.G.; Ferreira, L.R.; Perazzo, F.F.; Carvalho, J.C.T. Anti-inflammatory and ulcerogenic effects of indomethacin and tenoxicam in combination with cimetidine. Inflammopharmacology 2004, 2, 203–210. [Google Scholar] [CrossRef]
- Jarosz, M.; Szkaradek, N.; Marona, H.; Nowak, G.; Młyniec, K.; Librowski, T. Evaluation of anti-inflammatory and ulcerogenic potential of zinc–ibuprofen and zinc–naproxen complexes in rats. Inflammopharmacology 2017, 25, 653–663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cocco, M.; Pellegrini, C.; Martínez-Banaclocha, H.; Giorgis, M.; Marini, E.; Costale, A.; Miglio, G.; Fornai, M.; Antonioli, L.; López-Castejón, G.; et al. Development of an Acrylate Derivative Targeting the NLRP3 Inflam-masome for the Treatment of Inflammatory Bowel Disease. J. Med. Chem. 2017, 60, 3656–3671. [Google Scholar] [CrossRef] [Green Version]
- Posadas, I.; Bucci, M.; Roviezzo, F.; Rossi, A.; Parente, L.; Sautebin, L.; Cirino, G. Carrageenan-induced mouse paw oedema is biphasic, age-weight dependent and displays differential nitric oxide cyclooxygenase-2 expression. Br. J. Pharmacol. 2004, 142, 331–338. [Google Scholar] [CrossRef]
- Hokanson, G. Acetic acid for analgesic screening. J. Nat. Prod. 1978, 41, 497–498. [Google Scholar]
- Chan, C.C.; Boyce, S.; Brideau, C.; Ford-Hutchinson, A.W.; Gordon, R.; Guay, D.; Hill, R.G.; Li, C.S.; Mancini, J.; Penneton, M.; et al. Pharmacology of a selective cyclooxygenase-2 inhibitor, L-745,337: A novel nonsteroidal anti-inflammatory agent with an ulcerogenic sparing effect in rat and nonhuman primate stomach. J. Pharmacol. Exp. Ther. 1995, 3, 1531–1537. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Lipophilicity | Solubility at 25 °C (mM) c | |||
|---|---|---|---|---|---|
| ClogP a | logD7.4 b | Water | SGF pH 1.2 | PBS pH 7.4 | |
| IbuGAL | 1.92 | 1.73 ± 0.04 | 1.94 | 2.53 | 2.93 |
| Ibuprofen | 3.68 | 0.82 ± 0.04 | 0.22 | 0.58 | 23.5 |
| OkyGAL | 1.00 | 0.92 ± 0.01 | 23.36 | 8.34 | 5.53 |
| Ketoprofen | 2.76 | −0.22 ± 0.03 | 0.46 | 0.21 | 7.71 |
| FluGAL | 1.99 | 1.89 ± 0.09 | 5.21 | 1.44 | 1.01 |
| Flurbiprofen | 3.75 | 0.81 ± 0.01 | 0.07 | 0.02 | 5.43 |
| IndoGAL | 2.42 | 2.05 ± 0.06 | 0.07 | 0.08 | 0.13 |
| Indomethacin | 4.18 | 0.91 ± 0.01 | 0.07 | <LOQ d | 2.25 |
| Compound | Stability at 37 °C % Compounds after 24 h of Incubation (Mean ± SD) | Serum Protein Binding % Bound a | ||
|---|---|---|---|---|
| SGF pH 1.2 a | PBS pH 7.4 a | Human Serum a | ||
| IbuGAL | 98.3 ± 1.0 | 81.2 ± 2.5 | 54.1 ± 2.2 | 97.2 ± 0.4 |
| Ibuprofen (IbuGAL) | 3.4 ± 1.1 | 9.4 ± 1.5 | 40.8 ± 6.2 | |
| Ibuprofen | stable | stable | stable | 99.98 ± 0.01 |
| OkyGAL | 97.9 ± 0.7 | 91.0 ± 0.2 | 63.0 ± 8.7 | 95.0 ± 1.6 |
| Ketoprofen (OkyGAL) | 1.4 ± 0.5 | 9.6 ± 0.8 | 30.4 ± 7.7 | |
| Ketoprofen | stable | stable | stable | 99.98 ± 0.01 |
| FluGAL | 98.9 ± 0.1 | 87.3 ± 2.1 | 57.3 ± 1.4 | 96.1 ± 0.5 |
| Flurbiprofen (FluGAL) | 1.1 ± 0.4 | 11.6 ± 0.8 | 44.5 ± 2.4 | |
| Flurbiprofen | stable | stable | stable | 99.98 ± 0.01 |
| IndoGAL | 97.8 ± 0.4 | 89.0 ± 1.6 | 82.3 ± 2.2 | 99.9 ± 0.1 |
| Indomethacin (IndoGAL) | 2.6 ± 0.5 | 11.5 ± 1.5 | 17.8 ± 4.6 | |
| Indomethacin | stable | stable | stable | 99.98 ± 0.01 |
| Compound | Fraction Absorbed (Fa) ± SE | ||
|---|---|---|---|
| Duodenum | Jejunum | Ileum | |
| IbuGAL | 0.013 ± 0.006 | 0.026 ± 0.014 | 0.031 ± 0.016 |
| Ibuprofen (IbuGAL) | 0.043 ± 0.015 | 0.028 ± 0.011 | 0.036 ± 0.011 |
| Ibuprofen | 0.94 ± 0.2 | 0.84 ± 0.11 | 0.98 ± 0.10 |
| OkyGAL | 0.038 ± 0.012 | 0.015 ± 0.012 | 0.018 ± 0.006 |
| Ketoprofen (OkyGAL) | 0.10 ± 0.004 | 0.060 ± 0.018 | 0.076 ± 0.007 |
| Ketoprofen | 0.53 ± 0.02 | 0.48 ± 0.03 | 0.46 ± 0.02 |
| FluGAL | 0.060 ± 0.014 | 0.031 ± 0.015 | 0.033 ± 0.008 |
| Flurbiprofen (FluGAL) | 0.13 ± 0.01 | 0.075 ± 0.023 | 0.096 ± 0.009 |
| Flurbiprofen | 0.69 ± 0.02 | 0.62 ± 0.04 | 0.57 ± 0.04 |
| IndoGAL | Not Evaluable | Not Evaluable | Not Evaluable |
| Indomethacin (IndoGAL) | |||
| Indomethacin | 0.040 ± 0.005 | 0.047 ± 0.012 | 0.094 ± 0.017 |
| Compounds | Oral Dose (mg/kg) | Gastric Lesion Score (4 h) a | Gastric Lesion Score (48 h) a |
|---|---|---|---|
| Control (CMC 0.5%) | - | 0 | 0 |
| Ibuprofen | 10 | 2 ## | 1.5 |
| IbuGAL | 17.8 | 0 ** | 1 |
| Ketoprofen | 10 | 2.5 ### | 1.5 |
| OkyGAL | 16.4 | 0 **** | 1 |
| Flurbiprofen | 10 | 2 ## | 1.5 |
| FluGAL | 16.7 | 0 ** | 0.5 |
| Indomethacin | 10 | 3 #### | 2 ## |
| IndoGAL | 14.5 | 0 **** | 1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sodano, F.; Cristiano, C.; Rolando, B.; Marini, E.; Lazzarato, L.; Cuozzo, M.; Albrizio, S.; Russo, R.; Rimoli, M.G. Galactosylated Prodrugs: A Strategy to Improve the Profile of Nonsteroidal Anti-Inflammatory Drugs. Pharmaceuticals 2022, 15, 552. https://doi.org/10.3390/ph15050552
Sodano F, Cristiano C, Rolando B, Marini E, Lazzarato L, Cuozzo M, Albrizio S, Russo R, Rimoli MG. Galactosylated Prodrugs: A Strategy to Improve the Profile of Nonsteroidal Anti-Inflammatory Drugs. Pharmaceuticals. 2022; 15(5):552. https://doi.org/10.3390/ph15050552
Chicago/Turabian StyleSodano, Federica, Claudia Cristiano, Barbara Rolando, Elisabetta Marini, Loretta Lazzarato, Mariarosaria Cuozzo, Stefania Albrizio, Roberto Russo, and Maria Grazia Rimoli. 2022. "Galactosylated Prodrugs: A Strategy to Improve the Profile of Nonsteroidal Anti-Inflammatory Drugs" Pharmaceuticals 15, no. 5: 552. https://doi.org/10.3390/ph15050552
APA StyleSodano, F., Cristiano, C., Rolando, B., Marini, E., Lazzarato, L., Cuozzo, M., Albrizio, S., Russo, R., & Rimoli, M. G. (2022). Galactosylated Prodrugs: A Strategy to Improve the Profile of Nonsteroidal Anti-Inflammatory Drugs. Pharmaceuticals, 15(5), 552. https://doi.org/10.3390/ph15050552