1. Introduction
N-methyl-D-aspartate receptors (NMDARs) are ionotropic receptors gated by two ligands, glycine (or D-serine) and glutamate, and by the electric potential of the cell membrane. At resting membrane potential, Mg
2+ is situated in the channel pore formed by the four NMDAR subunits. When NMDAR channels are coincidentally in the open structural conformation and free from Mg
2+, they allow tightly regulated, subtype-specific Ca
2+ influx. The co-occurrence of open conformation and Mg
2+ release can be elicited by excitatory synaptic transmission with presynaptic glutamate release. When glutamate binds to NMDARs, changing their conformation from closed to open, alpha-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid receptors (AMPARs), also activated by glutamate, determine AMPAR-mediated membrane depolarization and Mg
2+ release from the NMDAR channel pore, allowing Ca
2+ influx. This phasic, time-controlled, subtype-specific Ca
2+ signaling via NMDARs determines downstream effects that modulate synapses, forming the basis of structural and functional memory [
1,
2]. Tonic NMDAR-mediated Ca
2+ currents at resting membrane potential also require the coincident opening of the NMDAR structural conformation and release of Mg
2+ from the channel pore. The physiological role of tonic NMDAR-mediated Ca
2+ currents is increasingly recognized [
3,
4]. Excessive tonic Ca
2+ influx mediated by NMDARs may impair the availability of synaptic proteins [
3], interfering with stimulus-evoked memory formation induced by phasic NMDAR activation. NMDAR channel blockers reduce excessive tonic Ca
2+ influx mediated by NMDARs and restore synaptic protein availability, enabling neural plasticity elicited by action potential-mediated, phasic Ca
2+ influx in animal models of depressive-like behavior [
5,
6,
7,
8,
9]. NMDAR heterotetramers are composed of two constitutively expressed glycine-binding NR1 subunits, necessary for membrane expression of the functional channel, and two regulatory glutamate-binding NR2 subunits, named 2A, 2B, 2C, and 2D, which differentially regulate Ca
2+ influx. NMDARs regulate Ca
2+ influx during phasic and tonic receptor activity. NMDARs containing the 3A and 3B subunits bind only glycine and not glutamate and are not blocked by Mg
2+ [
10], and therefore they were not included in our study.
The uncompetitive NMDAR blockers memantine, esketamine, and the dextromethorphan-quinidine combination are approved by the U.S. Food and Drug Administration (FDA) to treat Alzheimer’s disease, treatment-resistant depression (TRD), and pseudobulbar affect (PBA), respectively. Differences in clinical effects and indications among uncompetitive NMDAR channel blockers appear to be linked to unique receptor-ligand kinetic properties [
1,
4,
11]. Therefore, advances in characterization of the pharmacological properties of therapeutically effective and clinically tolerated uncompetitive NMDAR blockers at the different NMDAR channel subtypes may improve our understanding of NMDAR-related pathophysiology in different disease states, potentially expanding therapeutic options.
REL-1017 (esmethadone HCl; dextromethadone HCl), the d-isomer of racemic methadone, is a low-affinity, low-potency, uncompetitive NMDAR channel blocker, devoid of meaningful opioid agonist effects [
12] with experimental evidence for producing brain-derived neurotrophic factor (BDNF) and target of rapamycin complex 1 (TORC1)–dependent antidepressant-like effects [
9], similarly to ketamine [
5]. In a double-blind, placebo-controlled Phase 2 trial in patients with major depressive disorder (MDD) and inadequate response to standard serotonergic antidepressants, the addition of REL-1017 showed rapid, robust, and sustained antidepressant effects without dissociative side effects [
13]. Phase 3 trials of REL-1017 as adjunctive treatment and as monotherapy in patients with MDD are currently ongoing (ClinicalTrials.gov Identifiers: NCT04855747; NCT04688164; NCT05081167). In the context of this work, the current studies aim to characterize the properties of REL-1017 and other uncompetitive NMDAR blockers on cell lines over-expressing specific human heterodimeric NMDAR channel subtypes by fluorometric imaging plate reader (FLIPR) Ca
2+ assay and by automated and manual patch-clamp electrophysiology. Additionally, we performed [Piperidyl-3,4-(N)]-(N-(1-[2-thienyl]cyclohexyl)3,4-piperidine ([
3H]TCP) radioligand binding displacement studies with the same molecules in the cerebral cortex of Wistar rats. Finally, we performed in silico studies with REL-1017, esketamine, and arketamine on the four NMDAR 2A-D subtypes, in closed and open conformational states.
3. Discussion
NMDAR channel pores of NR1-2A-D subunit compositions are gated into their open configuration upon the binding of two molecules of glycine or D-serine to the agonist domain on each NR1 subunit and the concurrent binding of two L-glutamate molecules to the agonist domain on each NR2 subunit. During phasic channel activation, presynaptic glutamate release induces AMPAR-mediated membrane depolarization leading to the release of Mg
2+ from the NMDAR channel pore, initiating time-controlled, subtype-specific influx of Ca
2+. Stimulus-evoked, action potential-mediated Ca
2+ signaling through NMDARs directs cellular signaling cascades that modulate synapses, forming memory [
1,
2]. The physiological role of tonic Ca
2+ signaling has been related to local availability of synaptic proteins and neuronal maturation [
1,
3,
4,
6,
7,
8]. Well-regulated tonic Ca
2+ currents, which are graded by “spontaneous” NMDAR-mediated miniature excitatory postsynaptic currents (NMDAR-mEPSCs), may be essential to ensure availability of synaptic proteins necessary for neural plasticity elicited by stimulus-evoked postsynaptic currents (NMDAR-eEPSCs) [
3,
6,
7,
8]. Excessive tonic NMDAR-mediated Ca
2+ currents can be downregulated by uncompetitive NMDAR antagonists, such as ketamine and esmethadone, restoring neural plasticity and resolving depressive-like behavior in animal models [
5,
6,
7,
8,
9] and in patients [
13,
20]. The uncompetitive NMDAR channel blocker dextromethorphan has also shown efficacy without psychotomimetic effects in a Phase 2 trial of patients with MDD [
21].
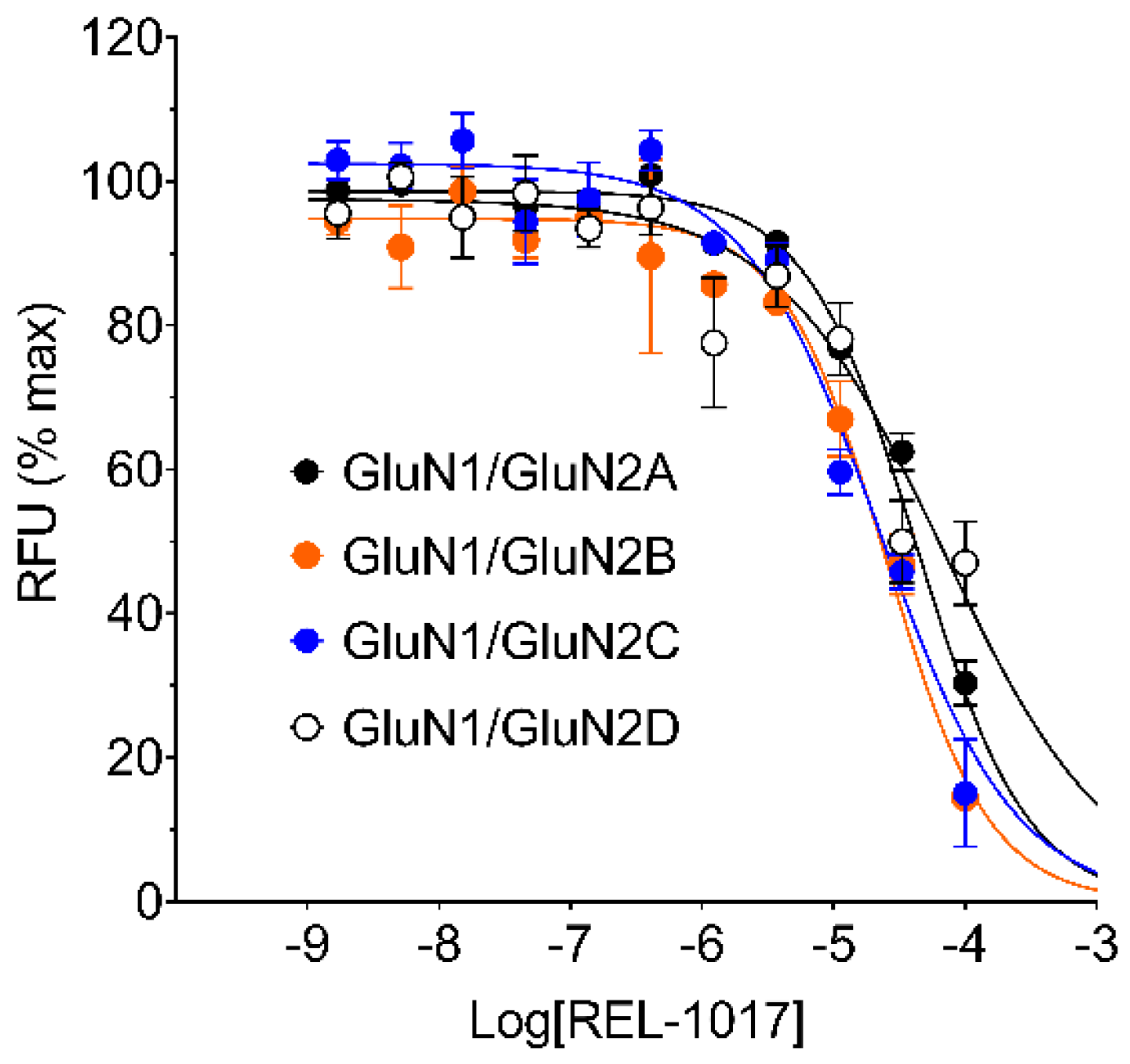
In this study, we performed in vitro and in silico characterizations of REL-1017 and other clinically relevant uncompetitive NMDAR antagonists in CHO cells over-expressing NR1-2A, NR1-2B, NR1-2C, or NR1-2D subtypes, in rat cortical membranes, and in computational models. As demonstrated in FLIPR assays, low glutamate concentrations were most active on NR1-2D subtypes (
Table S2a), in agreement with prior studies [
22]. As expected, REL-1017 and the other tested uncompetitive antagonists were able to antagonize NMDAR-mediated Ca
2+ influx triggered by glutamate. In the absence of physiological Mg
2+, REL-1017, dextromethorphan, memantine, and (±)-ketamine showed some degree of preference for NR1-2C subtypes, as shown by FLIPR assay. However, the subtype preference for REL-1017 was generally low.
In FLIPR assays, REL-1017 showed calculated affinity that was lower compared to the other tested compounds, in alignment with the results of the [
3H]TCP radioligand binding displacement study in the cerebral cortex of Wistar rats. FLIPR assays were conducted in the absence of Mg
2+ to allow cytosolic measurements of NMDAR-dependent Ca
2+ influx; however, Mg
2+ is physiologically present at the synaptic sites. In the presence of physiological concentrations of Mg
2+ at resting membrane potentials, persistent tonic Ca
2+ currents are seen preferentially via NR1-2C and NR1-2D subtypes [
1,
17] due to their lower affinity for Mg
2+ compared to NR1-2A and NR1-2B subtypes [
23].
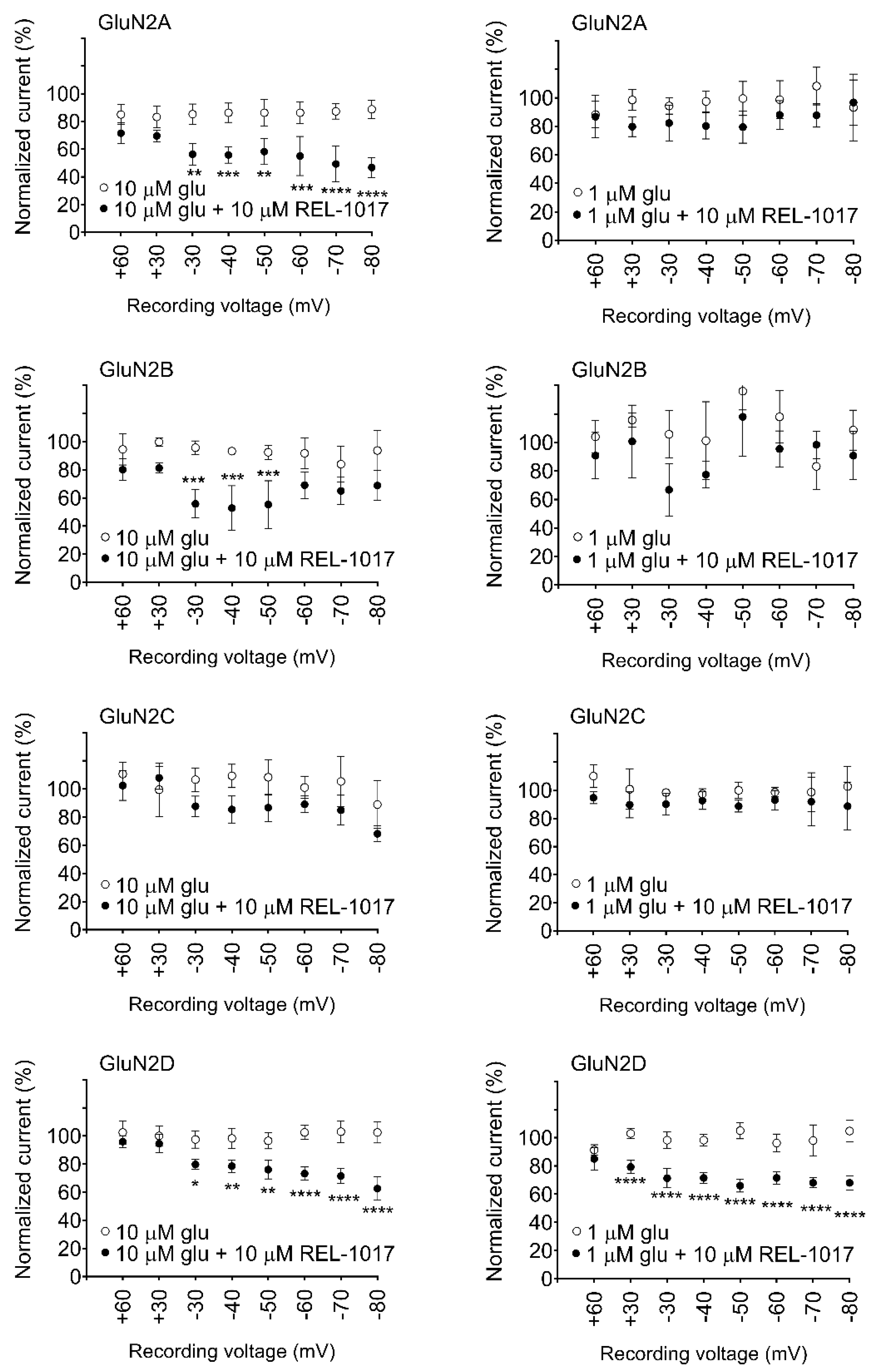
In automated and manual patch-clamp electrophysiological recordings, in the presence of physiological Mg
2+ and low glutamate concentrations, REL-1017 preferentially blocked Ca
2+ currents mediated by NR1-2D in a voltage-dependent manner. These results are consistent with earlier findings for memantine and (±)-ketamine [
24]. Ca
2+ signaling at resting membrane potential is distinct from stimulus-evoked, AMPAR-mediated, phasic activation [
1,
4,
25]. Tonic Ca
2+ currents via NR1-2C and NR1-2D subtypes constitute a physiological signaling pathway important for neuronal maturation and plasticity [
3,
4]. Excess ambient glutamate can cause pathological tonic hyperactivity of NR1-2D subtypes (
Table S2a), the subtype preferentially targeted by REL-1017 at physiological Mg
2+ concentrations.
When experiments are conducted in the presence of Mg
2+, (±)-ketamine and memantine have also shown similar subtype preference [
24]. Furthermore, REL-1017 is positively charged at a physiological pH [
26], and therefore it is not surprising that its block of NMDARs is voltage dependent, similar to Mg
2+, as seen in the automated (
Figure S7) and manual patch experiments. The voltage-dependent antagonism exerted by REL-1017 may help to explain the lack of dissociative side effects: when membrane potential shifts toward positive, such as during AMPAR-mediated depolarization, the positively charged REL-1017 molecule is less likely to be docked at its binding site within the channel pore. The voltage-dependent block exerted by REL-1017 does not cause dissociative side effects [
13], potentially because it does not interfere with phasic NMDAR-mediated Ca
2+ signaling at activation membrane potential. Following the completion of a phasic activation, once the membrane repolarizes with negative intracellular potential, Mg
2+ and REL-1017 may exert their tonic blockade at the channel pore at resting or near-resting membrane potentials (tonic channel blockade), causing their physiological and therapeutic effects, respectively.
NR1-2D subtypes have the highest affinity for glutamate (
Table S2a), which makes these subtypes more likely to be in the open configuration due to the effect of ambient glutamate. NR1-2D subtypes have a relatively low affinity for Mg
2+ and therefore the highest probability of being coincidentally in the open configuration and free from the Mg
2+ block [
17]. NMDARs require glutamate binding for the change into the open conformation and must be concurrently free from Mg
2+ before allowing Ca
2+ influx during phasic and tonic activity. NR1-2D subtypes remain open for several seconds rather than a few hundred milliseconds or less of other NMDAR subtypes [
1]. This extended Ca
2+ permeability time after activation may increase the probability for the docking of uncompetitive channel blockers, such as REL-1017, into the NR1-2D subtype channel pore. Therefore, in the presence of physiological Mg
2+ concentrations, compared to other receptor subtypes, the NR1-2D subtype may be the preferential target for REL-1017 and other clinically tolerated uncompetitive channel blockers.
The IC
50 values determined by radioligand binding assays in this study (3.52 nM for MK-801, 0.55 μM for (±)-ketamine, 0.79 μM for memantine, 2.06 μM for dextromethorphan, and 6.21 μM for REL-1017) may be informative for determining clinical tolerability, with a lower IC
50 likely indicative of a higher probability of dissociative effects. However, other characteristics, such as trapping [
11], may help explain clinically meaningful pharmacodynamic differences among uncompetitive NMDAR blockers with similar NMDAR affinity. Low trapping is considered the reason for the lack of cognitive side effects of memantine, despite its relatively high NMDAR affinity [
11]. This low trapping of memantine may also determine its lack of efficacy in the treatment of MDD [
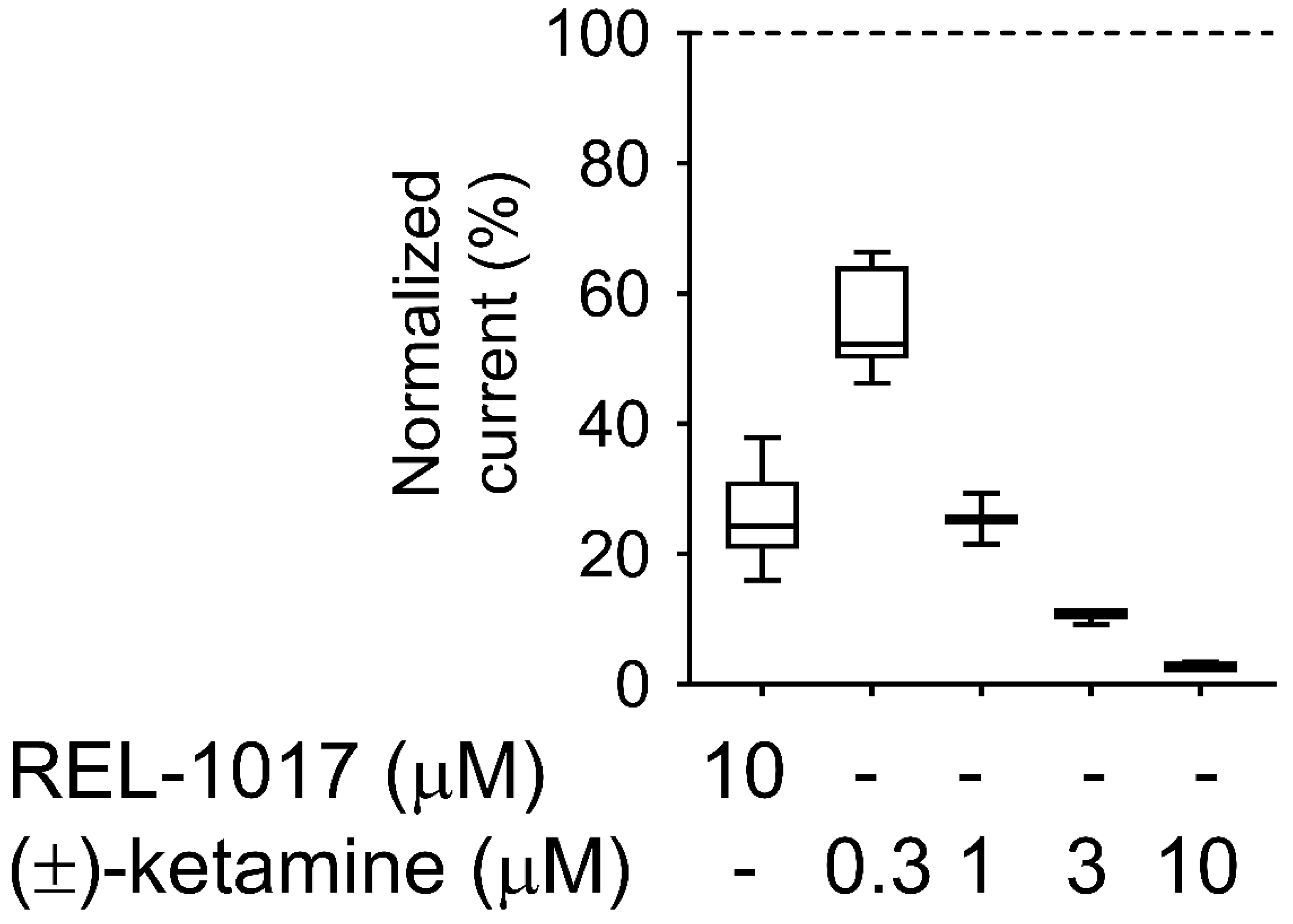
27], despite the relatively high (±)-ketamine-like affinity for NMDARs, which was confirmed in our radioligand displacement assay and FLIPR assay. The degree of trapping of REL-1017 was similar to (±)-ketamine in our experimental conditions. The relatively low NMDAR affinity of REL-1017, compared to the tested uncompetitive NMDAR channel blockers, and its relatively high (±)-ketamine-like trapping, could help to explain the absence of dissociative side effects and the robust, rapid, and sustained antidepressant effects [
13]. The lack of neuropathological changes in cortical neurons of rats exposed to high doses of REL-1017 [
28], in contrast with other uncompetitive NMDAR antagonists known to produce Olney’s lesions [
28,
29,
30,
31,
32], may also be related to the relatively lower affinity of REL-1017 at the NMDAR, confirmed in our radioligand binding assays, FLIPR assays, and automated and manual patch assays.
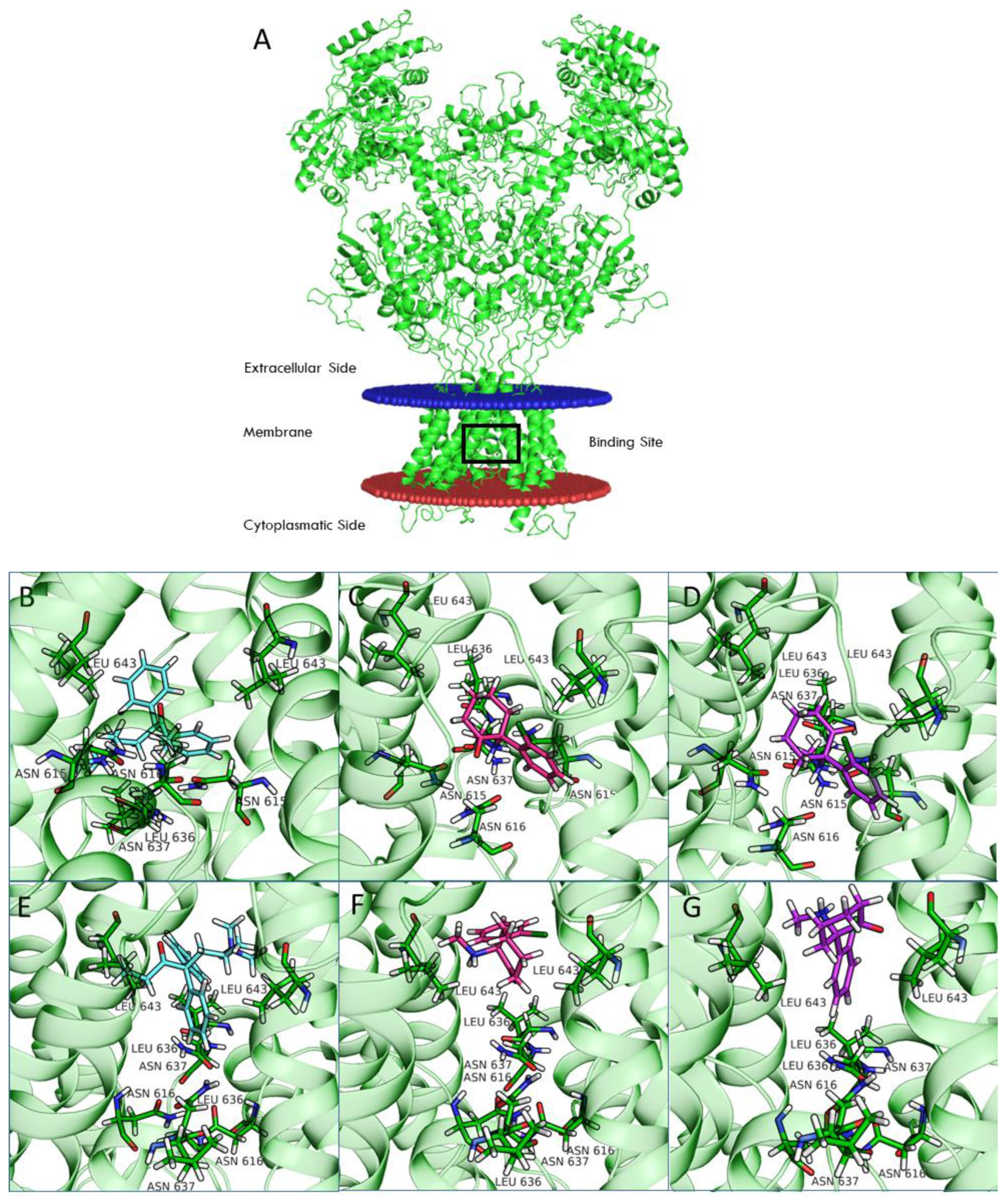
Computational studies of ketamine were recently performed for the NR1-2A and NR1-2B subtypes [
33]. In this study, we included the NR1-2C and NR1-2D subtypes in our computational analysis. Compared to NR1-2A and NR1-2B subtypes, NR1-2C and NR1-2D subtypes have a higher probability of being antagonized by uncompetitive NMDAR channel blockers at concentrations potentially therapeutic for MDD and other disorders [
24]. Our studies suggest that binding of both REL-1017 and esketamine in the open conformation of NMDARs was deeper in the channel compared to the binding position for the closed conformation. Furthermore, the binding site was deeper in the channel for esketamine compared to REL-1017, suggesting a higher propensity of REL-1017 compared to esketamine to undock from the open conformation. These computational findings, which may also be related to the larger size of REL-1017 compared to esketamine, could also help to explain the lack of dissociative effects of REL-1017, in contrast with esketamine and racemic ketamine. The importance of trapping and voltage dependence for NMDAR blockers has been underscored by Bolshakov and colleagues [
34].
Recent data for REL-1017, including clinical data [
13,
35], experimental preclinical data [
9], and the present pharmacological characterization of NMDAR channel blockers, support the hypothesis that excessive Ca
2+ signaling via tonically and pathologically hyperactive NR1-2D subtypes may in part drive MDD. NR1-2D is the same receptor subtype that is preferentially activated by ambient glutamate and preferentially targeted by REL-1017 and other clinically relevant uncompetitive channel blockers. We therefore hypothesize that, when excessive tonic Ca
2+ influx is corrected by an uncompetitive channel blocker with NR1-2D preference, such as REL-1017, the resolution of MDD symptomatology [
13] may result from normalization of synaptic proteins that allow normal neural plasticity, as has been demonstrated in experimental models of depressive-like behavior using ketamine and REL-1017 [
5,
6,
7,
8,
9]. Kotermanski and Johnson [
24] have demonstrated that uncompetitive NMDAR blockers such as ketamine and memantine may also have a preference for the NR1-2D channel subtype in the presence of physiological Mg
2+ concentrations. Therefore, the hypothesized mechanism of action of REL-1017 in the treatment of MDD appears to be similar to the mechanisms hypothesized for ketamine in animal models [
5,
6,
7,
8,
9] and in patients with MDD [
36].
Importantly, the approximately 10-fold higher affinity of ketamine at NMDARs, confirmed in the current FLIPR, electrophysiological, and radioligand studies, narrows the safety window of ketamine. Although temporary dissociative effects occur in most patients treated with racemic ketamine and esketamine at doses currently used to treat MDD, the current consensus is that induction of dissociative effects is not necessary for the rapid antidepressant effects of ketamine [
37,
38]. The low affinity of REL-1017 for the NMDAR spares the block of phasic NMDAR activity, as evidenced by its lack of dissociative effects at therapeutic exposures [
13,
39]. Despite its low affinity, REL-1017 remains trapped in the NMDAR selectivity pore similarly to ketamine and is therefore able to reduce excessive tonic Ca
2+ currents, with downstream effects that restore synaptic proteins [
40] and exert antidepressant-like effects in animal models [
9], increase serum BDNF [
35], and rapidly improve MDD in patients [
13]. The importance of tonic blockade by uncompetitive NMDARs in determining synaptic protein availability, synaptic enhancement, and resolution of depressive-like behavior in animal models has been anticipated by other authors [
5,
6,
7,
8]. The importance of the NR1-2D subtype in the sustained antidepressant actions of arketamine, a less potent NMDAR blocker compared to esketamine, has been suggested by experimental work showing that the sustained antidepressant-like effects of arketamine are lost in NR1-2D subunit knockout mice [
41].
The preferential activity for NR1-2D subtypes at resting membrane potential in the presence of physiological Mg2+ and the sensitivity of this NMDAR subtype to low-concentration glutamate may explain the downregulating effects of uncompetitive NMDAR channel blockers on tonic Ca2+ currents via preferential block of hyperactivated NR1-2D subtypes. In particular, the effects of uncompetitive NMDAR channel blockers at NR1-2D subtypes may be seen when pathologically elevated ambient glutamate induces a higher probability of the open configuration preferentially for the NR1-2D subtype, as shown in this report.
Future studies of REL-1017 blocking effects on NMDAR-mEPSCs in cortical brain slices may add informative data to these results.
4. Materials and Methods
4.1. Drugs and Reagents
All chemicals were of analytical grade. Dextromethorphan and (±)-ketamine were from Merck Sigma-Aldrich (Milan, Italy). Memantine and (+)-MK-801 were from Bio-Techne/Tocris (Milan, Italy). REL-1017 (dextromethadone HCl) was from SpecGx (Webster Groves, MO, USA).
Cell culture consumables were from Gibco Life Technologies (Milan, Italy), including Dulbecco’s Modified Eagle Medium/F12 (DMEM/F12; Cat # 21331), heat-inactivated fetal bovine serum (FBS; Cat # 10500), penicillin-streptomycin (Cat # 15140), L-glutamine (Cat # 25030), MEM nonessential amino acids (NEAAs; Cat # 11140), blasticidin (Cat # A11139), G418 (Cat # 10131), Zeocin™ (Cat # R250), and TrypLE™ enzyme (Cat # 12604). Hygromycin B (Cat# 10687) and Fluo-4 (F14202) were from Invitrogen (Waltham, MA, USA).
4.2. Cell Lines
CHO cell lines stably over-expressing heterodimeric NMDARs composed of human NR1 and 2 subunits were generated by Aptuit, an Evotec company (Verona, Italy). Cell line details are available at ExPASy Cellosaurus database. Research Resource Identifiers (RRIDs) are CVCL_B3TY, CVCL_B3TZ, CVCL_B3U0, and CVCL_B3U1 for CHO-hNR1-h2A, CHO-hNR1-h2B, CHO-hN1-h2C, and CHO-hN1-h2D, respectively. Protein accession numbers were NP_015566, NP_000824, NP_000825, NP_000826, and NP_000827 for NR1, 2A, 2B, 2C, and 2D, respectively. NP_015566 corresponds to the NR1-1a splice variant [
42] lacking the N1 cassette but including C1 and C2 cassettes. The four different heterodimeric NMDARs studied were NR1-2A, NR1-2B, NR1-2C, and NR1-2D. Cell lines were cultured for a maximum of 75 passages.
Cells were grown at 37˚C, 5% CO2 in DMEM/F12, added with 10% FBS, 1% penicillin-streptomycin, 2 mM L-glutamine, 1% MEM-NEAA, 500 µM ketamine, and selected antibiotics. Selected antibiotics were 10 µg/mL blasticidin, 400 µg/mL G418, 400 µg/mL hygromycin B (except for NR1-2B-CHO), and 300 µg/mL Zeocin™ (for NR1-2B-CHO cells only).
4.3. FLIPR Assay
FLIPR assay allowed for indirect measurement of intracellular Ca2+ concentration by means of a Ca2+-sensitive fluorescent dye (Fluo-4) using the FLIPR instrument (Molecular Devices, San Jose, CA, USA). For these assays, 40 mM stock solutions of the test items were prepared in 100% dimethyl sulfoxide (DMSO), while glutamate and glycine 4 mM stock solutions were prepared in water. 384-well compound plates containing 400 × concentrated test item, glutamate, and glycine were prepared using Labcyte Echo acoustic dispensing system and stored at −20 °C until the day of the experiment. Then, 4 × assay plates were prepared by the addition of 30 µL/well of assay buffer in the presence of 0.5% (v/v) poloxamer 188 solution (Sigma-Aldrich code P5556) on experiment day.
Cells were plated in 384 black/clear bottom plates, at a density of 15.000/well, in the presence of 500 µM ketamine and 10 µg/mL tetracycline to induce receptor expression (except for NR1-2B-CHO cells, which showed NMDAR constitutive expression) and then kept at 30 °C in 5% CO2 incubator for 24 h. Plated cells were preloaded for 1 h with 2 μM Fluo-4 Ca2+-sensitive dye in presence of 2.5 mM probenecid, an inhibitor of nonspecific anion transport, and 500 μM ketamine to avoid cytotoxicity and then washed with a three-cycle wash in assay buffer. This wash procedure removed all ketamine, as assessed by a consistent full response elicited in positive control wells of every plate by 10 µM glutamate plus 10 µM glycine. Assay buffer composition was 145 mM NaCl, 5 mM KCl, 2 mM CaCl2, 1 g/liter D-(+)-glucose, and 20 mM HEPES (pH was adjusted to 7.3 with NaOH). Intracellular Ca2+ level was monitored by FLIPR (excitation wavelength at 470–495 nm, emission wavelength at 515–575 nm) for 10 s before and 5 min after the addition of test items. All test items, including L-glutamate (when present) and glycine, were added simultaneously. Glycine 10 μM was always included in every test item addition.
In FLIPR CRC experiments, every test item was assessed at 11 final concentrations: 100 μM, 33 μM, 11 μM, 3.7 μM, 1.2 μM, 412 μM, 137 nM, 46 nM, 15 nM, 5.1 nM, and 1.7 nM. L-glutamate and glycine were both used at a 10 μM final concentration. In FLIPR mode of action experiments, every test item was assessed at six final concentrations: 50 μM, 12.5 μM, 3.13 μM, 0.781 μM, 0.195 μM, and 0.049 μM. L-glutamate was assessed at 11 final concentrations: 100 mM, 1 mM, 100 μM, 10 μM, 3.3 μM, 1.1 μM, 370 nM, 123 nM, 41 nM, 13.7 nM, and 4.6 nM. Glycine was kept at a constant concentration of 10 μM during experiments. Fluorescence was measured as the area under the curve (AUC) during the 5 min immediately after the addition of the test item. FLIPR data were normalized to intracellular Ca2+ levels in the presence of 10 μM L-glutamate and 10 μM glycine as 100%, while in the presence of assay buffer as 0% response.
4.4. Whole-Cell Automated Patch-Clamp Assay
QPatch HTX (Sophion Bioscience, Denmark) automated planar whole-cell patch-clamp platform was used to assess the effects of REL-1017 on L-glutamate–elicited current on heterodimeric NMDARs in the presence of physiological Mg2+. For QPatch experiments, cells were grown in T-flasks first at 37 °C, 5% CO2 for 3 to 4 h, then at 30 °C, 5% CO2 for 18 to 24 h in DMEM/F12 medium with added 500 μM ketamine and 10 μg/mL tetracycline (except for NR1-2B-CHO cells) without additional antibiotics. Cells were manually detached from the culture dish and automatically dispensed in QPatch wells with extracellular solution in the presence of 1 mM Mg2+ and 500 μM ketamine. Extracellular solution composition was 155 mM NaCl, 3 mM KCl, 1.5 mM CaCl2, 1 mM MgCl2, 10 mM HEPES, and 10 mM D-glucose, adjusted to pH 7.4 with NaOH. Intracellular solution composition was 80 mM CsF, 50 mM CsCl, 0.5 mM CaCl2, 10 mM HEPES, and 11 mM EGTA, adjusted to pH 7.25 with CsOH.
Clamped cells were extensively washed in extracellular buffer solution to eliminate endogenous L-glutamate and ketamine, which had been previously added to block excitotoxicity. NMDAR-mediated currents elicited by L-glutamate in the presence of 10 μM glycine and 1 mM MgCl
2 were recorded at different voltages. Holding potential was −80 mV, and voltage protocol included a depolarizing 2-s step pulse to +60 mV to check the quality of the seal and cell NMDAR expression level, followed by a 2-s ramp-up to holding potential. L-glutamate 10 μM or 1 μM was added during the pulse to +60 mV and washed away at the end of the ramp (
Figure S6). L-glutamate addition was repeated in two consecutive voltage protocols, and 10 μM REL-1017 was present during the second L-glutamate addition in the treated group but not in the control group. MgCl
2 1 mM was present throughout the L-glutamate addition.
4.5. Whole-Cell Manual Patch-Clamp Assay
Cells were grown on poly-D-lysine coated glass coverslips for manual patch-clamp whole-cell recording. Intracellular solution composition was 80 mM CsF, 50 mM CsCl, 0.5 mM CaCl2, 10 mM HEPES, and 11 mM EGTA, adjusted to pH 7.25 with CsOH. Extracellular solution composition for on-/off-rate and trapping assays was 155 mM NaCl, 3 mM KCl, 1.5 mM CaCl2, 10 mM HEPES, and 10 mM D-glucose, adjusted to pH 7.4 with NaOH. Extracellular solution for REL-1017 CRC also contained 1 mM MgCl2. Whole-cell recordings occurred at −70 mV fixed voltage equal to holding potential for onset and offset kinetic and trapping assays using NR1-2C-CHO cells. Whole-cell recordings occurred at −60 mV for REL-1017 CRCs.
The data capture and primary analysis were accomplished using HEKA Elektronik’s PATCHMASTER software (v2.53). Data were filtered at 1 kHz and sampled at 2 kHz. Rapid agonist or agonist-antagonist application was accomplished using a BioLogic RSC-160 perfusion device (BioLogic, Seyssinet-Pariset, France). The system consisted of a custom-made manifold of three tubes, whose tip was placed at around 100 μm from the patch-clamped cell under study.
4.6. NMDAR Binding Assay in Rat Brain Cortical Membranes
Multiple concentrations of (+)-MK-801, (±)-ketamine, memantine, dextromethorphan, and REL-1017 were tested in displacement binding assays in rat cortical membranes by Eurofins Panlabs Discovery Services Taiwan (Taipei City, Taiwan). The tritiated [3H]TCP ligand was selected for its ability to bind the pore of NMDARs (specific binding 94%, Kd 8.40 nM, and Bmax 0.78 pmol/mg).
Wistar rat cortical membranes were obtained as previously described [
43]. A 6.3-mg aliquot was incubated with 4 nM [
3H]TCP for 45 min at 25 °C. Nonspecific binding was defined by 1 μM dizocilpine ((+)-MK-801). Each tested compound was evaluated at the following concentrations: 1 μM, 3 μM, 10 μM, 30 μM, 100 μM, 3 nM, 10 nM, 30 nM, 100 nM, and 300 nM in 10 mM Tris-HCl, pH 7.4, as an incubation buffer. Membranes were filtered and washed, and the filters were then counted to determine [
3H]TCP specifically bound, allowing us to calculate IC
50 and K
i for each tested compound.
IC
50 values were determined by a nonlinear, least squares regression analysis using MathIQ
TM (ID Business Solutions Ltd., UK). The K
i values were calculated using the Cheng and Prusoff equation [
44], using the observed IC
50 of the tested compound and the concentration of radioligand employed in the assay.
4.7. Additional Radioligand Displacement Binding Assays
(±)-Ketamine, memantine, dextromethorphan, and REL-1017 were also submitted to Eurofins Panlabs Discovery Services Taiwan for single-concentration (10 μM) binding assays for the following receptors under the following conditions:
Glutamate, AMPA. Source: Wistar rat cerebral cortex. Ligand: 5.0 nM [3H]AMPA. Nonspecific ligand: 1.0 mM L-glutamic acid. Incubation time/temp: 90 min/4 °C. Specific binding: 90%. Incubation buffer: 50 mM Tris-HCl, pH 7.4, 200 mM KSCN.
Glutamate, kainate. Source: Wistar rat brain cortex. Ligand: 6.0 nM [3H]kainic acid. Nonspecific ligand: 1.0 mM L-glutamic acid. Incubation time/temp: 2 h/4 °C. Specific binding: 80%. Incubation buffer: 50 mM Tris-HCl, pH 7.4.
Glutamate, metabotropic, mGlu2 human: Source: Human recombinant Chem-1 cells. Ligand: 2.0 nM [3H]LY341495. Nonspecific ligand: 5.0 μM LY-354740. Incubation time/temp: 60 min/25 °C. Specific binding: 90%. Incubation buffer: 10 mM KH2PO4, 100 mM KBr, pH 7.6 (pH adjusted with KOH).
Glutamate, metabotropic, mGlu5 human: Source: Human recombinant CHO-K1 cells. Ligand: 30 nM [3H]quisqualic acid. Nonspecific ligand: 1.0 mM L-glutamic acid. Incubation time/temp: 2 h/25 °C. Specific binding: 85%. Incubation buffer: 25 mM HEPES, 1 mM MgCl2, 2.5 mM CaCl2, pH 7.4.
Glutamate, NMDA, agonism: Source: Wistar rat cerebral cortex. Ligand: 2.0 nM [3H]CGP-39653. Nonspecific ligand: 1.0 mM L-glutamic acid. Incubation time/temp: 20 min/4 °C. Specific binding: 90%. Incubation buffer: 50 mM Tris-HCl, pH 7.4.
Glutamate, NMDA, glycine: Source: Wistar rat cerebral cortex. Ligand: 0.33 nM [3H]MDL 105,519. Nonspecific ligand: 10.0 μM MDL 105,519. Incubation time/temp: 30 min/4 °C. Specific binding: 85%. Incubation buffer: 50 mM HEPES, pH 7.7.
Glutamate, NMDA, polyamine: Source: Wistar rat cerebral cortex. Ligand: 2.0 nM [3H]ifenprodil. Nonspecific ligand: 10.0 μM ifenprodil. Incubation time/temp: 2 h/4 °C. Specific binding: 80%. Incubation buffer: 50 mM Tris-HCl, pH 7.4.
Glycine, strychnine sensitive: Source: Wistar rat spinal cord. Ligand: 10.0 nM [3H]strychnine. Nonspecific ligand: 1.0 mM glycine. Incubation time/temp: 10 min/4 °C. Specific binding: 77%. Incubation buffer: 50 mM KH2PO4, 200 mM NaCl, pH 7.1.
For each assay, quantitation was performed by radioligand binding, and significance was considered when ≥50% of max stimulation or inhibition was attained.
4.8. Computational Model of Four NMDAR Subtypes in the Open and Closed Conformational States
Structural models were generated for the four NMDAR subtypes using the homology modeling pipeline and implemented the Schrödinger suite for molecular modeling, leaving all the options to their default values.
Amino acid sequences of the subunits were retrieved from UniProt (Q12879, Q13224, Q14957, and O15399 for 2A, 2B, 2C, and 2D, respectively). The structure of the NR1b-2B subtype from
Rattus norvegicus recently solved by Chou and colleagues [
19] in the open (PDB 6WHT) and closed (PDB 6WHS) conformation was used as a template. Finally, all generated models were optimized using the protein preparation wizard procedure implemented by the Schrödinger suite.
Ligand docking calculation was performed with Glide [
45,
46], centering the grid on the QRN site [
33] (i.e., the asparagine residues that correspond to position 615 in the template structures) and using the standard precision (SP) mode. All ligand structures were prepared by LigPrep software. In the case of ketamine, docking calculations were performed on both enantiomers (arketamine and esketamine).
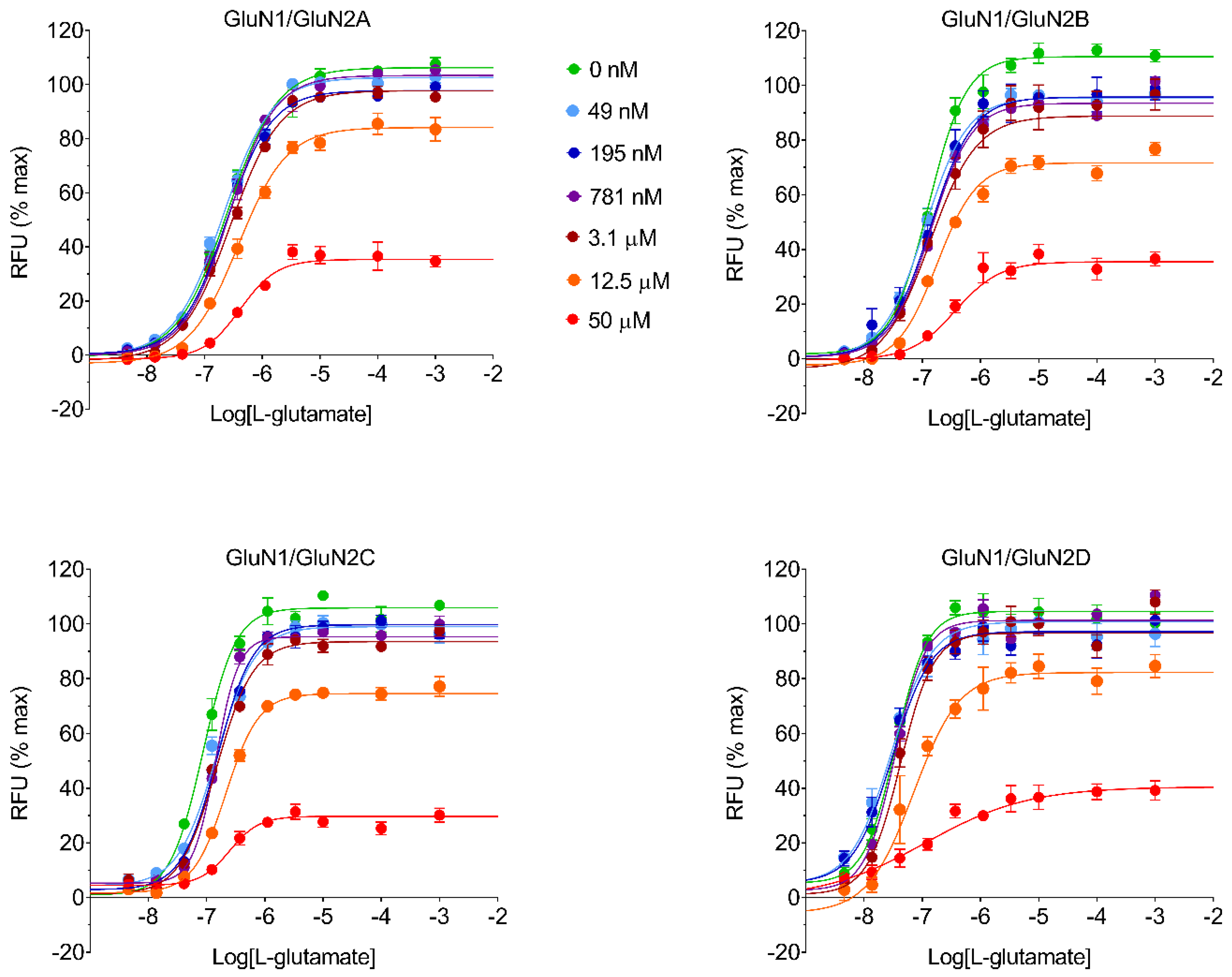
4.9. Operational Equation for KB Estimation
The operational equation for allosteric modulators [
14,
15,
16] was created in GraphPad Prism v8.0 software to estimate K
B and α parameters for every test item with the assumption that, as a channel blocker, every test item would be able to produce a compete blockade of agonist response at sufficiently high concentration:
where Y is the % effect of L-glutamate, [A] is the L-glutamate molar concentration; E
MAX is the maximal possible L-glutamate effect estimated from the four-parameter logistic equation; EC
50 is the half maximal effective L-glutamate concentration estimated from the four-parameter logistic equation; τ is an arbitrary L-glutamate efficacy value at NMDAR set at τ = 100; [B] is the test item molar concentration; K
B is the estimated test item equilibrium dissociation constant; and α is the estimated cooperativity term, which indicates the effect of the test item on the L-glutamate dissociation equilibrium constant for the receptor (i.e., α is the estimated ratio between the L-glutamate equilibrium dissociation constant in the absence and in the presence of the test item, and it is expected to be 0 < α ≤ 1 for a negative allosteric modulator affecting the agonist equilibrium dissociation constant). Calculated α values are reported in
Table S1. Percent affinity ratio was computed from estimated affinities, which are the reciprocal of K
B, and considering the highest affinity for an NMDAR subtype as 100%.
4.10. Onset and Offset Kinetic Calculation
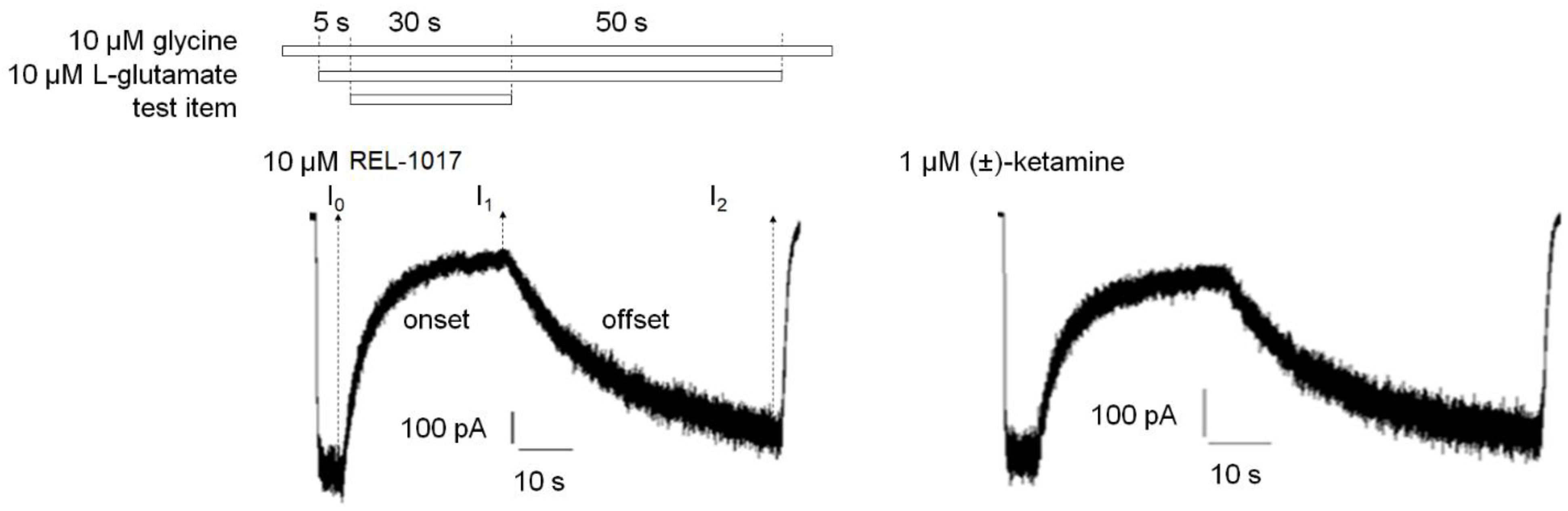
At least
n = 10 independent cells were analyzed. For each cell, the current in the presence of 10 μM glycine only was set as 0%, while the steady-state current induced after 5 s of 10 μM L-glutamate and 10 μM glycine application was set as 100% (
Figures S4 and S5). The time constants of onset (tau on, seconds) and offset (tau off, seconds) of test item inhibition of glutamate-induced current were calculated using a first order exponential equation:
First order equation for test item onset: I(t) = I1 + (I0 − I1) × e−t/τon
First order equation for test item offset: I(t) = I1 + (I2 − I1) × (1 − e−t/τoff)
I(t) is the current at time t. t is the time (seconds) after test item application or removal in the onset or offset equation, respectively. I0 is the current after 5 s of 10 μM L-glutamate and 10 μM glycine application prior to test item application. I1 is the current 30 s after application of the test item in the presence of 10 μM L-glutamate and 10 μM glycine. I2 is the current 50 s after removal of the test item in the continuous presence of 10 μM L-glutamate and 10 μM glycine. τon (tau on) is the time constant (seconds) of onset. τoff (tau off) is the time constant (seconds) of offset.
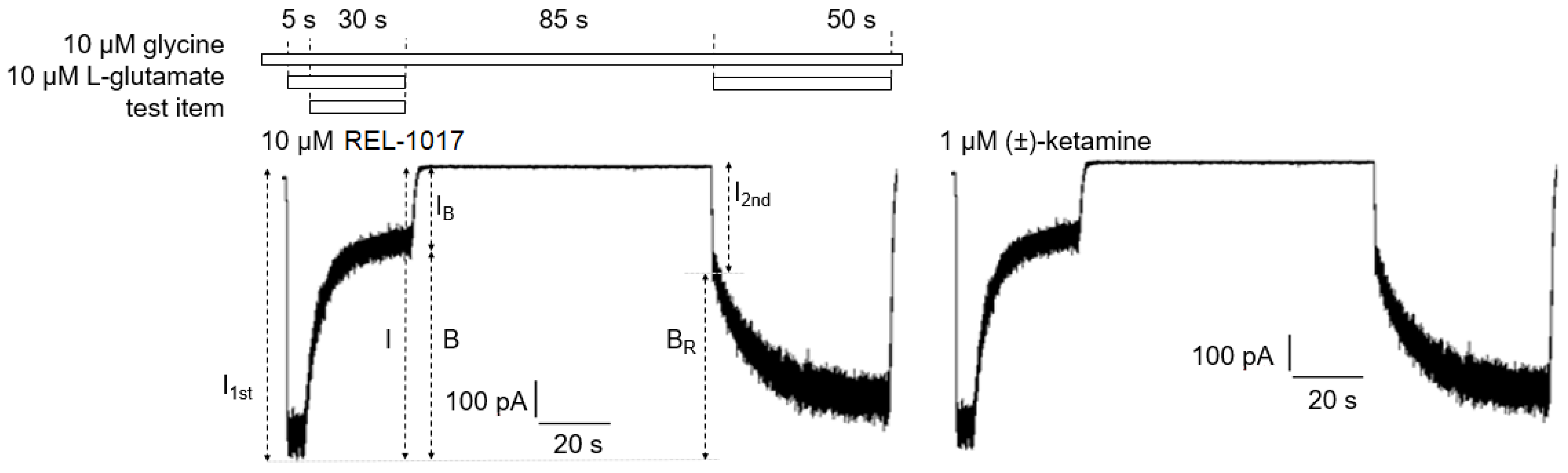
4.11. Trapping Calculation
Trapping was calculated as previously described [
18]. Blocks of 10/10 μM L-glutamate/glycine–evoked currents were calculated according to the formula
B = [(
I −
IB)/
I] × 100, where I was determined as the current value derived from a linear extrapolation to the end of the L-glutamate antagonist co-application, and IB was the current measured at the end of L-glutamate/blocker co-application.
The residual block of L-glutamate–evoked currents was calculated according to the formula BR = [(I1t − I2st)/I1st] × 100 (2), where I1st was the maximal current measured 1 s after onset of the first L-glutamate exposure, and I2nd was the maximal current measured 1 s after onset of the delayed second L-glutamate exposure following washout of the blocker from the bath.
The block trapped (BT), or the amount of block remaining at the beginning of the second L-glutamate application as a percent of the initial block produced at the end of the previous L-glutamate/antagonist co-application, was calculated according to the formula
BT = BR/B × 100 (3), where B and BR were defined as above.
4.12. Statistical Analysis
The normality (Shapiro-Wilk) test was performed before any statistical analysis. Only data that passed the normality test (p ˃ 0.05) were considered for statistical analyses. Comparisons between groups were performed by Student’s t-test for unpaired data or one-way analysis of variance (ANOVA) followed by the Tukey’s post hoc multiple comparison test, when appropriate. In all the experiments, p < 0.05 was considered statistically significant.



 ,
, 









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}