1. Introduction
Virus-mediated infectious diseases, such as coronavirus-induced disease 2019 (COVID-19), caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), are global health challenges. Moreover, the treatment options for virus-mediated diseases are limited, so the development of well-tolerated, as well as efficient, antiviral therapies against emerging and re-emerging viruses is a high priority. Silvestrol could be a promising new broad-spectrum antiviral, since it inhibits the replication of Hepatitis E- [
1], Ebola- [
2], Zika- [
3] Chikungunya- [
4], Lassa-[
5], Crimean Congo hemorrhagic fever [
5], as well as subtypes of Picorna- [
6] and coronaviruses [
6].
Several viruses need the ATP-dependent DEAD-box RNA helicase elF4A for the translation of their mRNAs; therefore, the inhibition of this host enzyme by silvestrol is an interesting antiviral approach [
7,
8]. The advantages of targeting host factors include a decreased risk of escape mutations by the virus [
9,
10]. The disadvantages of such a strategy are possible pleiotropic side effects [
11]. However, the inhibition by silvestrol and other rocaglates of the viral 5′ mRNA structure unwinding activity of eIF4A appears to be highly specific, which should reduce the risk of side effects. Since silvestrol was initially described as an anti-cancer drug agent [
12,
13,
14], the assessment of the safety, off-target and pharmacokinetic profile is essential for its preclinical development as a broad-spectrum antiviral.
For the in vitro safety profile viability testing in various cancer cell lines, a micronucleus and an Ames test were performed. For detection of possible off-target effects mediated by GPCR signaling, a cyclic adenosine monophosphate (cAMP), an inositol 1,4,5-triphosphate (IP
3) and a Ca
2+-release assay were used. For the in vitro pharmacokinetic profile, a stability assay, a transport assay and a cellular uptake assay were used to gain initial insights into the expected bioavailability of silvestrol [
15,
16]. Here, these aspects were explored in standard cell culture test systems to assess the druggability of silvestrol.
3. Discussion
Silvestrol, originally described as a potent anti-tumor compound [
28], was recently identified as a promising antiviral drug candidate with broad-spectrum activity [
3,
6]. Initially, the antiviral activity of silvestrol was identified in primary human macrophages infected with the Zaire Ebolavirus (ZEBOV) [
3]. Importantly, mRNAs from EBOV are translated in a cap-dependent manner and harbor well-defined stable RNA secondary structures in their 5’-untranslated regions that need to be unwound to allow the binding of the 43S–preinitiation complex [
29]. Therefore, it was not unexpected that EBOV requires eIF4A for translation initiation and, consequently, for its replication [
3]. Usually, the next step on the path towards drug development includes prediction of potential bioavailability and an in vitro safety profile. Here, we performed relevant preclinical experiments in order to characterize the potential of silvestrol as an antiviral drug candidate in more detail. Our data indicate that silvestrol did not impair Caco-2 cell barrier integrity and has low permeability through Caco-2 and Calu-3 cell barriers. Silvestrol shows good stability in liver microsomes and good cellular uptake in cells expressing low levels of P-glycoprotein. Furthermore, we observed no off-target effects of silvestrol regarding GPCR signaling pathways, no mutagenic potential (Ames test) and a low genotoxic potential at higher concentrations. This apparently minor genotoxicity will need to be confirmed and characterized in further in vitro and in vivo studies. Regarding its cytotoxicity, silvestrol reduced cell viability in kidney cell lines HEK293T and Caki-2 with a CC
50 of 16 and 37 nM, respectively, whereas in Caco-2, HepG2 and Calu-3 cells, concentrations up to 1000 nM silvestrol reduce viability only by about 50%. In other studies, cell-type-dependent cytotoxicity with silvestrol has also been observed. For example, in the lung cancer cell line A549, the CC
50 value was 9.42 nM [
5] and in the colon cancer cell line HT-29, the CC
50 value was 0.7 nM [
17]. Importantly, in several non-transformed primary cells, silvestrol has no major cytotoxicity [
6,
20,
30].
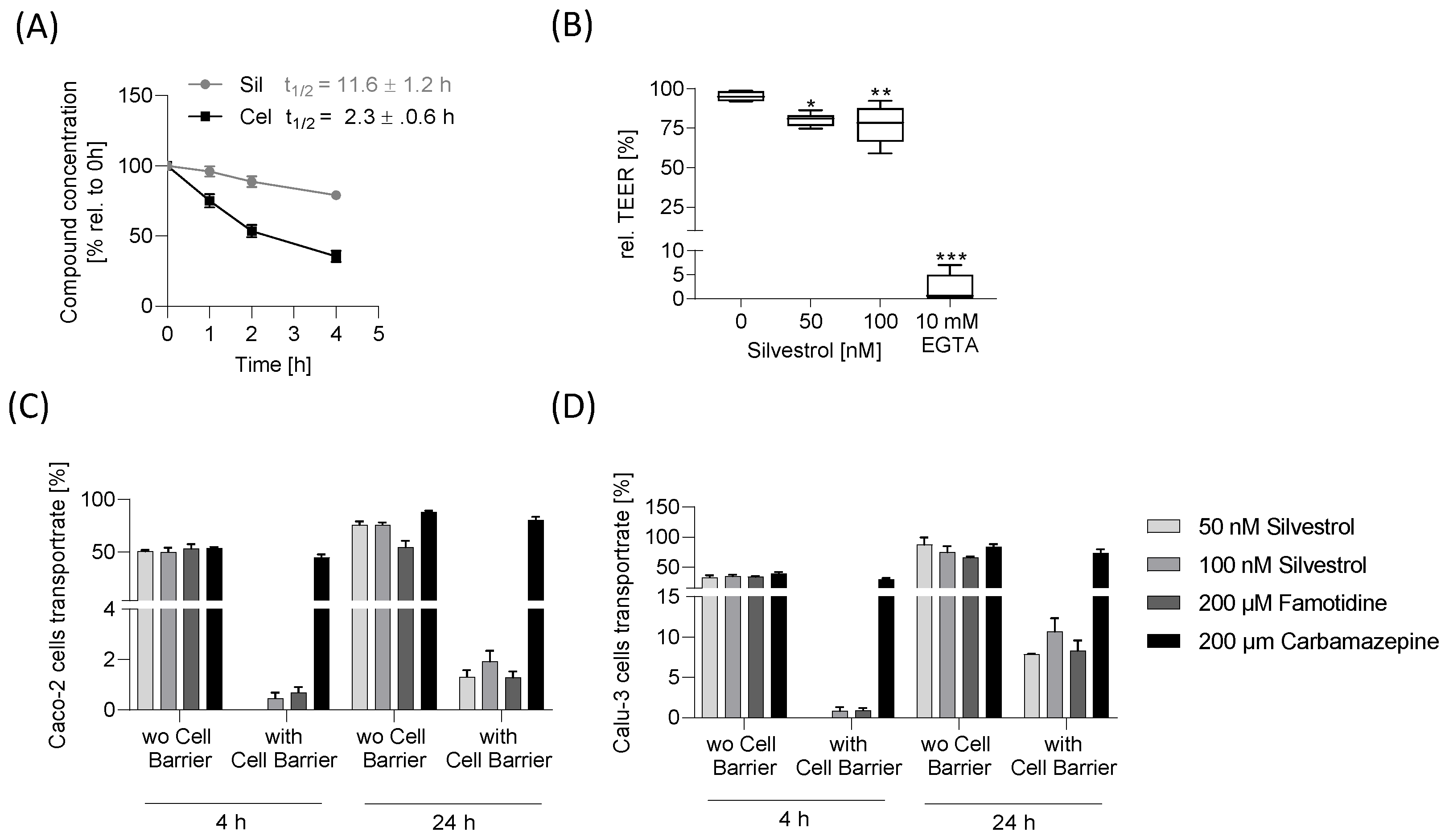
To estimate transport across the intestinal or lung epithelium, the Caco-2 and Calu-3 cell barrier model was used. Silvestrol showed a low transport rate in both barrier models, indicating low permeability. Our in vitro data, thereby, confirm the results of a previous pharmacokinetic study in mice, which revealed that the bioavailability of silvestrol depends on the administration route. In this earlier study, mice were treated with silvestrol formulated in hydroxypropyl-β-cyclodextrin via intravenous, intraperitoneal and oral routes. Intraperitoneal systemic availability was 100%, but oral administration resulted in only 1.7% bioavailability [
30]. The low oral bioavailability and the observed potential cytotoxic effects suggest that silvestrol would best be administered locally for treatment of infections caused by respiratory viruses via inhalation using a targeted aerosol formulation. This would have the advantage that the poor absorption through the intestine, transport via the bloodstream and, thus, cytotoxic effects on peripheral immune cells can all be avoided. Improvement in silvestrol’s bioavailability without affecting its antimicrobial efficacy by suitable formulations, such as solid dispersions [
31], may also be an option to increase the local concentration of silvestrol in the infected respiratory tract.
Surprisingly, the high level of P-glycoprotein in Caco-2 cells did not result in a higher transportation rate of silvestrol. In a previous study, it was shown that a Caco-2 cell monolayer exhibits apical efflux of drugs, indicating that substrates of P-glycoprotein are transported in the reverse direction back to the apical compartment [
22]. Therefore, instead of using the transcellular route to the basolateral compartment, P-glycoprotein may shuttle silvestrol back to the apical compartment.
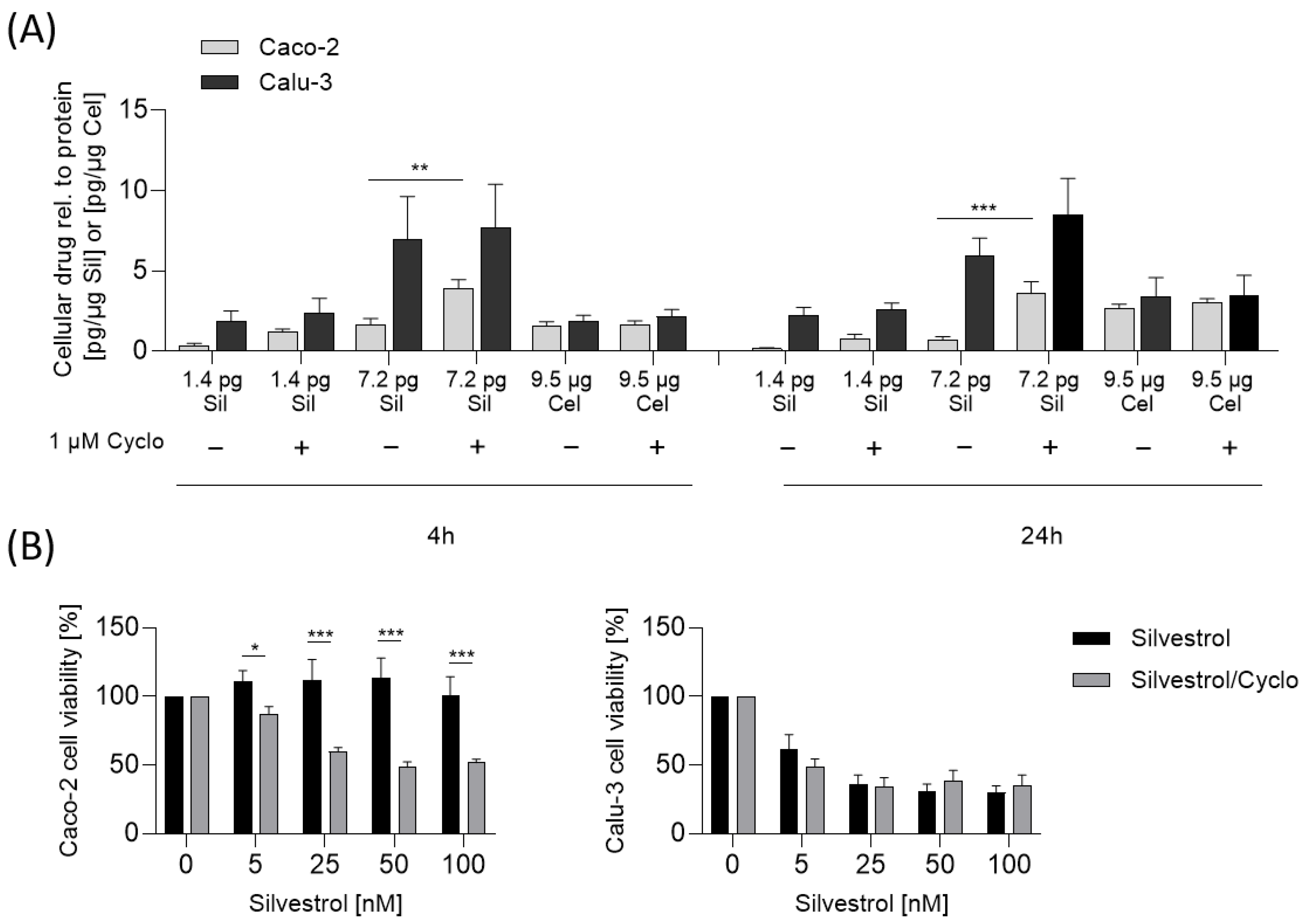
As mentioned, the concentration of silvestrol was higher in the lung cancer cell line Calu-3 than in the colon cancer cell line Caco-2. One explanation for the observed cell-type-dependent cytotoxicity could be that the cellular uptake and/or the efflux of silvestrol differs in these cell types due to different cellular P-glycoprotein levels. Calu-3 cells express lower levels of P-glycoprotein in comparison to Caco-2 cells, as shown by Hamilton et al. [
27] and confirmed in our experiments (see
Figure 5B). We assumed that the cell-type-dependent, intracellular silvestrol level depends on an increased efflux of silvestrol and not on different absorption rates. This would also fit with the observed time-dependent increase in uptake into Calu-3 compared to Caco-2 cells. In cells with increased efflux of silvestrol, the equilibrium between uptake and efflux is reached earlier and, consequently, no further increase in cellular silvestrol concentration can be observed. Moreover, celecoxib, which is not a substrate of P-glycoprotein [
32], showed similar uptake rates in Caco-2 and Calu-3 cells. Accordingly, the cytotoxicity of silvestrol correlates with the level of P-glycoprotein in cancer cell lines. In line with this, inhibition of P-glycoprotein by cyclosporine increased the intracellular silvestrol levels and, therefore, the sensitivity of Caco-2 cells towards the cytotoxicity of silvestrol. These data indicate that the cell-type-dependent cytotoxicity of silvestrol depends on its intracellular concentration, which is determined not only by cellular uptake but also by its efflux from the cell.
The CC
50 value of silvestrol increased in primary monocytes (CC
50 = 29 nM), M1 macrophages (CC
50 = 45.6 nM) and M2 macrophages (CC
50 > 100 nM), in that order [
9]. Interestingly, the P-glycoprotein level in U937 monocytes, M1 macrophages and M2 macrophages increased in the same order [
33]. These data indicate that P-glycoprotein level in primary monocytes, M1 and M2 macrophages could also be responsible for the different CC
50 values of silvestrol in these cell types. For T cells expressing high levels of P-glycoprotein, we found no cytotoxic effects up to 100 nM silvestrol [
9,
34]. Moreover, the genotoxic effects observed in HepG2 cells may also be caused by high intracellular silvestrol concentration, since HepG2 cells have about 10-fold lower P-glycoprotein levels compared to Caco-2 cells [
35]. Given the wide distribution of P-glycoprotein in the body [
36] and in immune cells (e.g., B cells, T cells, natural killer cells) [
34,
37], its relevance for the effects of silvestrol in vivo will require careful consideration.
The cytotoxicity of a drug can be considered tolerable if the CC
50 is at least 50-fold higher than the effective concentration (EC
50) [
16]. In our study, we observed a cell-type-dependent cytotoxic effect, with the lowest CC
50 value of 16 nM in HEK293T cells. However, cytotoxicity in cancer cells, but not in healthy cells, is a prerequisite for an anti-cancer compound like silvestrol. Of course, in several primary cells, the selectivity index (SI) of silvestrol (SI calculated by CC
50/EC
50), regarding its antiviral activity, can reach 1000 and virus titers can be reduced by up to 4 log phases [
5]. Therefore, silvestrol is a very promising candidate for further antiviral drug development. In accordance with this, the synthetic rocaglate zotatifin has already reached the clinic in a dose-escalation study in patients with mild or moderate COVID-19 [
38].
Like silvestrol, zotatifin also induces a G2 cell cycle block and apoptosis at 100 nM in the MDA-MB-231 cell line [
39], indicating that the inhibition of the target elF4A mediates cytotoxic effects. However, in primary immune cells, zotatifin mediates lower cytotoxicity in comparison to silvestrol, whereas CR31B (-), another synthetic rocaglate, has comparable cytotoxic effects to those of silvestrol [
9]. Since rocaglates are highly specific eIF4A inhibitors, our data indicate that the inhibition of elF4A is mainly responsible for the observed cell-type-dependent cytotoxic effects. However, additional secondary effects based on reduced translation of cellular mRNAs should also be considered. It can be expected that different rocaglates have different effects on the pool of mRNAs in a given cell. For example, only the natural compound silvestrol contains a dioxane moiety that could promote polypurine-independent RNA-clamping, whereas synthetic rocaglates without the dioxane moiety need a polypurine stretch to clamp the RNA substrate efficiently onto the surface of eIF4A [
5].
Interestingly, silvestrol reduces energy metabolism [
40] and cancer cell lines have a higher energy requirement than primary cells due their higher proliferation rate [
41]. In line with this, several proto-oncogenes have complex 5’-UTRs with stable RNA secondary structures that need to be unwound by eIF4A during translation initiation. For example, eIF4A-dependent mRNAs, including critical Kirsten rat sarcoma virus (KRAS) signaling molecules, such as phosphoinositide 3-kinase (PI3K), Ras-related protein (RalA), Ras-related C3 botulinum toxin substrate 2 (Rac2) and MYC, were identified in pancreatic adenocarcinoma by transcriptome-scale ribosome footprinting [
42]. These data further indicate that the effects of rocaglates on proliferation could be mediated by the translation inhibition of proto-oncogenic mRNAs, which drive the cell cycle. Nevertheless, more extensive global analyses of rocaglate-treated primary cells by proteomics and/or ribosome profiling approaches should be performed to gain further information on eIF4A-dependent mRNAs in infection-relevant cell systems.
The main therapeutic indicator from the studies discussed is that bioavailability of silvestrol is rather low, indicating that oral application should be avoided and local administration, for example, via inhalation, should be clearly preferred. A further important finding of our study is that silvestrol shows minor genotoxicity at higher concentrations. These potential side effect should be addressed in further studies. The cell-type-dependent effect could be relevant for side effects in cancer and infection treatment, since cells with high P-glycoprotein expression are potentially protected from side effects, but possibly also excluded from treatment effects.
Taken together, our findings indicate that silvestrol, due to its cell-type-dependent cytotoxicity and low permeability, should be administered directly into the respiratory tract at least to combat respiratory viruses.
4. Materials and Methods
4.1. Cells and Reagents
Caki-2 cells were cultured in McCoy’s 5A (modified) medium supplemented with 10% FCS, Calu-3 cells were cultured in MEM with 10% FCS and 1x non-essential amino acids and both obtained from ATCC (Manassas, VA, USA). HEK293T cells were cultured in DMEM supplemented with GlutaMAX, 10% heat-inactivated FCS. Caco-2 cells were from Sigma Aldrich (Schnelldorf, Germany) and were cultured in EMEM medium supplemented with 10% FCS, L-glutamine and non-essential amino acids (M7145, Sigma Aldrich, Schnelldorf, Germany). HepG2 was obtained from Sigma Aldrich (Schnelldorf, Germany) and cultured in DMEM supplemented with GlutaMAX and 10% heat-inactivated FCS. All media contained 1% penicillin/streptomycin and the cells were cultured at 37 °C in a 5% CO2 atmosphere. Silvestrol was dissolved in DMSO and further diluted in media (cstock = 6 mM, maximal DMSO concentration during experiments 0.1% v/v). EGTA, famotidine and carbamazepine were from Sigma Aldrich (Schnellendorf, Germany). Lucifer Yellow (sc-215269) was obtained from Santa Cruz. Celecoxib was synthesized by WITEGA Laboratorien Berlin-Adlershof GmbH (Berlin, Germany). Forskolin and ionomycin were obtained from Sigma Aldrich (Schnelldorf, Germany). Silvestrol was provided by the Sarawak Biodiversity Centre (Kuching, North-Borneo, Malaysia; purity > 99%) and zotatifin was bought from MedChemExpress (USA, purity: 98%).
4.2. Cell Viability/Proliferation Assays
The OranguTM kit for viability testing was performed as described by the supplier. Briefly, 2.0 × 104 Caco-2 cells, 2.0 × 104 Calu-3 cells, 2.0 × 104 HEK293T cells or 1.0 × 101 Caki-2 cells were incubated for 24 h at 37 °C. Silvestrol (0–500 nM or 0–5000 nM (Caco-2 cells)) and control (DMSO) were added to the cells and mixed. After 48 h, 10 µL Orangu (WST-8) reagent (Cell Guidance System, Cambridge, UK) was added, mixed and incubated at 37 °C for 60 min. An EnSpire plate reader (PerkinElmer, Waltham, MA, USA) was used to measure absorbance at 450 nm and at 650 nm (reference). The absorbance at 450 nm was normalized with the absorbance at 650 nm. The sample values were corrected with the background wells (wells with medium and without cells). The absorbance of DMSO-treated cells was set to 100% and the silvestrol samples were correlated to the absorbance value of DMSO-treated samples to calculate cell viability.
4.3. Ames Test
The Ames MPF 98/100 test (Xenometrix, Allschwil, Switzerland) was performed as recommended by the supplier and recently published [
43]. Briefly, two
Salmonella typhimurium strains TA98 and TA100 (incapable to produce histidine) were used with or without liver S9 homogenate to simulate the metabolic conversions of silvestrol with liver enzymes. After exposure to increasing concentrations of silvestrol or positive controls (2 μg/mL of 2-NF (TA98), 0.1 μg/mL of 4-NQO (TA100), 2.5 μg/mL (TA100) and 1.0 µg/mL (TA98) of 2-AA), the cultures were diluted in pH indicator medium lacking histidine and incubated for two days. Cells that had undergone reversion to amino acid prototrophy grew into colonies and bacterial metabolism reduces the pH of the medium, changing the color of that well. The number of wells containing revertant colonies were counted and compared to a negative control (DMSO). The experiment was performed once in triplicate. The Ames MPF calculation sheet provided by Xenometrix (Allschwil, Switzerland) was used for data analysis. Fold induction over the baseline was taken as the ratio of the mean number of positive wells for the dose concentration divided by the baseline. The baseline was obtained by adding one standard deviation to the mean number of positive wells of the solvent control. Compounds inducing revertant numbers above the baseline were characterized as substances with mutagenic potential.
4.4. Micronuclei Assay
The In Vitro Micronuclei Plus assay (Becton Dickinson, Heidelberg, Germany) was performed as described by the supplier. The assay was performed with HepG2 cell line since Valentin-Severin et al. identified this cell line as a suitable tool to study genotoxicity [
44]. Briefly, 2.0 × 10
4 HepG2 cells were seeded for 24 h and incubated with increasing concentrations of silvestrol and zotatifin for 4 h. After a washing step with PBS, fresh medium (without test substance) was added and cells were incubated for 44 h. The assay was stopped by placing the plate on ice for 20 min and the medium was carefully removed, taking care not to disturb the cell surface. Further, 50 µL of Complete Nucleic Acid Dye A Solution was added and the plate was placed on ice and exposed to visible light for 30 min. The Nucleic Acid Dye A solution was removed and the cells were washed with 0.15 mL of cold 1x Buffer Solution. Then, 100 µL Complete Lysis Solution 1 was added, followed by gently mixing the plate for 5 s and incubation of 1 h in the dark at 37 °C. After this, 100 µL Complete Lysis Solution 2 was added and incubated for 30 min in the dark at room temperature. The cells were analyzed by flow cytometry with the MACSQuant 10 (Miltenyi GmbH, Bergisch Gladbach, Germany).
4.5. Analysis of Intracellular Ca2+ Levels
Next, 50,000 HEK293T cells were seeded in a 96-well poly-D-lysine-coated plate and incubated at 37 °C for 24 h. Cells were incubated with 4 µM Fluo-8-AM in HBSS for 1 h at 37 °C. After 1 h, the Fluo-8/HBSS was replaced by 100 µL HBSS. Five images per second were taken using an ImageXpress Micro Confocal High Content Imaging System (Molecular Device, San Jose, CA, USA). Silvestrol (10 nM, 50 nM, 100 nM), 5 µM ionomycin (Sigma Aldrich; positive control) or DMSO (negative control) was added to the cells. An image was taken every second for the next 20 s. The MetaXpress Software Version 6.7.1.157 (San Jose, CA, USA) was used to analyze the data. A threshold of fluorescence intensity was defined using cells before treatment and all cells with a fluorescence signal above the threshold level were counted and related to all cells.
4.6. Analysis of cAMP Synthesis
The cAMP assay was performed as recommended by the supplier. HEK293T cells were transfected with pGloSensor-22F cAMP plasmid (Promega, Walldorf, Germany) using the turbofect reagent (Thermo Fisher Scientific, Dreieich, Germany). As such, 8.0 × 104 of transfected cells were seeded on a 96-well poly-d-lysine plate and incubated for 24 h at 37 °C. Then, 100 µL DMEM supplemented with 2% pGlo Sensor cAMP Reagent Solution (Promega, Walldorf, Germany) was added. Background luminescence (relative light units) before addition of compounds was determined with the EnSpire plate reader (PerkinElmer, Waltham, MA, USA). To investigate whether silvestrol induces cAMP synthesis, silvestrol (0 nM, 10 nM, 50 nM, 100 nM) or 5 µM forskolin (positive control) was added and the luminescence was detected.
4.7. Stability Assay
To 0.5 mg/mL human liver microsomes (50 donor pool; GIBCO) diluted in 0.1 M potassium phosphate buffer (0.1 M K
2HPO
4, 0.1 M KH
2PO
4, ph 7.4), 100 nM silvestrol or 10 µM celecoxib was added. The reaction was started with 1 mM NADPH diluted in potassium phosphate buffer. As control, samples without NAPDH were used. After various time points (0 h, 1 h, 2 h, 4 h), the reaction was stopped by adding ice-cold acetonitrile. The samples were centrifuged (5 min, 4 °C, 10,000×
g) and supernatant was stored at −80 °C until the remaining drug concentration was determined by LC-MS/MS as described in detail below. The concentrations of the drugs were plotted against time and the elimination constant (k) was calculated from the slope (m):
Equation to determine half-life:
Equation to determine intrinsic clearance:
4.8. Cell Barrier Model
The cell barrier model was set up as described previously [
43]. Briefly, 2.0 × 10
4 Caco-2 or 5.0 × 10
4 Calu-3 cells in 300 µL medium were seeded on 24-well ThinCerts (Greiner Bio-One GmbH, Frickenhausen, Germany) (pre-coated with FCS for 30 min) and in the lower compartment, 1 mL culture medium per well was added. The Caco-2 cell barrier was generated over 20 days and the Calu-3 barrier was generated over 14 days. To the apical compartment, silvestrol (0 nM, 50 nM, 100 nM) diluted in medium was added. Further, 10 mM of EGTA served as control. The transepithelial electrical resistance (TEER) was documented for 24 h every 30 min. To calculate the relative TEER % value, TEER values at the time point at which the compound solution (t
0 h) was added were related to the TEER value obtained after 20 h. For the transport assay, the Caco-2 and Calu-3 cell barrier was generated as mentioned above. The compounds carbamazepine and famotidine were used as high- and low-permeability controls according to FDA guidelines for bioavailability (FDA, 2017). Silvestrol (0 nM, 50 nM, 100 nM), 200 µM famotidine or 200 µM carbamazepine diluted in medium was added to the apical compartment and incubated for 4 h or 24 h. The basolateral and apical medium was collected and stored at −20 °C and analyzed by LC-MS/MS as detailed below.
4.9. Cellular Uptake Assay for Silvestrol and Celecoxib
The cellular uptake assay protocol of Bosnar et al. [
45] was modified. Briefly, 2.0 × 10
5 Caco-2 or Calu-3 cells per well were seeded in 24-well plates and incubated for 24 h. Cells were washed with 500 µL PBS, silvestrol (5 nM, 25 nM), celecoxib (25 µM) or vehicle diluted in medium, with 1 µM cyclosporine or without, were added and incubated for various time points (2 h, 4 h, 24 h). After incubation, medium was removed and cells were washed four times with 500 µL ice-cold PBS. Cells were harvested, centrifuged (300×
g, 5 min, 4 °C) and medium was removed. Protein concentration was determined by Bradford assay (Sigma Aldrich, Schnelldorf, Germany). Silvestrol and celecoxib concentration in cells was determined by LC-MS/MS analysis as described below.
4.10. Determination of P-glycoprotein Abundance
Caco-2 cells, Caki-2 cells, HepG2 cells, Calu-3 cells and HEK293T cells were harvested and lysed in RIPA-buffer (25 mM Tris-HCl (pH7.6), 1% Sodium deoxycholate, 0.1% SDS, 1% IPEGAL, 150 mM NaCl, Roche cOmplete™ Mini tablets (Sigma Aldrich, Schnelldorf, Germany)). A bicinchoninic acid assay (Thermo Fisher Scientific, Schwerte, Germany) was used to assess protein concentrations.
Moreover, 100 µg (Caco-2 cells, Caki-2 cells, HepG2 cells, Calu-3 cells, HEK293T cells) of total protein extract was prepared for Western blot analysis without denaturation by heating, then separated electrophoretically by 8% SDS-PAGE and electroblotted onto nitrocellulose membranes (Amersham Life Science, Freiburg, Germany). EveryBlot Blocking Buffer (Bio-Rad Laboratories, Feldkirchen, Germany) was used to block membranes. All antibodies were diluted in TBS with 1% BSA and 0.1% Tween 20. Membranes were incubated with the respective P-glycoprotein (1:1000, overnight at 4 °C) and GAPDH (1:5000, 2 h at room temperature) primary antibodies, washed three times with 0.05% Tween 20 in TBS, incubated with an anti-rabbit AF488 or an anti-mouse AF546 antibody (1:10,000 each) in TBS with 1% BSA and 0.1% Tween 20 for 60 min and washed again three times with 0.05% Tween 20 in TBS. The ChemiDoTM MP Imaging System from Bio-Rad Laboratories (Feldkirchen, Germany) was used to detect the fluorescence signals. The rabbit monoclonal anti-P-glycoprotein antibody and the mouse monoclonal anti-GAPDH were purchased from Abcam (Berlin, Germany) and the anti-rabbit AF488 and anti-mouse AF546 antibodies from Thermo Scientific (Schwerte, Germany).
4.11. Determination of Silvestrol, Carbamazepine, Famotidine and Celecoxib via LC-MS/MS
The concentrations of carbamazepine, silvestrol, famotidine and celecoxib were determined using three different LC-MS methods. Carbamazepine and silvestrol were determined using the same LC-MS method but with different sample pre-treatment protocols.
For the analysis of carbamazepine, 50 µL medium was mixed with 20 µL of the internal standard (IS) solution (carbamazepine-d10, 5 ng/mL in methanol) and 320 µL methanol. For calibration standards and quality control samples, 50 µL blank medium was spiked with 20 µL of standard working solutions and processed like the samples. After vortexing and centrifugation, 100 µL of the liquid phase was transferred to glass vials.
For the analysis of silvestrol, 50 µL medium was mixed with 20 µL of the IS solution (carbamazepine-d10, 5 ng/mL in methanol) and 250 µL methanol. For calibration standards and quality control samples, 50 µL blank supernatant was spiked with 50 µL of standard working solutions and processed like the samples. After vortexing and centrifugation, the liquid phase was removed and evaporated at a temperature of 45 °C under a gentle stream of nitrogen. The residues were reconstituted with 50 μL of methanol.
The LC-MS/MS analysis of silvestrol and carbamazepine was carried out using an Agilent 1290 Infinity LC system (Agilent, Waldbronn, Germany) coupled to a hybrid triple quadrupole linear ion trap mass spectrometer QTRAP 6500+ (Sciex, Darmstadt, Germany) equipped with a Turbo-V-source operating in positive electrospray ionization mode. The chromatographic separation was carried out using a Zorbax C8 Eclipse Plus RRHD column (50 × 2.1 mm, 1.8 µm particle size and 95 Å pore size; Agilent, Santa Clara, CA, USA), maintained at 40 °C (
Figure S4). A gradient program was employed at a flow rate of 300 µL. Mobile phase A was 0.1% formic acid + 10 mM ammonium formate and mobile phase B was acetonitrile + 0.0025% formic acid.
For the analysis of famotidine, 40 µL sample was spiked with 20 µL methanol, 20 µL IS (famotidine-d4, 100 ng/mL in methanol), 280 µL H2O and 40 µL methanol. For calibration standards and quality control samples, 40 µL blank medium was spiked with 20 µL standard working solution, 20 µL IS, 280 µL H2O and 40 µL methanol. After mixing and centrifugation, the supernatant was transferred to glass vials. The analysis was performed with an Agilent 1200 Infinity LC system (Agilent, Waldbronn, Germany) coupled to a hybrid triple quadrupole linear ion trap mass spectrometer QTRAP 5500 (Sciex, Darmstadt, Germany) equipped with a Turbo-V-source operating in positive electrospray ionization mode. The autosampler was a CTC PAL (CTC Analytics AG, Zwingen, Switzerland). The chromatographic separation was carried out using a Synergi Hydro RP coloumn (150 × 2 mm, 4 µm; Phenomenex, Aschaffenburg, Deutschland), maintained at 50 °C. Mobile Phase A was 10 mM NH4Ac and mobile Phase B was acetonitrile.
For the analysis of celecoxib, 20 µL sample was spiked with 20 µL IS (celecoxib-d7, 50 ng/mL in ethanol) and 140 µL ethanol. For calibration curve and quality control samples, 20 µL standard working solution was spiked with 20 µL IS, 20 µL PBS and 120 µL ethanol. After mixing and centrifugation, the supernatant was transferred to glass vials. The analysis of celecoxib was performed with an Agilent 1200 Infinity LC system (Agilent, Waldbronn, Germany) coupled to a hybrid triple quadrupole linear ion trap mass spectrometer QTRAP 5500 (Sciex, Darmstadt, Germany) equipped with a Turbo-V-source operating in negative electrospray ionization mode. The autosampler was a CTC PAL (CTC Analytics AG, Zwingen, Switzerland). The chromatographic separation was carried out using a Synergi Max RP coloumn (150 × 2 mm, 4 µm; Phenomenex, Aschaffenburg, Germany), maintained at 35 °C. Mobile Phase A was water and mobile Phase B was acetonitrile. The LC method was an isocratic method, which started at 80% B for 6 min at 300 µL/min. Further, 10 µL sample was injected.
For analysis and quantification of all compounds, Analyst Software 1.7.1 and Multiquant Software 3.0.3 (both Sciex, Darmstadt, Germany) were used. The precursor-to-product ion transitions used for quantification were: m/z 237.1 → m/z 194.1 for carbamazepine, m/z 672.3 → m/z 535.1 for silvestrol, m/z 338.1 → m/z 259.1 for famotidine and m/z 380.1 → m/z 315.9 for celecoxib. Calibration curves were constructed using linear regression with 1/x weighting. Variations in accuracy were less than 15% over the whole range of calibration, except for the lowest limit of quantification, where a variation in accuracy of 20% was accepted.
4.12. Statistics
Results are presented as means ± standard errors (SEM). The data were analyzed with two-way or one-way analysis of variance (ANOVA) and with Dunnett’s or Sidak’s multiple comparisons test. For all calculations and creation of graphs, GraphPad Prism v8 was used and p < 0.05 was considered the threshold for significance. The concentration–response data were fitted to a 4-parameter logistic fit using GraphPad Prism v8 to yield the CC50 value.


 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}





