
A Versatile Class of 1,4,4-Trisubstituted Piperidines Block Coronavirus Replication In Vitro

 , , ,
, , ,  , , , , and
, , , , and
Abstract
:1. Introduction
2. Results
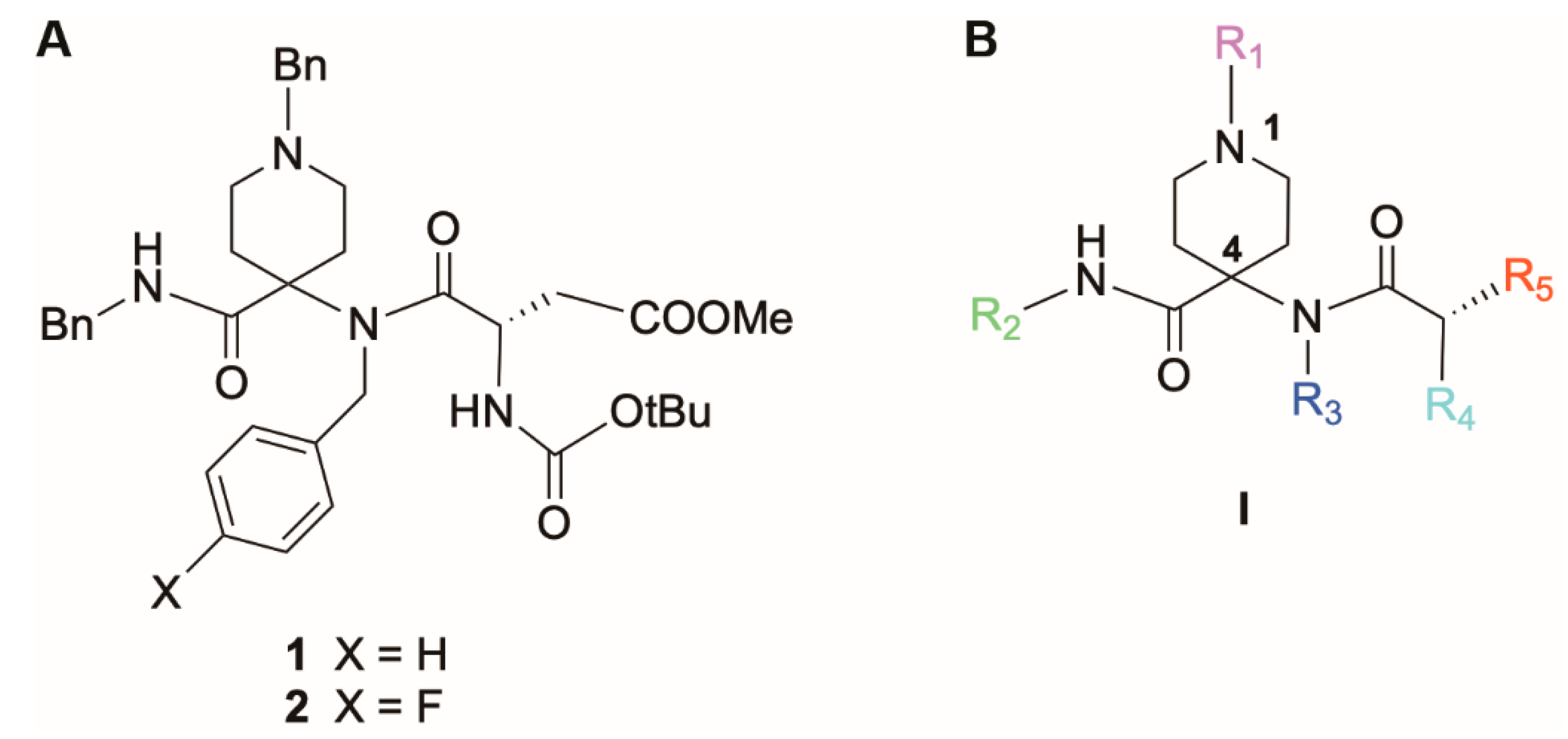
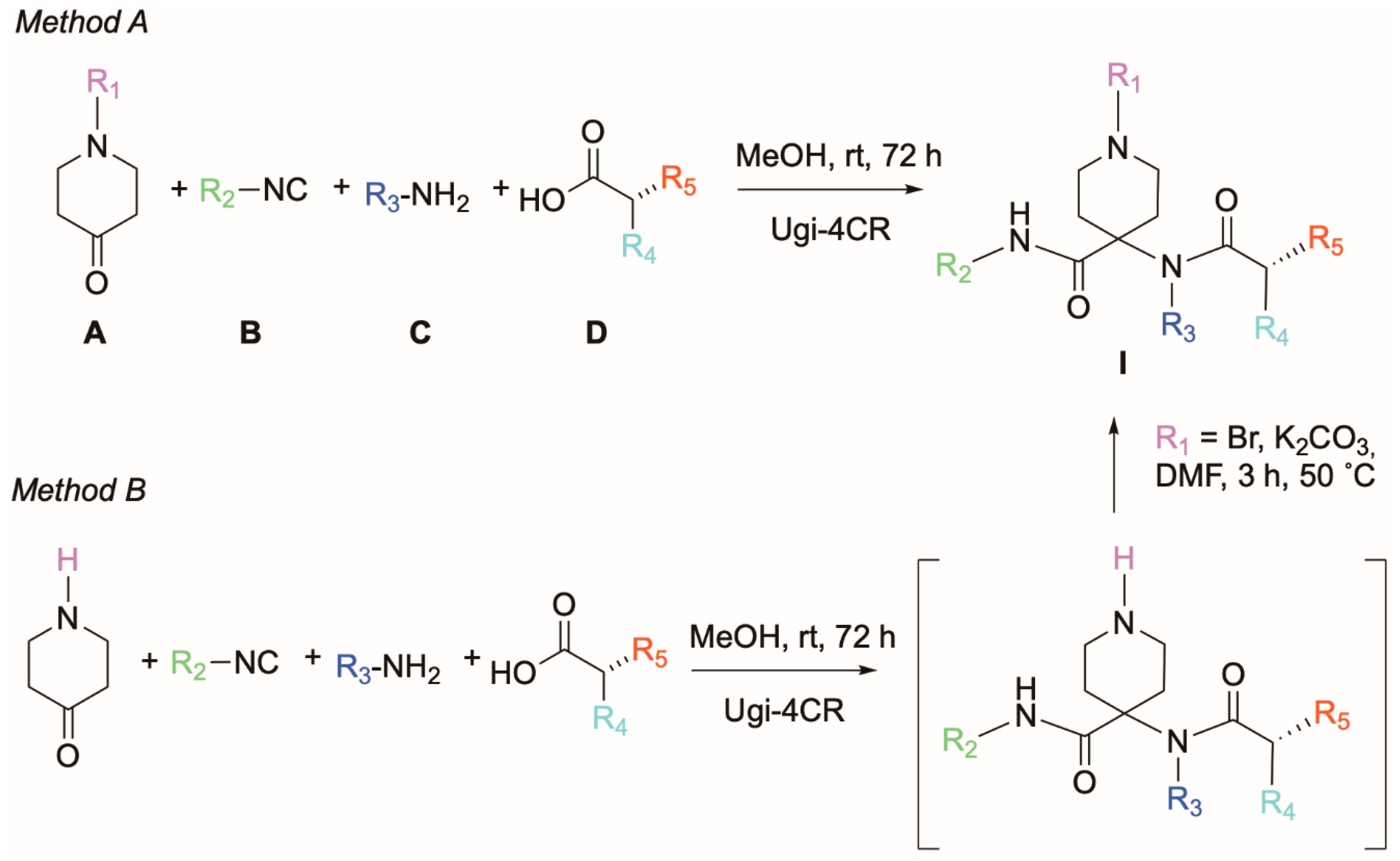
2.1. Chemical Synthesis
2.2. Biological Activity
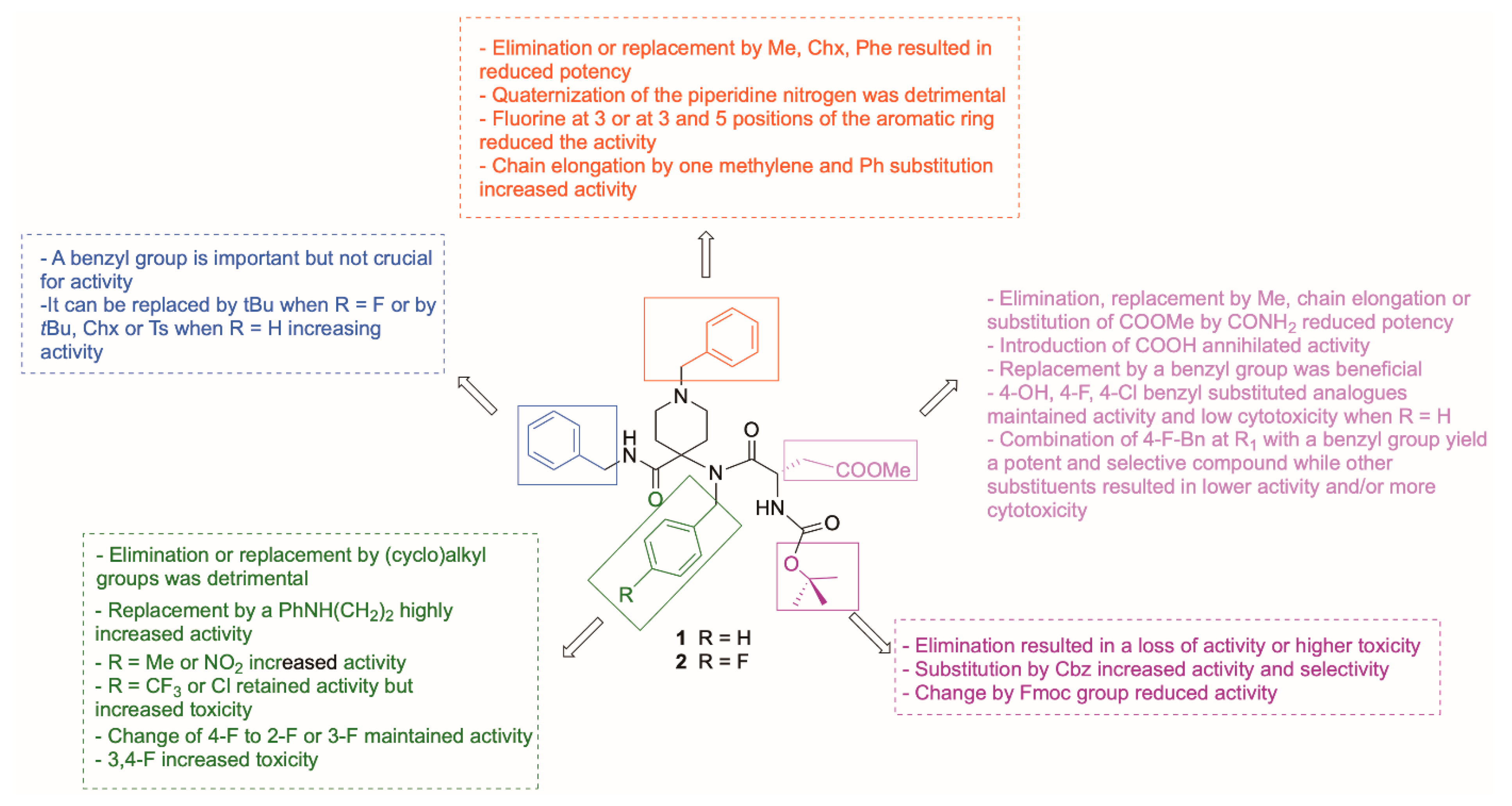
2.2.1. SAR Analysis for Antiviral Activity against HCoV-229E
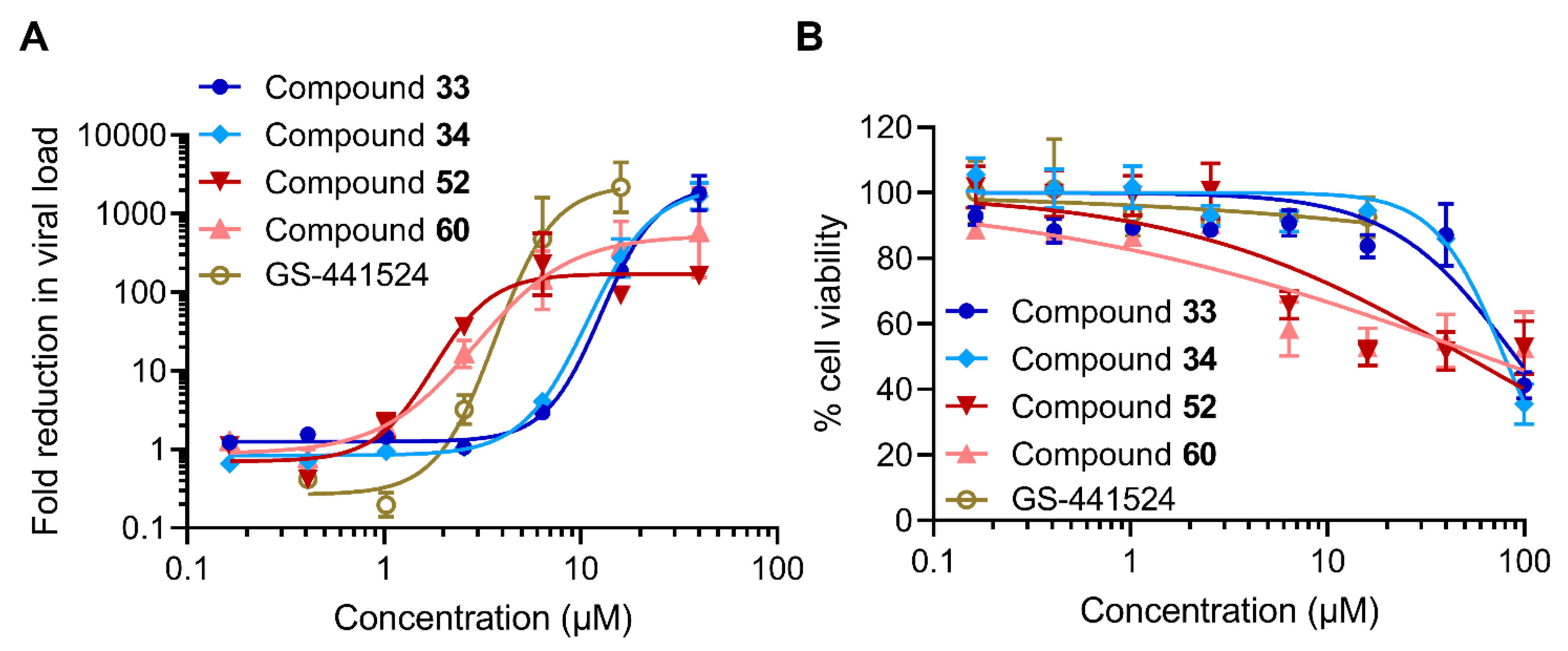
2.2.2. Inhibitory Activity against SARS-CoV-2
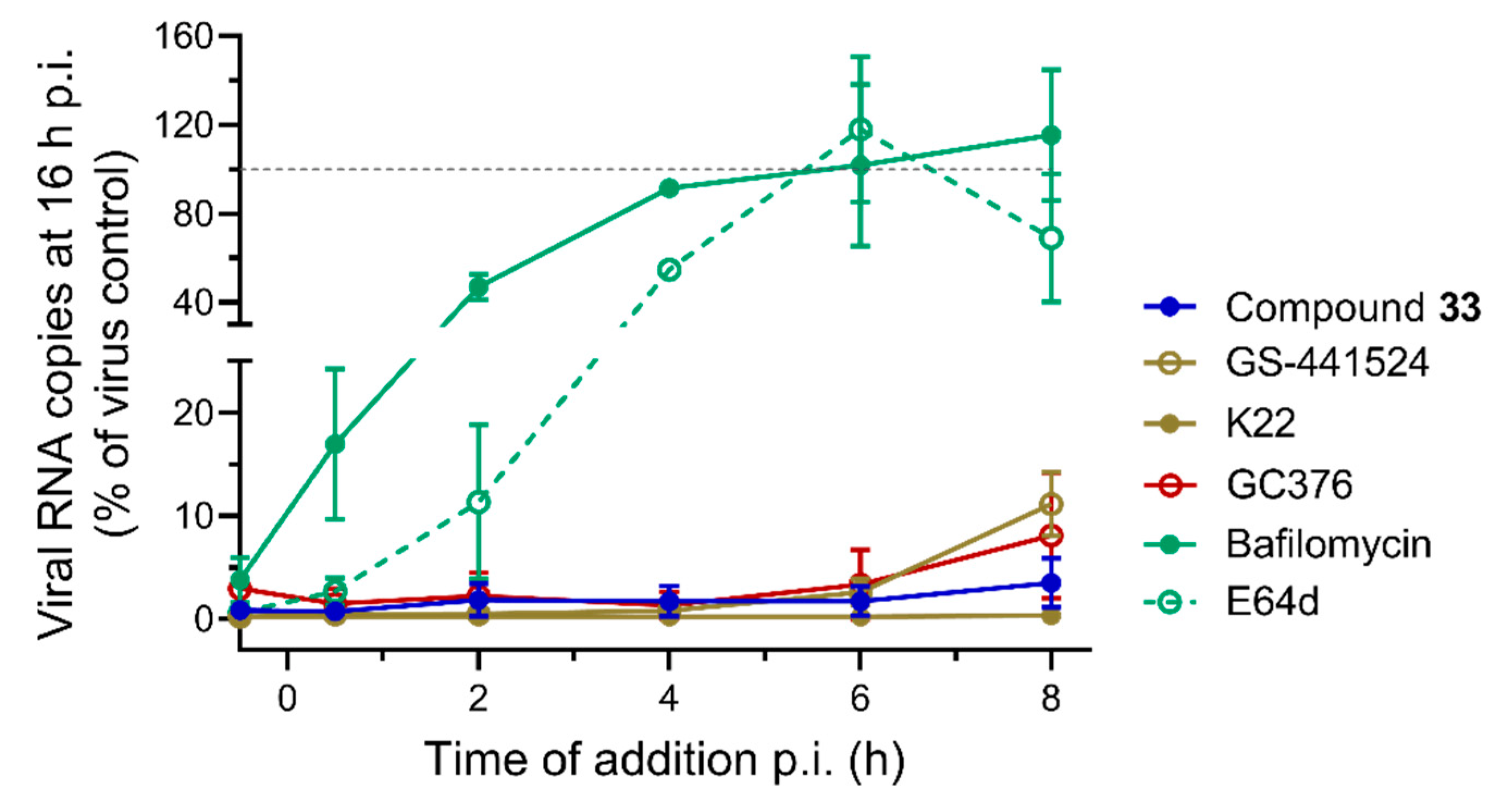
2.2.3. Time-of-Compound Addition Profile
2.2.4. Evaluation against Enzymes of the SARS-CoV-2 Replication–Transcription Complex and the Two Viral Proteases
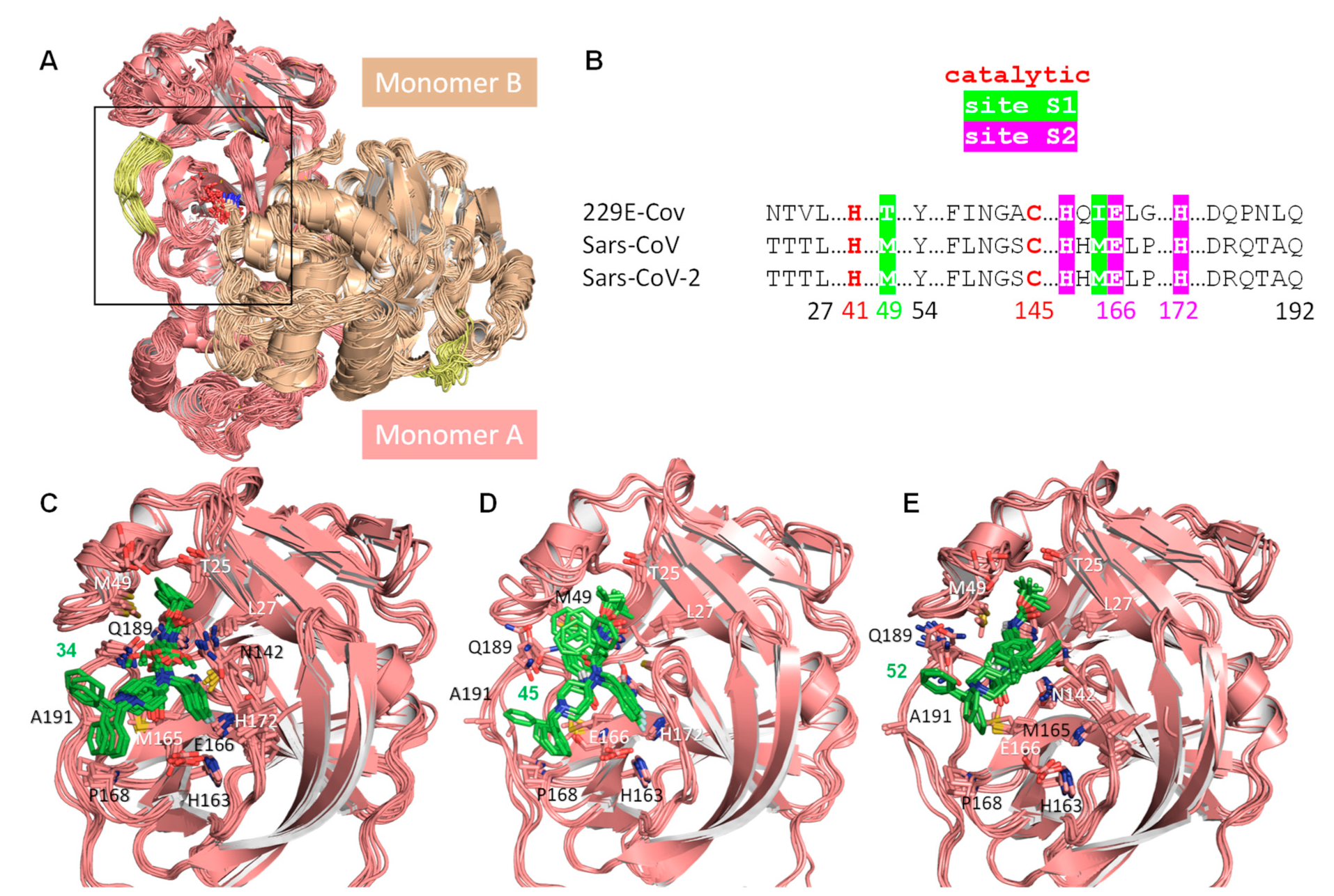
2.3. In Silico Modeling
3. Discussion
4. Materials and Methods
4.1. Chemical Synthesis
4.1.1. Instrumentation and Chemicals
4.1.2. General Procedure for the Ugi Reaction (Method A)
4.1.3. General Ugi Reaction Protocol followed by Alkylation (Method B)
4.2. Biological Procedures
4.2.1. Cytopathic Effect Reduction Assay with HCoV-229E
4.2.2. Time-Of-Compound Addition Assay with HCoV-229E
4.2.3. Antiviral Evaluation against SARS-CoV-2
4.2.4. Production of SARS-CoV-2 Recombinant Proteins
4.2.5. Enzymatic Assays
4.3. Computational Methods
4.3.1. Protein Models
4.3.2. Ligand Preparation
4.3.3. Automated Ligand Docking
4.3.4. Energy Minimization and Molecular Dynamics Simulations
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Abdelshaheed, M.M.; Fawzy, I.M.; El-Subbagh, H.I.; Youssef, K.M. Piperidine nucleus in the field of drug discovery. Future J. Pharm. Sci. 2021, 7, 188. [Google Scholar] [CrossRef]
- Vardanyan, R. Piperidine-Based Drug Discovery; Vardanyan, R., Ed.; Elsevier: Amsterdam, The Netherlands, 2017. [Google Scholar]
- Ardakani, L.S.; Arabmarkadeh, A.A.; Kazemi, M. Multicomponent synthesis of highly functionalized piperidines. Synth. Commun. 2021, 51, 856–879. [Google Scholar] [CrossRef]
- Graebin, C.S.; Ribeiro, F.V.; Rogério, K.R.; Kümmerle, A.E. Multicomponent reactions for the synthesis of bioactive compounds: A review. Curr. Org. Synth. 2019, 16, 855–899. [Google Scholar] [CrossRef] [PubMed]
- Dömling, A.; Wang, W.; Wang, K. Chemistry and biology of multicomponent reactions. Chem. Rev. 2012, 112, 3083–3135. [Google Scholar] [CrossRef]
- Ugi, I.; Meyr, R.; Fetzer, U.; Steinbrückner, C. Versuche mit Isonitrilen. Angew. Chem. 1959, 71, 386–387. [Google Scholar]
- Dömling, A.; Ugi, I.I. Multicomponent reactions with isocyanides. Angew. Chem. Int. Ed. Engl. 2000, 39, 3168–3210. [Google Scholar] [CrossRef]
- Fouad, M.A.; Abdel-Hamid, H.; Ayoup, M.S. Two decades of recent advances of Ugi reactions: Synthetic and pharmaceutical applications. RSC Adv. 2020, 10, 42644–42681. [Google Scholar] [CrossRef]
- de Castro, S.; Camarasa, M.J.; Balzarini, J.; Velazquez, S. Discovery and SAR studies of a novel class of cytotoxic 1,4-disubstituted piperidines via Ugi reaction. Eur. J. Med. Chem. 2014, 83, 174–189. [Google Scholar] [CrossRef]
- de Castro, S.; Ginex, T.; Vanderlinden, E.; Laporte, M.; Stevaert, A.; Cumella, J.; Gago, F.; Camarasa, M.J.; Luque, F.J.; Naesens, L.; et al. N-benzyl 4,4-disubstituted piperidines as a potent class of influenza H1N1 virus inhibitors showing a novel mechanism of hemagglutinin fusion peptide interaction. Eur. J. Med. Chem. 2020, 194, 112223. [Google Scholar] [CrossRef]
- Walsh, E.E.; Shin, J.H.; Falsey, A.R. Clinical impact of human coronaviruses 229E and OC43 infection in diverse adult populations. J. Infect. Dis. 2013, 208, 1634–1642. [Google Scholar] [CrossRef]
- Dijkman, R.; van der Hoek, L. Human coronaviruses 229E and NL63: Close yet still so far. J. Formos. Med. Assoc. 2009, 108, 270–279. [Google Scholar] [CrossRef]
- Hall, M.D.; Anderson, J.M.; Anderson, A.; Baker, D.; Bradner, J.; Brimacombe, K.R.; Campbell, E.A.; Corbett, K.S.; Carter, K.; Cherry, S.; et al. Report of the National Institutes of Health SARS-CoV-2 Antiviral Therapeutics Summit. J. Infect. Dis. 2021, 224, S1–S21. [Google Scholar] [CrossRef] [PubMed]
- Sendi, P.; Razonable, R.R.; Nelson, S.B.; Soriano, A.; Gandhi, R.T. First-generation oral antivirals against SARS-CoV-2. Clin. Microbiol. Infect. 2022, in press. [CrossRef] [PubMed]
- Owen, D.R.; Allerton, C.M.N.; Anderson, A.S.; Aschenbrenner, L.; Avery, M.; Berritt, S.; Boras, B.; Cardin, R.D.; Carlo, A.; Coffman, K.J.; et al. An oral SARS-CoV-2 M(pro) inhibitor clinical candidate for the treatment of COVID-19. Science 2021, 374, 1586–1593. [Google Scholar] [CrossRef] [PubMed]
- Najjar-Debbiny, R.; Gronich, N.; Weber, G.; Khoury, J.; Amar, M.; Stein, N.; Goldstein, L.H.; Saliba, W. Effectiveness of paxlovid in reducing severe COVID-19 and mortality in high risk patients. Clin. Infect. Dis. 2022, in press.
- Hammond, J.; Leister-Tebbe, H.; Gardner, A.; Abreu, P.; Bao, W.; Wisemandle, W.; Baniecki, M.; Hendrick, V.M.; Damle, B.; Simón-Campos, A.; et al. Oral nirmatrelvir for high-risk, nonhospitalized adults with Covid-19. N. Engl. J. Med. 2022, 386, 1397–1408. [Google Scholar] [CrossRef] [PubMed]
- Beigel, J.H.; Tomashek, K.M.; Dodd, L.E.; Mehta, A.K.; Zingman, B.S.; Kalil, A.C.; Hohmann, E.; Chu, H.Y.; Luetkemeyer, A.; Kline, S.; et al. Remdesivir for the treatment of Covid-19—Final report. N. Engl. J. Med. 2020, 383, 1813–1826. [Google Scholar] [CrossRef]
- Beigel, J.H. What is the role of remdesivir in patients with COVID-19? Curr. Opin. Crit. Care 2021, 27, 487–492. [Google Scholar] [CrossRef]
- Gottlieb, R.L.; Vaca, C.E.; Paredes, R.; Mera, J.; Webb, B.J.; Perez, G.; Oguchi, G.; Ryan, P.; Nielsen, B.U.; Brown, M.; et al. Early remdesivir to prevent progression to severe Covid-19 in outpatients. N. Engl. J. Med. 2022, 386, 305–315. [Google Scholar] [CrossRef]
- Jayk Bernal, A.; Gomes da Silva, M.M.; Musungaie, D.B.; Kovalchuk, E.; Gonzalez, A.; Delos Reyes, V.; Martin-Quiros, A.; Caraco, Y.; Williams-Diaz, A.; Brown, M.L.; et al. Molnupiravir for oral treatment of Covid-19 in nonhospitalized patients. N. Engl. J. Med. 2022, 386, 509–520. [Google Scholar] [CrossRef]
- Arribas, J.R.; Bhagani, S.; Lobo, S.M.; Khaertynova, I.; Mateu, L.; Fishchuk, R.; Park, W.Y.; Hussein, K.; Kim, S.W.; Ghosn, J.; et al. Randomized trial of molnupiravir or placebo in patients hospitalized with Covid-19. NEJM Evid. 2022, 1, EVIDoa2100044. [Google Scholar] [CrossRef]
- Finberg, R.W.; Ashraf, M.; Julg, B.; Ayoade, F.; Marathe, J.G.; Issa, N.C.; Wang, J.P.; Jaijakul, S.; Baden, L.R.; Epstein, C. US201 study: A Phase 2, randomized proof-of-concept trial of favipiravir for the treatment of COVID-19. Open Forum Infect. Dis. 2021, 8, ofab563. [Google Scholar] [CrossRef] [PubMed]
- Hung, D.T.; Ghula, S.; Aziz, J.M.A.; Makram, A.M.; Tawfik, G.M.; Abozaid, A.A.; Pancharatnam, R.A.; Ibrahim, A.M.; Shabouk, M.B.; Turnage, M.; et al. The efficacy and adverse effects of favipiravir on patients with COVID-19: A systematic review and meta-analysis of published clinical trials and observational studies. Int. J. Infect. Dis. 2022, 120, 217–227. [Google Scholar] [CrossRef]
- Seeking to Combat COVID-19 with an Oral RNA Viral Polymerase Inhibitor. Available online: https://ateapharma.com/covid-19/bemnifosbuvir/ (accessed on 22 July 2022).
- Shannon, A.; Fattorini, V.; Sama, B.; Selisko, B.; Feracci, M.; Falcou, C.; Gauffre, P.; El Kazzi, P.; Delpal, A.; Decroly, E.; et al. A dual mechanism of action of AT-527 against SARS-CoV-2 polymerase. Nat. Commun. 2022, 13, 621. [Google Scholar] [CrossRef] [PubMed]
- Uraki, R.; Kiso, M.; Imai, M.; Yamayoshi, S.; Ito, M.; Fujisaki, S.; Takashita, E.; Ujie, M.; Furusawa, Y.; Yasuhara, A.; et al. Therapeutic efficacy of monoclonal antibodies and antivirals against SARS-CoV-2 Omicron BA.1 in Syrian hamsters. Nat. Microbiol. 2022, 7, 1252–1258. [Google Scholar] [CrossRef] [PubMed]
- Cameroni, E.; Bowen, J.E.; Rosen, L.E.; Saliba, C.; Zepeda, S.K.; Culap, K.; Pinto, D.; VanBlargan, L.A.; De Marco, A.; di Iulio, J.; et al. Broadly neutralizing antibodies overcome SARS-CoV-2 Omicron antigenic shift. Nature 2022, 602, 664–670. [Google Scholar] [CrossRef]
- Corman, V.M.; Muth, D.; Niemeyer, D.; Drosten, C. Hosts and sources of endemic human coronaviruses. Adv. Virus Res. 2018, 100, 163–188. [Google Scholar]
- V’Kovski, P.; Kratzel, A.; Steiner, S.; Stalder, H.; Thiel, V. Coronavirus biology and replication: Implications for SARS-CoV-2. Nat. Rev. Microbiol. 2021, 19, 155–170. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Lin, D.; Sun, X.; Curth, U.; Drosten, C.; Sauerhering, L.; Becker, S.; Rox, K.; Hilgenfeld, R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science 2020, 368, 409–412. [Google Scholar] [CrossRef]
- Malone, B.; Urakova, N.; Snijder, E.J.; Campbell, E.A. Structures and functions of coronavirus replication-transcription complexes and their relevance for SARS-CoV-2 drug design. Nat. Rev. Mol. Cell Biol. 2022, 23, 21–39. [Google Scholar] [CrossRef]
- Bouvet, M.; Debarnot, C.; Imbert, I.; Selisko, B.; Snijder, E.J.; Canard, B.; Decroly, E. In vitro reconstitution of SARS-coronavirus mRNA cap methylation. PLoS Pathog. 2010, 6, e1000863. [Google Scholar] [CrossRef]
- Subissi, L.; Posthuma, C.C.; Collet, A.; Zevenhoven-Dobbe, J.C.; Gorbalenya, A.E.; Decroly, E.; Snijder, E.J.; Canard, B.; Imbert, I. One severe acute respiratory syndrome coronavirus protein complex integrates processive RNA polymerase and exonuclease activities. Proc. Natl. Acad. Sci. USA 2014, 111, E3900–E3909. [Google Scholar] [CrossRef] [PubMed]
- Eydoux, C.; Fattorini, V.; Shannon, A.; Le, T.T.; Didier, B.; Canard, B.; Guillemot, J.C. A fluorescence-based high throughput-screening assay for the SARS-CoV RNA synthesis complex. J. Virol. Methods 2021, 288, 114013. [Google Scholar] [CrossRef]
- Lundin, A.; Dijkman, R.; Bergstrom, T.; Kann, N.; Adamiak, B.; Hannoun, C.; Kindler, E.; Jonsdottir, H.R.; Muth, D.; Kint, J.; et al. Targeting membrane-bound viral RNA synthesis reveals potent inhibition of diverse coronaviruses including the Middle East respiratory syndrome virus. PLoS Pathog. 2014, 10, e1004166. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 2020, 181, 271–280.e8. [Google Scholar] [CrossRef]
- de Vries, M.; Mohamed, A.S.; Prescott, R.A.; Valero-Jimenez, A.M.; Desvignes, L.; O’Connor, R.; Steppan, C.; Devlin, J.C.; Ivanova, E.; Herrera, A.; et al. A comparative analysis of SARS-CoV-2 antivirals characterizes 3CL(pro) inhibitor PF-00835231 as a potential new treatment for COVID-19. J. Virol. 2021, 95, e01819–e01820. [Google Scholar] [CrossRef]
- Laporte, M.; Raeymaekers, V.; Van Berwaer, R.; Vandeput, J.; Marchand-Casas, I.; Thibaut, H.J.; Van Looveren, D.; Martens, K.; Hoffmann, M.; Maes, P.; et al. The SARS-CoV-2 and other human coronavirus spike proteins are fine-tuned towards temperature and proteases of the human airways. PLoS Pathog. 2021, 17, e1009500. [Google Scholar] [CrossRef]
- Kawase, M.; Shirato, K.; Matsuyama, S.; Taguchi, F. Protease-mediated entry via the endosome of human coronavirus 229E. J. Virol. 2009, 83, 712–721. [Google Scholar] [CrossRef]
- Kim, Y.; Mandadapu, S.R.; Groutas, W.C.; Chang, K.O. Potent inhibition of feline coronaviruses with peptidyl compounds targeting coronavirus 3C-like protease. Antivir. Res. 2013, 97, 161–168. [Google Scholar] [CrossRef]
- Fu, L.; Ye, F.; Feng, Y.; Yu, F.; Wang, Q.; Wu, Y.; Zhao, C.; Sun, H.; Huang, B.; Niu, P.; et al. Both Boceprevir and GC376 efficaciously inhibit SARS-CoV-2 by targeting its main protease. Nat. Commun. 2020, 11, 4417. [Google Scholar] [CrossRef]
- Pitts, J.; Li, J.; Perry, J.K.; Du Pont, V.; Riola, N.; Rodriguez, L.; Lu, X.; Kurhade, C.; Xie, X.; Camus, G.; et al. Remdesivir and GS-441524 retain antiviral activity against Delta, Omicron, and other emergent SARS-CoV-2 variants. Antimicrob. Agents Chemother. 2022, 66, e0022222. [Google Scholar] [CrossRef]
- Rappe, J.C.F.; de Wilde, A.; Di, H.; Müller, C.; Stalder, H.; V’Kovski, P.; Snijder, E.; Brinton, M.A.; Ziebuhr, J.; Ruggli, N.; et al. Antiviral activity of K22 against members of the order Nidovirales. Virus Res. 2018, 246, 28–34. [Google Scholar] [CrossRef]
- Ahmed-Belkacem, R.; Hausdorff, M.; Delpal, A.; Sutto-Ortiz, P.; Colmant, A.M.G.; Touret, F.; Ogando, N.S.; Snijder, E.J.; Canard, B.; Coutard, B.; et al. Potent inhibition of SARS-CoV-2 nsp14 N7-methyltransferase by sulfonamide-based bisubstrate analogues. J. Med. Chem. 2022, 65, 6231–6249. [Google Scholar] [CrossRef]
- Anand, K.; Palm, G.J.; Mesters, J.R.; Siddell, S.G.; Ziebuhr, J.; Hilgenfeld, R. Structure of coronavirus main proteinase reveals combination of a chymotrypsin fold with an extra alpha-helical domain. EMBO J. 2002, 21, 3213–3224. [Google Scholar] [CrossRef]
- Gurard-Levin, Z.A.; Liu, C.; Jekle, A.; Jaisinghani, R.; Ren, S.; Vandyck, K.; Jochmans, D.; Leyssen, P.; Neyts, J.; Blatt, L.M.; et al. Evaluation of SARS-CoV-2 3C-like protease inhibitors using self-assembled monolayer desorption ionization mass spectrometry. Antivir. Res. 2020, 182, 104924. [Google Scholar] [CrossRef] [PubMed]
- Tang, T.; Bidon, M.; Jaimes, J.A.; Whittaker, G.R.; Daniel, S. Coronavirus membrane fusion mechanism offers a potential target for antiviral development. Antivir. Res. 2020, 178, 104792. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Zhu, Y.; Liu, X.; Jin, Z.; Duan, Y.; Zhang, Q.; Wu, C.; Feng, L.; Du, X.; Zhao, J.; et al. Structural basis for replicase polyprotein cleavage and substrate specificity of main protease from SARS-CoV-2. Proc. Natl. Acad. Sci. USA 2022, 119, e2117142119. [Google Scholar] [CrossRef] [PubMed]
- Hilgenfeld, R. From SARS to MERS: Crystallographic studies on coronaviral proteases enable antiviral drug design. FEBS J. 2014, 281, 4085–4096. [Google Scholar] [CrossRef]
- Gao, K.; Wang, R.; Chen, J.; Tepe, J.J.; Huang, F.; Wei, G.W. Perspectives on SARS-CoV-2 main protease inhibitors. J. Med. Chem. 2021, 64, 16922–16955. [Google Scholar] [CrossRef]
- Anand, K.; Ziebuhr, J.; Wadhwani, P.; Mesters, J.R.; Hilgenfeld, R. Coronavirus main proteinase (3CLpro) structure: Basis for design of anti-SARS drugs. Science 2003, 300, 1763–1767. [Google Scholar] [CrossRef]
- Greasley, S.E.; Noell, S.; Plotnikova, O.; Ferre, R.; Liu, W.; Bolanos, B.; Fennell, K.; Nicki, J.; Craig, T.; Zhu, Y.; et al. Structural basis for the in vitro efficacy of nirmatrelvir against SARS-CoV-2 variants. J. Biol. Chem. 2022, 298, 101972. [Google Scholar] [CrossRef] [PubMed]
- Kneller, D.W.; Li, H.; Phillips, G.; Weiss, K.L.; Zhang, Q.; Arnould, M.A.; Jonsson, C.B.; Surendranathan, S.; Parvathareddy, J.; Blakeley, M.P.; et al. Covalent narlaprevir- and boceprevir-derived hybrid inhibitors of SARS-CoV-2 main protease. Nat. Commun. 2022, 13, 2268. [Google Scholar] [CrossRef]
- Yamane, D.; Onitsuka, S.; Re, S.; Isogai, H.; Hamada, R.; Hiramoto, T.; Kawanishi, E.; Mizuguchi, K.; Shindo, N.; Ojida, A. Selective covalent targeting of SARS-CoV-2 main protease by enantiopure chlorofluoroacetamide. Chem. Sci. 2022, 13, 3027–3034. [Google Scholar] [CrossRef] [PubMed]
- Ge, R.; Shen, Z.; Yin, J.; Chen, W.; Zhang, Q.; An, Y.; Tang, D.; Satz, A.L.; Su, W.; Kuai, L. Discovery of SARS-CoV-2 main protease covalent inhibitors from a DNA-encoded library selection. SLAS Discov. Adv. Life Sci. R D 2022, 27, 79–85. [Google Scholar] [CrossRef] [PubMed]
- Stevaert, A.; Krasniqi, B.; Van Loy, B.; Nguyen, T.; Thomas, J.; Vandeput, J.; Jochmans, D.; Thiel, V.; Dijkman, R.; Dehaen, W.; et al. Betulonic acid derivatives interfering with human coronavirus 229E replication via the nsp15 endoribonuclease. J. Med. Chem. 2021, 64, 5632–5644. [Google Scholar] [CrossRef]
- Szűcs, Z.; Naesens, L.; Stevaert, A.; Ostorházi, E.; Batta, G.; Herczegh, P.; Borbás, A. Reprogramming of the antibacterial drug vancomycin results in potent antiviral agents devoid of antibacterial activity. Pharmaceuticals 2020, 13, 139. [Google Scholar] [CrossRef]
- Hoffman, R.L.; Kania, R.S.; Brothers, M.A.; Davies, J.F.; Ferre, R.A.; Gajiwala, K.S.; He, M.; Hogan, R.J.; Kozminski, K.; Li, L.Y.; et al. Discovery of ketone-based covalent inhibitors of coronavirus 3CL proteases for the potential therapeutic treatment of COVID-19. J. Med. Chem. 2020, 63, 12725–12747. [Google Scholar] [CrossRef] [PubMed]
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C.; et al. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature 2020, 582, 289–293. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- PDBe-KB: Collaboratively defining the biological context of structural data. Nucleic Acids Res. 2022, 50, D534–D542. [CrossRef]
- Douangamath, A.; Fearon, D.; Gehrtz, P.; Krojer, T.; Lukacik, P.; Owen, C.D.; Resnick, E.; Strain-Damerell, C.; Aimon, A.; Ábrányi-Balogh, P.; et al. Crystallographic and electrophilic fragment screening of the SARS-CoV-2 main protease. Nat. Commun. 2020, 11, 5047. [Google Scholar] [CrossRef]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge Structural Database. Acta Crystallogr. Sect. B Struct. Sci. Cryst. Eng. Mater. 2016, 72, 171–179. [Google Scholar] [CrossRef]
- Liao, G.P.; Abdelraheem, E.M.; Neochoritis, C.G.; Kurpiewska, K.; Kalinowska-Tłuścik, J.; McGowan, D.C.; Dömling, A. Versatile multicomponent reaction macrocycle synthesis using α-isocyano-ω-carboxylic acids. Org. Lett. 2015, 17, 4980–4983. [Google Scholar] [CrossRef] [PubMed]
- Walker, R.C.; Crowley, M.F.; Case, D.A. The implementation of a fast and accurate QM/MM potential method in Amber. J. Comput. Chem. 2008, 29, 1019–1031. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Eberhardt, J.; Santos-Martins, D.; Tillack, A.F.; Forli, S. AutoDock Vina 1.2.0: New docking methods, expanded force field, and Python bindings. J. Chem. Inf. Model. 2021, 61, 3891–3898. [Google Scholar] [CrossRef]
- Case, D.A.; Cheatham, T.E., 3rd; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M., Jr.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The Amber biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Kokubo, H. Prediction of ligand binding mode among multiple cross-docking poses by molecular dynamics simulations. J. Comput. Aided Mol. Des. 2020, 34, 1195–1205. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Compound | R1 | R2 | R3 | R4 | R5 | Antiviral EC50 (µM) a | Cytotoxicity (µM) b | Selectivity Index c | |||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Microscopy | MTS | MCC | CC50 | Microscopy | MTS | ||||||
| 1d | Bn | Bn | Bn | NHBoc | CH2COOMe | >100 | >100 | >100 | >100 | ||
| 2d | Bn | Bn | 4-F-Bn | NHBoc | CH2COOMe | 7.8 ± 2.7 | 7.4 ± 2.5 | 100 | 44 ± 8 | 13 | 6 |
| Subseries 1: modifications at R1 | |||||||||||
| 3e | - | - | - | - | - | >100 | >100 | >100 | >100 | ||
| 4e | H | Bn | 4-F-Bn | NHBoc | CH2COOMe | 50 ± 18 | 78 ± 14 | 100 | 66 ± 7 | ||
| 5e | Me | Bn | 4-F-Bn | NHBoc | CH2COOMe | 22 ± 9 | 22 ± 7 | >100 | 92 ± 4 | ||
| 6e | Chx | Bn | 4-F-Bn | NHBoc | CH2COOMe | 22 ± 9 | 14 ± 1 | 100 | 85 ± 9 | ||
| 7e | Ph | Bn | 4-F-Bn | NHBoc | CH2COOMe | 43 ± 3 | >100 | >100 | >100 | ||
| 8e | Bn(Me) piperidinium salt | Bn | 4-F-Bn | NHBoc | CH2COOMe | >100 | >100 | >100 | >100 | ||
| 9e | 3-F-Bn | Bn | 4-F-Bn | NHBoc | CH2COOMe | >100 | >100 | >100 | >100 | ||
| 10e | 3,5-diF-Bn | Bn | 4-F-Bn | NHBoc | CH2COOMe | 15 ± 1 | >100 | 70 | 10 ± 2 | ||
| 11e | (CH2)2Ph | Bn | 4-F-Bn | NHBoc | CH2COOMe | 3.2 ± 0.1 | 4.4 ± 0.8 | 100 | 38 ± 2 | 31 | |
| Subseries 2: modifications at R2 | |||||||||||
| 12e | Bn | tBu | 4-F-Bn | NHBoc | CH2COOMe | 11 | 11 | ≥100 | 77 | ||
| 13e | Bn | Chx | 4-F-Bn | NHBoc | CH2COOMe | >100 | >100 | >100 | >100 | ||
| 14e | Bn | CH2SO2Ph-4-Me | 4-F-Bn | NHBoc | CH2COOMe | 57 ± 18 | 30 ± 6 | 100 | 78 ± 11 | ||
| 15 | Bn | tBu | Bn | NHBoc | CH2COOMe | 25 ± 0 | 25 ± 0 | >100 | 78 ± 7 | ||
| 16 | Bn | Chx | Bn | NHBoc | CH2COOMe | 16 ± 3 | 10 ± 0 | ≥40 | 42 ± 4 | ||
| 17d | Bn | CH2COOMe | Bn | NHBoc | CH2COOMe | 68 ± 3 | 75 ± 8 | >100 | >100 | ||
| Subseries 3: modifications at R3 and/or R4 | |||||||||||
| 18d | Bn | Bn | H | NHBoc | CH2COOMe | 55 ± 16 | >100 | ≥100 | 48 ± 8 | ||
| 19d | Bn | Bn | Me | NHBoc | CH2COOMe | 14 ± 1 | >100 | 100 | 18 ± 2 | ||
| 20d | Bn | Bn | CH(Me)(Et) | NHBoc | CH2COOMe | 14 ± 1 | 13 ± 1 | 100 | 31 ± 3 | ||
| 21d | Bn | Bn | Cyclopropyl | NHBoc | CH2COOMe | 12 ± 0.4 | > 100 | 50 | 14 ± 3 | ||
| 22e | Bn | Bn | 4-Me-Bn | NHBoc | CH2COOMe | 3.1 ± 0.0 | 4.3 ± 0.6 | 75 | 31 ± 10 | 24 | |
| 23e | Bn | Bn | 4-NO2-Bn | NHBoc | CH2COOMe | 3.1 ± 0.0 | 4.0 ± 0.6 | 75 | 49 ± 6 | 24 | 12 |
| 24e | Bn | Bn | 4-CF3-Bn | NHBoc | CH2COOMe | 3.1 ± 0.0 | 4.2 ± 1.0 | 25 | 11 ± 1 | ||
| 25e | Bn | Bn | 4-Cl-Bn | NHBoc | CH2COOMe | 3.1 ± 0.0 | 3.2 ± 0.2 | 50 | 12 ± 0 | ||
| 26e | Bn | Bn | 2-F-Bn | NHBoc | CH2COOMe | 12 ± 1 | 15 ± 2 | 100 | 44 ± 1 | ||
| 27e | Bn | Bn | 3-F-Bn | NHBoc | CH2COOMe | 10 ± 3 | 14 ± 1 | 100 | 46 ± 4 | 10 | |
| 28e | Bn | Bn | 3,4-F-Bn | NHBoc | CH2COOMe | 6.0± 2.6 | 3.7 ± 1.0 | 44 | 23 ± 11 | ||
| 29d | Bn | Bn | PhNH(CH2)2 | NHBoc | CH2COOMe | 3.0 ± 0.1 | 3.2 ± 0.4 | 75 | 36 ± 9 | 25 | 11 |
| 30e | Bn | Bn | 4-F-Bn | H | CH2COOMe | 13 + 0 | 13 ± 2 | 100 | 64 ± 3 | ||
| 31e | Bn | Bn | 4-F-Bn | NH2 | CH2COOMe | 3.3 ± 0.2 | 2.8 ± 0.5 | 11 | 4.9 ± 1.5 | ||
| 32e | Bn | Bn | Bn | NH2 | CH2COOMe | 55 ± 3 | 62 ± 7 | >100 | >100 | ||
| 33d | Bn | Bn | Bn | NHCbz | CH2COOMe | 3.0 ± 0.1 | 3.3 ± 0.4 | >100 | >100 | >33 | >30 |
| 34e | Bn | Bn | 4-F-Bn | NHCbz | CH2COOMe | 3.1 ± 0.1 | 3.4 ± 0.4 | 81 | 75 ± 18 | 26 | 22 |
| 35d | Bn | Bn | Bn | NHFmoc | CH2COOMe | 36 ± 14 | 26 ± 9 | >100 | >100 | ||
| 36d | Bn | Bn | CH(Me)(Et) | NHCbz | CH2COOMe | 9.2 ± 0.5 | 6.2 ± 0.8 | 40 | 27 ± 1 | ||
| 37d | Bn | Bn | Cyclopropyl | NHCbz | CH2COOMe | 3.0 ± 0.1 | >100 | 11 | 3.6 ± 0.1 | ||
| Subseries 4: modifications at R5 | |||||||||||
| 38e | Bn | Bn | 4-F-Bn | NHBoc | CH3 | 12 ± 1 | 15 ± 3 | 100 | 48 ± 2 | ||
| 39d | Bn | Bn | Bn | NHBoc | H | 11 ± 0 | 15 ± 2 | 100 | 49 ± 1 | ||
| 40e | Bn | Bn | 4-F-Bn | NHBoc | CH2CH2COOMe | 4.8 ± 1.7 | 11 ± 4 | 100 | 50 ± 3 | 21 | |
| 41e | Bn | Bn | 4-F-Bn | NHBoc | CH2CONH2 | 54 ± 4 | 64 ± 8 | >100 | 82 ± 13 | ||
| 42d | Bn | Bn | Bn | NHBoc | CH2CONH2 | 13 ± 0 | 16 ± 3 | 100 | 40 ± 6 | ||
| 43e | Bn | Bn | 4-F-Bn | NHBoc | CH2COOH | >100 | >100 | >100 | >100 | ||
| 44d | Bn | Bn | Bn | NHBoc | CH2COOH | >100 | >100 | >100 | >100 | ||
| 45e | Bn | Bn | 4-F-Bn | NHBoc | CH2Ph | 0.85 ± 0.07 | 1.1 ± 0.2 | 50 | 4.4 ± 0.8 | ||
| 46e | Bn | Bn | Bn | NHBoc | CH2Ph | 7.4 ± 1.3 | 4.5 ± 0.2 | ≥100 | 75 ± 0 | ≥14 | 17 |
| 47e | Bn | Bn | Bn | NHBoc | CH2Indolyl | 57 ± 17 | 49 ± 12 | >100 | >100 | ||
| 48d | Bn | Bn | Bn | NHBoc | CH2Ph-4-OH | 11 ± 1 | 9.3 ± 0 | >100 | >100 | >11 | |
| 49 | Bn | Bn | Bn | NHBoc | CH2Ph-4-F | 10 ± 0 | 8.4 ± 0 | ≥100 | >100 | ≥10 | >12 |
| 50 | Bn | Bn | Bn | NHBoc | CH2Ph-4-Cl | 10 ± 0 | 9.7 ± 0.4 | ≥100 | >100 | ≥10 | ≥10 |
| 51 | Bn | Bn | Bn | NHBoc | CH2Ph-4-OMe | 8.2 ± 1.9 | 4.8 ± 0.4 | ≥100 | >100 | ≥12 | ≥21 |
| 52 | Bn | Bn | Bn | NHBoc | CH2Ph-4-Me | 6.6 ± 1.8 | 4.7 ± 0.6 | ≥100 | >100 | ≥15 | >21 |
| Subseries 5: combination of modifications at R1-R5 | |||||||||||
| 53 | (CH2)2Ph | Bn | Bn | NHCbz | CH2COOMe | 6.1 ± 2.0 | 5.8 ± 1.9 | 40 | 24 ± 2 | ||
| 54 | (CH2)2Ph-4-Me | Bn | Bn | NHCbz | CH2COOMe | 10 ± 0 | 8.4 ± 0.5 | 40 | 24 ± 2 | ||
| 55 | (CH2)2Ph-4NO2 | Bn | Bn | NHCbz | CH2COOMe | 10 ± 0 | 7.7 ± 0.9 | 60 | >100 | >13 | |
| 56 | (CH2)2Ph-4-F | Bn | Bn | NHCbz | CH2COOMe | 7.6 ± 1.8 | 6.0 ± 1.9 | 32 | 23 ± 3 | ||
| 57 | Bn | Bn | PhNH(CH2)2 | NHCbz | CH2COOMe | 10 ± 0 | 7.1 ± 0.9 | 40 | 51 ± 9 | ||
| 58 | (CH2)2Ph | Bn | PhNH(CH2)2 | NHCbz | CH2COOMe | 7.9 ± 1.9 | 5.4 ± 1.4 | 24 | 20 ± 4 | ||
| 59 | (CH2)2Ph | Bn | 4-Me-Bn | NHCbz | CH2COOMe | 8.8 ± 0.7 | 5.1 ± 1.0 | 16 | 15 ± 5 | ||
| 60 | 4-F-Bn | Bn | Bn | NHBoc | CH2Ph | 4.5 ± 0.2 | 4.0 ± 0.1 | ≥100 | >100 | ≥22 | >25 |
| 61 | 4-Cl-Bn | Bn | Bn | NHBoc | CH2Ph | 36 ± 9 | 30 ± 7 | >100 | >100 | ||
| 62 | 4-OMe-Bn | Bn | Bn | NHBoc | CH2Ph | 5.9 ± 1.8 | 4.3 ± 0.4 | 40 | 28 ± 0 | ||
| 63 | 4-NO2-Bn | Bn | Bn | NHBoc | CH2Ph | 82 ± 15 | 77 ± 9 | ≥100 | >100 | ||
| Reference compounds | |||||||||||
| GS-441524 | 3.5 ± 0.0 | 2.3 ± 0.2 | >100 | >100 | >29 | >43 | |||||
| K22 [36] | 2.6 ± 0.5 | 3.6 ± 0.6 | 50 | 28 ± 3 | 19 | ||||||
| Compound | Antiviral activity (µM) | Cytotoxicity b (µM) | |
|---|---|---|---|
| EC90 a | EC99 a | CC50 | |
| 33 | 9.3 | 14 | 90 |
| 34 | 8.0 | 13 | 79 |
| 52 | 1.7 | 3.9 | 51 |
| 60 | 2.2 | 5.2 | 63 |
| GS-441524 | 3.2 | 4.6 | >16 |
| Compound | IC50 (µM) |
|---|---|
| 1 | 161 ± 35 |
| 33 | 64 ± 8 |
| 34 | 38 ± 5 |
| 45 | 15 ± 2 |
| 46 | 22 ± 2 |
| 52 | 14 ± 4 |
| 63 | 196 ± 39 |
| 60 | 68 ± 21 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Castro, S.; Stevaert, A.; Maldonado, M.; Delpal, A.; Vandeput, J.; Van Loy, B.; Eydoux, C.; Guillemot, J.-C.; Decroly, E.; Gago, F.; et al. A Versatile Class of 1,4,4-Trisubstituted Piperidines Block Coronavirus Replication In Vitro. Pharmaceuticals 2022, 15, 1021. https://doi.org/10.3390/ph15081021
De Castro S, Stevaert A, Maldonado M, Delpal A, Vandeput J, Van Loy B, Eydoux C, Guillemot J-C, Decroly E, Gago F, et al. A Versatile Class of 1,4,4-Trisubstituted Piperidines Block Coronavirus Replication In Vitro. Pharmaceuticals. 2022; 15(8):1021. https://doi.org/10.3390/ph15081021
Chicago/Turabian StyleDe Castro, Sonia, Annelies Stevaert, Miguel Maldonado, Adrien Delpal, Julie Vandeput, Benjamin Van Loy, Cecilia Eydoux, Jean-Claude Guillemot, Etienne Decroly, Federico Gago, and et al. 2022. "A Versatile Class of 1,4,4-Trisubstituted Piperidines Block Coronavirus Replication In Vitro" Pharmaceuticals 15, no. 8: 1021. https://doi.org/10.3390/ph15081021
APA StyleDe Castro, S., Stevaert, A., Maldonado, M., Delpal, A., Vandeput, J., Van Loy, B., Eydoux, C., Guillemot, J. -C., Decroly, E., Gago, F., Canard, B., Camarasa, M. -J., Velázquez, S., & Naesens, L. (2022). A Versatile Class of 1,4,4-Trisubstituted Piperidines Block Coronavirus Replication In Vitro. Pharmaceuticals, 15(8), 1021. https://doi.org/10.3390/ph15081021