
Binding of Natural Inhibitors to Respiratory Complex I


Abstract
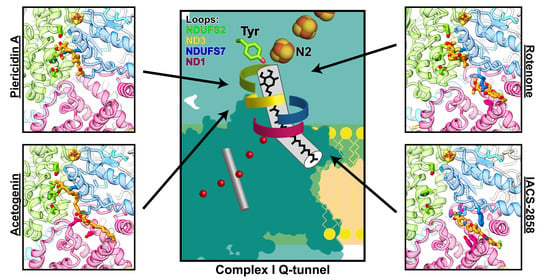
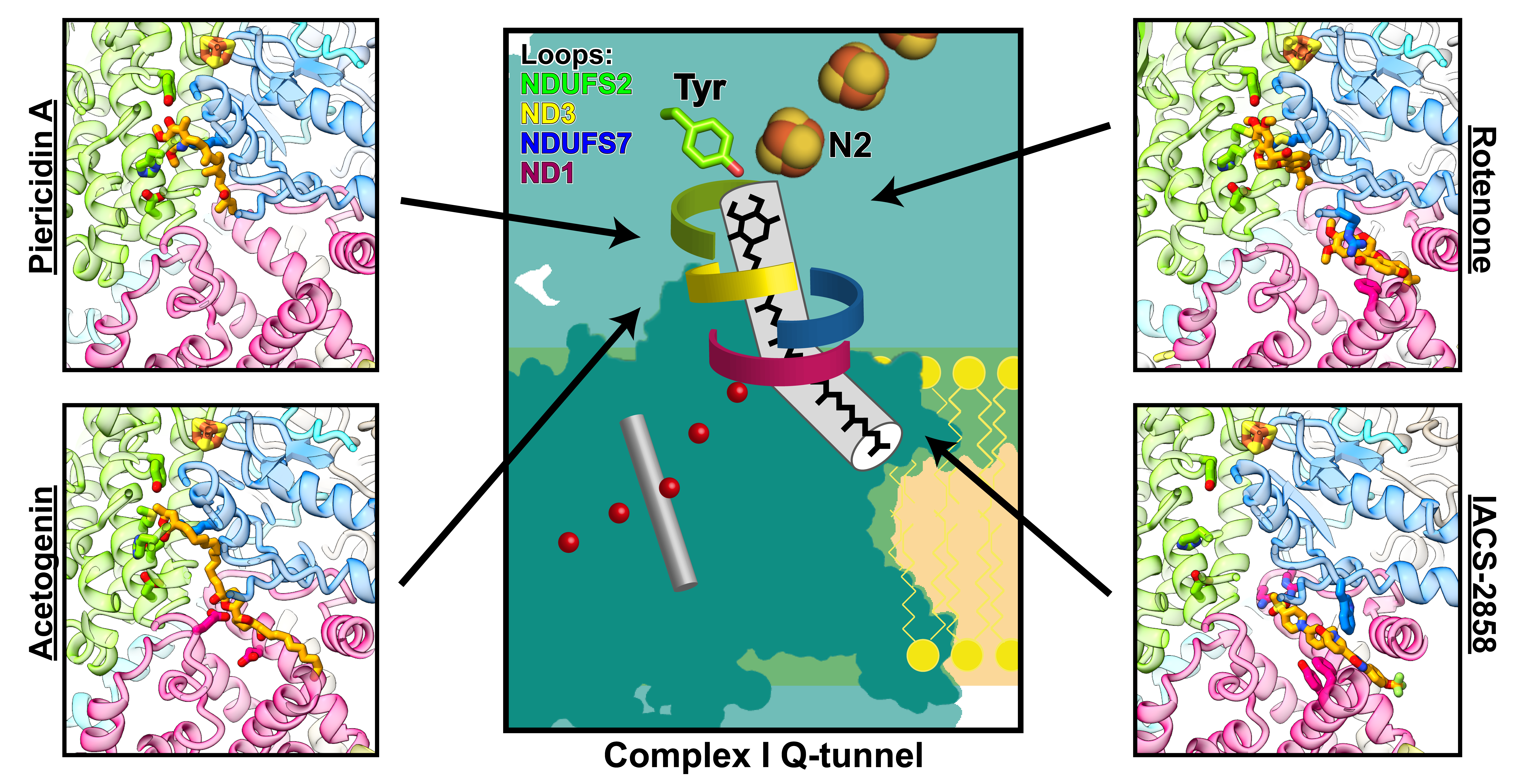
:
1. Introduction
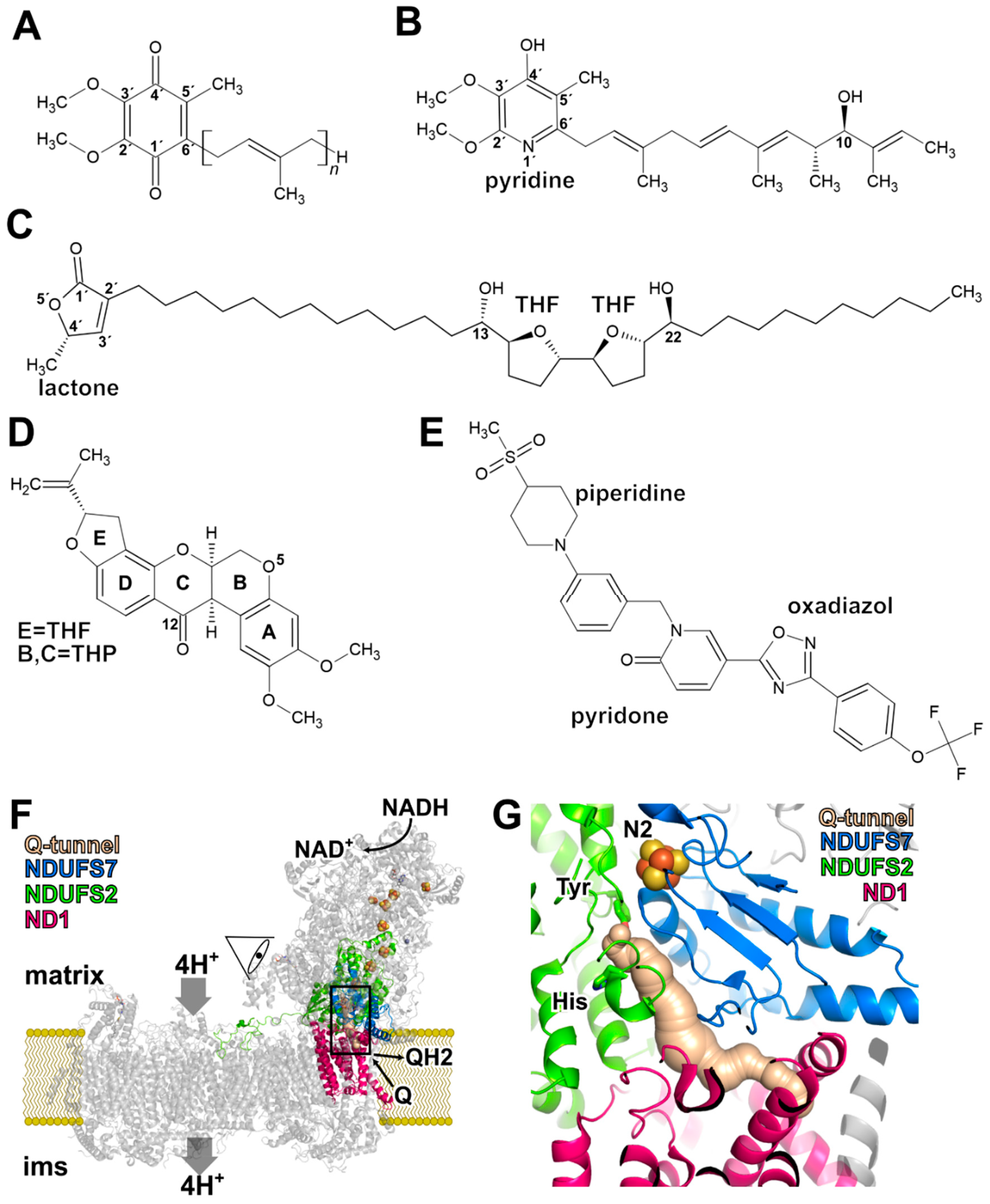
2. Rotenone
3. Piericidin A
4. Annonaceous Acetogenins
5. Binding of Q in the Active Site and in the Access Pathway Connecting It to the Membrane
6. Binding Sites of Natural Inhibitors
7. Binding of a Synthetic Anti-Cancer Compound
8. Conclusions
9. Materials and Methods
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hirst, J. Mitochondrial complex I. Annu. Rev. Biochem. 2013, 82, 551–575. [Google Scholar] [CrossRef] [PubMed]
- Galemou Yoga, E.; Angerer, H.; Parey, K.; Zickermann, V. Respiratory complex I—Mechanistic insights and advances in structure determination. Biochim. Biophys. Acta 2020, 1861, 148153. [Google Scholar] [CrossRef] [PubMed]
- Kampjut, D.; Sazanov, L.A. Structure of respiratory complex I—An emerging blueprint for the mechanism. Curr. Opin. Struct. Biol. 2022, 74, 102350. [Google Scholar] [CrossRef] [PubMed]
- Rodenburg, R.J. Mitochondrial complex I-linked disease. Biochim. Biophys. Acta 2016, 1857, 938–945. [Google Scholar] [CrossRef]
- Hock, D.H.; Robinson, D.R.L.; Stroud, D.A. Blackout in the powerhouse: Clinical phenotypes associated with defects in the assembly of OXPHOS complexes and the mitoribosome. Biochem. J. 2020, 477, 4085–4132. [Google Scholar] [CrossRef]
- Fiedorczuk, K.; Sazanov, L.A. Mammalian Mitochondrial Complex I Structure and Disease-Causing Mutations. Trends Cell Biol. 2018, 28, 835–867. [Google Scholar] [CrossRef]
- Abramov, A.Y.; Angelova, P.R. Cellular mechanisms of complex I-associated pathology. Biochem. Soc. Trans. 2019, 47, 1963–1969. [Google Scholar] [CrossRef]
- Chouchani, E.T.; Pell, V.R.; Gaude, E.; Aksentijevic, D.; Sundier, S.Y.; Robb, E.L.; Logan, A.; Nadtochiy, S.M.; Ord, E.N.J.; Smith, A.C.; et al. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature 2014, 515, 431–435. [Google Scholar] [CrossRef]
- Parey, K.; Wirth, C.; Vonck, J.; Zickermann, V. Respiratory complex I—Structure, mechanism and evolution. Curr. Opin. Struct. Biol. 2020, 63, 1–9. [Google Scholar] [CrossRef]
- Baradaran, R.; Berrisford, J.M.; Minhas, G.S.; Sazanov, L.A. Crystal structure of the entire respiratory complex I. Nature 2013, 494, 443–448. [Google Scholar] [CrossRef] [Green Version]
- Fedor, J.G.; Jones, A.J.Y.; Di Luca, A.; Kaila, V.R.I.; Hirst, J. Correlating kinetic and structural data on ubiquinone binding and reduction by respiratory complex I. Proc. Natl. Acad. Sci. USA 2017, 114, 12737–12742. [Google Scholar] [CrossRef] [PubMed]
- Chung, I.; Wright, J.J.; Bridges, H.R.; Ivanov, B.S.; Biner, O.; Pereira, C.S.; Arantes, G.M.; Hirst, J. Cryo-EM structures define ubiquinone-10 binding to mitochondrial complex I and conformational transitions accompanying Q-site occupancy. Nat. Commun. 2022, 13, 2758. [Google Scholar] [CrossRef]
- Warnau, J.; Sharma, V.; Gamiz-Hernandez, A.P.; Di Luca, A.; Haapanen, O.; Vattulainen, I.; Wikstrom, M.; Hummer, G.; Kaila, V.R.I. Redox-coupled quinone dynamics in the respiratory complex I. Proc. Natl. Acad. Sci. USA 2018, 115, E8413–E8420. [Google Scholar] [CrossRef] [PubMed]
- Haapanen, O.; Reidelbach, M.; Sharma, V. Coupling of quinone dynamics to proton pumping in respiratory complex I. Biochim. Biophys. Acta 2020, 1861, 148287. [Google Scholar] [CrossRef] [PubMed]
- Hoias Teixeira, M.; Menegon Arantes, G. Balanced internal hydration discriminates substrate binding to respiratory complex I. Biochim. Biophys. Acta 2019, 1860, 541–548. [Google Scholar] [CrossRef]
- Tocilescu, M.A.; Zickermann, V.; Zwicker, K.; Brandt, U. Quinone binding and reduction by respiratory complex I. Biochim. Biophys. Acta 2010, 1797, 1883–1890. [Google Scholar] [CrossRef]
- Gutierrez-Fernandez, J.; Kaszuba, K.; Minhas, G.S.; Baradaran, R.; Tambalo, M.; Gallagher, D.T.; Sazanov, L.A. Key role of quinone in the mechanism of respiratory complex I. Nat. Commun. 2020, 11, 4135. [Google Scholar] [CrossRef]
- Kampjut, D.; Sazanov, L.A. The coupling mechanism of mammalian respiratory complex I. Science 2020, 370, eabc4209. [Google Scholar] [CrossRef]
- Parey, K.; Haapanen, O.; Sharma, V.; Kofeler, H.; Zullig, T.; Prinz, S.; Siegmund, K.; Wittig, I.; Mills, D.J.; Vonck, J.; et al. High-resolution cryo-EM structures of respiratory complex I: Mechanism, assembly, and disease. Sci. Adv. 2019, 5, eaax9484. [Google Scholar] [CrossRef]
- Parey, K.; Lasham, J.; Mills, D.J.; Djurabekova, A.; Haapanen, O.; Yoga, E.G.; Xie, H.; Kühlbrandt, W.; Sharma, V.; Vonck, J.; et al. High-resolution structure and dynamics of mitochondrial complex I-Insights into the proton pumping mechanism. Sci. Adv. 2021, 7, eabj3221. [Google Scholar] [CrossRef]
- Gu, J.; Liu, T.; Guo, R.; Zhang, L.; Yang, M. The coupling mechanism of mammalian mitochondrial complex I. Nat. Struct. Mol. Biol. 2022, 29, 172–182. [Google Scholar] [CrossRef]
- Parey, K.; Brandt, U.; Xie, H.; Mills, D.J.; Siegmund, K.; Vonck, J.; Kühlbrandt, W.; Zickermann, V. Cryo-EM structure of respiratory complex I at work. Elife 2018, 7, e39213. [Google Scholar] [CrossRef]
- Galemou Yoga, E.; Schiller, J.; Zickermann, V. Ubiquinone Binding and Reduction by Complex I-Open Questions and Mechanistic Implications. Front. Chem. 2021, 9, 672851. [Google Scholar] [CrossRef]
- Murai, M.; Miyoshi, H. Current topics on inhibitors of respiratory complex I. Biochim. Biophys. Acta 2016, 1857, 884–891. [Google Scholar] [CrossRef]
- Degli Esposti, M. Inhibitors of NADH-ubiquinone reductase: An overview. Biochim. Biophys. Acta 1998, 1364, 222–235. [Google Scholar] [CrossRef]
- Degli Esposti, M.; Ghelli, A.; Ratta, M.; Cortes, D.; Estornell, E. Natural substances (acetogenins) from the family Annonaceae are powerful inhibitors of mitochondrial NADH dehydrogenase (Complex I). Biochem. J. 1994, 301 Pt 1, 161–167. [Google Scholar] [CrossRef]
- Moghadamtousi, S.Z.; Fadaeinasab, M.; Nikzad, S.; Mohan, G.; Ali, H.M.; Kadir, H.A. Annona muricata (Annonaceae): A Review of Its Traditional Uses, Isolated Acetogenins and Biological Activities. Int. J. Mol. Sci. 2015, 16, 15625–15658. [Google Scholar] [CrossRef]
- Radad, K.; Al-Shraim, M.; Al-Emam, A.; Wang, F.; Kranner, B.; Rausch, W.D.; Moldzio, R. Rotenone: From modelling to implication in Parkinson’s disease. Folia Neuropathol. 2019, 57, 317–326. [Google Scholar] [CrossRef]
- Lümmen, P. Complex I inhibitors as insecticides and acaricides. Biochim. Biophys. Acta 1998, 1364, 287–296. [Google Scholar] [CrossRef]
- Langston, J.W. The MPTP Story. J. Parkinsons Dis. 2017, 7, S11–S19. [Google Scholar] [CrossRef] [Green Version]
- Johnson, M.E.; Bobrovskaya, L. An update on the rotenone models of Parkinson’s disease: Their ability to reproduce the features of clinical disease and model gene-environment interactions. Neurotoxicology 2015, 46, 101–116. [Google Scholar] [CrossRef]
- Owen, M.R.; Doran, E.; Halestrap, A.P. Evidence that metformin exerts its anti-diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochem. J. 2000, 348 Pt 3, 607–614. [Google Scholar] [CrossRef]
- Bridges, H.R.; Jones, A.J.; Pollak, M.N.; Hirst, J. Effects of metformin and other biguanides on oxidative phosphorylation in mitochondria. Biochem. J. 2014, 462, 475–487. [Google Scholar] [CrossRef]
- Baccelli, I.; Gareau, Y.; Lehnertz, B.; Gingras, S.; Spinella, J.F.; Corneau, S.; Mayotte, N.; Girard, S.; Frechette, M.; Blouin-Chagnon, V.; et al. Mubritinib Targets the Electron Transport Chain Complex I and Reveals the Landscape of OXPHOS Dependency in Acute Myeloid Leukemia. Cancer Cell 2019, 36, 84–99.e88. [Google Scholar] [CrossRef]
- Urra, F.A.; Munoz, F.; Lovy, A.; Cardenas, C. The Mitochondrial Complex(I)ty of Cancer. Front. Oncol. 2017, 7, 118. [Google Scholar] [CrossRef]
- Ellinghaus, P.; Heisler, I.; Unterschemmann, K.; Haerter, M.; Beck, H.; Greschat, S.; Ehrmann, A.; Summer, H.; Flamme, I.; Oehme, F.; et al. BAY 87-2243, a highly potent and selective inhibitor of hypoxia-induced gene activation has antitumor activities by inhibition of mitochondrial complex I. Cancer Med. 2013, 2, 611–624. [Google Scholar] [CrossRef]
- Stephenson, Z.A.; Harvey, R.F.; Pryde, K.R.; Mistry, S.; Hardy, R.E.; Serreli, R.; Chung, I.; Allen, T.E.; Stoneley, M.; MacFarlane, M.; et al. Identification of a novel toxicophore in anti-cancer chemotherapeutics that targets mitochondrial respiratory complex I. Elife 2020, 9, e55845. [Google Scholar] [CrossRef]
- Chouchani, E.T.; Methner, C.; Nadtochiy, S.M.; Logan, A.; Pell, V.R.; Ding, S.; James, A.M.; Cocheme, H.M.; Reinhold, J.; Lilley, K.S.; et al. Cardioprotection by S-nitrosation of a cysteine switch on mitochondrial complex I. Nat. Med. 2013, 19, 753–759. [Google Scholar] [CrossRef]
- Chouchani, E.T.; Pell, V.R.; James, A.M.; Work, L.M.; Saeb-Parsy, K.; Frezza, C.; Krieg, T.; Murphy, M.P. A Unifying Mechanism for Mitochondrial Superoxide Production during Ischemia-Reperfusion Injury. Cell Metab. 2016, 23, 254–263. [Google Scholar] [CrossRef]
- Brand, M.D.; Goncalves, R.L.; Orr, A.L.; Vargas, L.; Gerencser, A.A.; Borch Jensen, M.; Wang, Y.T.; Melov, S.; Turk, C.N.; Matzen, J.T.; et al. Suppressors of Superoxide-H2O2 Production at Site IQ of Mitochondrial Complex I Protect against Stem Cell Hyperplasia and Ischemia-Reperfusion Injury. Cell Metab. 2016, 24, 582–592. [Google Scholar] [CrossRef] [Green Version]
- Murphy, M.P.; Hartley, R.C. Mitochondria as a therapeutic target for common pathologies. Nat. Rev. Drug Discov. 2018, 17, 865–886. [Google Scholar] [CrossRef]
- Zickermann, V.; Wirth, C.; Nasiri, H.; Siegmund, K.; Schwalbe, H.; Hunte, C.; Brandt, U. Structural biology. Mechanistic insight from the crystal structure of mitochondrial complex I. Science 2015, 347, 44–49. [Google Scholar] [CrossRef]
- Bridges, H.R.; Fedor, J.G.; Blaza, J.N.; Di Luca, A.; Jussupow, A.; Jarman, O.D.; Wright, J.J.; Agip, A.A.; Gamiz-Hernandez, A.P.; Roessler, M.M.; et al. Structure of inhibitor-bound mammalian complex I. Nat. Commun. 2020, 11, 5261. [Google Scholar] [CrossRef]
- Grba, D.N.; Blaza, J.N.; Bridges, H.R.; Agip, A.A.; Yin, Z.; Murai, M.; Miyoshi, H.; Hirst, J. Cryo-electron microscopy reveals how acetogenins inhibit mitochondrial respiratory complex I. J. Biol. Chem. 2022, 298, 101602. [Google Scholar] [CrossRef]
- Chung, I.; Serreli, R.; Cross, J.B.; Di Francesco, M.E.; Marszalek, J.R.; Hirst, J. Cork-in-bottle mechanism of inhibitor binding to mammalian complex I. Sci. Adv. 2021, 7, eabg4000. [Google Scholar] [CrossRef]
- Laforge, F.B.; Haller, H.L.; Smith, L.E. The determination of the structure of rotenone. Chem. Rev. 1933, 12, 181–212. [Google Scholar] [CrossRef]
- Lindahl, P.E.; Oberg, K.E. The effect of rotenone on respiration and its point of attack. Exp. Cell Res. 1961, 23, 228–237. [Google Scholar] [CrossRef]
- Burgos, J.; Redfearn, E.R. The inhibition of mitochondrial reduced nicotinamide-adenine dinucleotide oxidation by rotenoids. Biochim. Biophys. Acta 1965, 110, 475–483. [Google Scholar] [CrossRef]
- Betarbet, R.; Sherer, T.B.; MacKenzie, G.; Garcia-Osuna, M.; Panov, A.V.; Greenamyre, J.T. Chronic systemic pesticide exposure reproduces features of Parkinson’s disease. Nat. Neurosci. 2000, 3, 1301–1306. [Google Scholar] [CrossRef]
- Brinkley, B.R.; Barham, S.S.; Barranco, S.C.; Fuller, G.M. Rotenone inhibition of spindle microtubule assembly in mammalian cells. Exp. Cell Res. 1974, 85, 41–46. [Google Scholar] [CrossRef]
- Bisbal, M.; Sanchez, M. Neurotoxicity of the pesticide rotenone on neuronal polarization: A mechanistic approach. Neural. Regen. Res. 2019, 14, 762–766. [Google Scholar] [CrossRef]
- Choi, W.S.; Palmiter, R.D.; Xia, Z. Loss of mitochondrial complex I activity potentiates dopamine neuron death induced by microtubule dysfunction in a Parkinson’s disease model. J. Cell Biol. 2011, 192, 873–882. [Google Scholar] [CrossRef]
- Gao, H.M.; Liu, B.; Hong, J.S. Critical role for microglial NADPH oxidase in rotenone-induced degeneration of dopaminergic neurons. J. Neurosci. 2003, 23, 6181–6187. [Google Scholar] [CrossRef]
- Zhou, X.; Fenical, W. The unique chemistry and biology of the piericidins. J. Antibiot. 2016, 69, 582–593. [Google Scholar] [CrossRef]
- Kroiss, J.; Kaltenpoth, M.; Schneider, B.; Schwinger, M.G.; Hertweck, C.; Maddula, R.K.; Strohm, E.; Svatos, A. Symbiotic streptomycetes provide antibiotic combination prophylaxis for wasp offspring. Nat. Chem. Biol. 2010, 6, 261–263. [Google Scholar] [CrossRef]
- Tamura, S.; Mori, R.; Miyamoto, S.; Takahashi, N.; Suzuki, S.; Nagatsu, J. Isolation and Physiological Activities of Piericidin a, a Natural Insecticide Produced by Streptomyces. Agric. Biol. Chem. Tokyo 1963, 27, 576–582. [Google Scholar] [CrossRef]
- Hall, C.; Wu, M.; Crane, F.L.; Takahashi, H.; Tamura, S.; Folkers, K. Piericidin a—A New Inhibitor of Mitochondrial Electron Transport. Biochem. Biophys. Res. Commun. 1966, 25, 373–377. [Google Scholar] [CrossRef]
- Gutman, M.; Singer, T.P.; Casida, J.E. Studies on the respiratory chain-linked reduced nicotinamide adenine dinucleotide dehydrogenase. XVII. Reaction sites of piericidin A and rotenone. J. Biol. Chem. 1970, 245, 1992–1997. [Google Scholar] [CrossRef]
- Hollerhage, M.; Deck, R.; De Andrade, A.; Respondek, G.; Xu, H.; Rosler, T.W.; Salama, M.; Carlsson, T.; Yamada, E.S.; Gad El Hak, S.A.; et al. Piericidin A aggravates Tau pathology in P301S transgenic mice. PLoS ONE 2014, 9, e113557. [Google Scholar] [CrossRef]
- Londershausen, M.; Leicht, W.; Lieb, F.; Moeschler, H.; Weiss, H. Molecular Mode of Action of Annonins. Pestic. Sci. 1991, 33, 427–438. [Google Scholar] [CrossRef]
- McLaughlin, J.L. Paw paw and cancer: Annonaceous acetogenins from discovery to commercial products. J. Nat. Prod. 2008, 71, 1311–1321. [Google Scholar] [CrossRef]
- Jacobo-Herrera, N.; Perez-Plasencia, C.; Castro-Torres, V.A.; Martinez-Vazquez, M.; Gonzalez-Esquinca, A.R.; Zentella-Dehesa, A. Selective Acetogenins and Their Potential as Anticancer Agents. Front. Pharmacol. 2019, 10, 783. [Google Scholar] [CrossRef]
- Hollerhage, M.; Matusch, A.; Champy, P.; Lombes, A.; Ruberg, M.; Oertel, W.H.; Hoglinger, G.U. Natural lipophilic inhibitors of mitochondrial complex I are candidate toxins for sporadic neurodegenerative tau pathologies. Exp. Neurol. 2009, 220, 133–142. [Google Scholar] [CrossRef]
- Lannuzel, A.; Hoglinger, G.U.; Verhaeghe, S.; Gire, L.; Belson, S.; Escobar-Khondiker, M.; Poullain, P.; Oertel, W.H.; Hirsch, E.C.; Dubois, B.; et al. Atypical parkinsonism in Guadeloupe: A common risk factor for two closely related phenotypes? Brain 2007, 130, 816–827. [Google Scholar] [CrossRef]
- Darrouzet, E.; Issartel, J.P.; Lunardi, J.; Dupuis, A. The 49-kDa subunit of NADH-ubiquinone oxidoreductase (Complex I) is involved in the binding of piericidin and rotenone, two quinone-related inhibitors. FEBS Lett. 1998, 431, 34–38. [Google Scholar] [CrossRef]
- Masuya, T.; Murai, M.; Ifuku, K.; Morisaka, H.; Miyoshi, H. Site-specific chemical labeling of mitochondrial respiratory complex I through ligand-directed tosylate chemistry. Biochemistry 2014, 53, 2307–2317. [Google Scholar] [CrossRef]
- Sharma, V.; Belevich, G.; Gamiz-Hernandez, A.P.; Rog, T.; Vattulainen, I.; Verkhovskaya, M.L.; Wikstrom, M.; Hummer, G.; Kaila, V.R. Redox-induced activation of the proton pump in the respiratory complex I. Proc. Natl. Acad. Sci. USA 2015, 112, 11571–11576. [Google Scholar] [CrossRef]
- Soufari, H.; Parrot, C.; Kuhn, L.; Waltz, F.; Hashem, Y. Specific features and assembly of the plant mitochondrial complex I revealed by cryo-EM. Nat. Commun. 2020, 11, 5195. [Google Scholar] [CrossRef]
- Klusch, N.; Senkler, J.; Yildiz, O.; Kühlbrandt, W.; Braun, H.P. A ferredoxin bridge connects the two arms of plant mitochondrial complex I. Plant Cell 2021, 33, 2072–2091. [Google Scholar] [CrossRef]
- Galemou Yoga, E.; Haapanen, O.; Wittig, I.; Siegmund, K.; Sharma, V.; Zickermann, V. Mutations in a conserved loop in the PSST subunit of respiratory complex I affect ubiquinone binding and dynamics. Biochim. Biophys. Acta 2019, 1860, 573–581. [Google Scholar] [CrossRef]
- Gamiz-Hernandez, A.P.; Jussupow, A.; Johansson, M.P.; Kaila, V.R.I. Terminal Electron-Proton Transfer Dynamics in the Quinone Reduction of Respiratory Complex I. J. Am. Chem. Soc. 2017, 139, 16282–16288. [Google Scholar] [CrossRef]
- Wang, P.; Dhananjayan, N.; Hagras, M.A.; Stuchebrukhov, A.A. Respiratory complex I: Bottleneck at the entrance of quinone site requires conformational change for its opening. Biochim. Biophys. Acta 2021, 1862, 148326. [Google Scholar] [CrossRef]
- Angerer, H.; Nasiri, H.R.; Niedergesass, V.; Kerscher, S.; Schwalbe, H.; Brandt, U. Tracing the tail of ubiquinone in mitochondrial complex I. Biochim. Biophys. Acta 2012, 1817, 1776–1784. [Google Scholar] [CrossRef]
- Roberts, P.G.; Hirst, J. The deactive form of respiratory complex I from mammalian mitochondria is a Na+/H+ antiporter. J. Biol. Chem. 2012, 287, 34743–34751. [Google Scholar] [CrossRef]
- Schrödinger, L.; DeLano, W. Pymol. 2020. Available online: http://www.pymol.org/pymol (accessed on 11 July 2022).
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Meng, E.C.; Couch, G.S.; Croll, T.I.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Structure visualization for researchers, educators, and developers. Protein. Sci. 2021, 30, 70–82. [Google Scholar] [CrossRef]
- Chovancova, E.; Pavelka, A.; Benes, P.; Strnad, O.; Brezovsky, J.; Kozlikova, B.; Gora, A.; Sustr, V.; Klvana, M.; Medek, P.; et al. CAVER 3.0: A tool for the analysis of transport pathways in dynamic protein structures. PLoS Comput. Biol. 2012, 8, e1002708. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | M. musculus (Mm) | B. taurus (Bt) | O. aries (Oa) | Y.lipolytica (Yl) | T. thermophilus (Tt) | |
|---|---|---|---|---|---|---|
| PDB ID | 6ZTQ,7PSA,7B93 | 7QSK | 6ZKN | 6RFR,7O6Y | 6I0D | |
| NDUFS2 | His | His 59 | His 59 | His 59 | His95 | His38 |
| Tyr | Tyr108 | Tyr108 | Tyr108 | Tyr144 | Tyr87 | |
| Met/Val | Met152 | Met152 | Met152 | Met188 | Val131 | |
| Thr/Ser | Thr156 | Thr156 | Thr156 | Ser193 | Thr135 | |
| Asp | Asp160 | Asp160 | Asp160 | Asp196 | Asp139 | |
| Phe1 | Phe167 | Phe167 | Phe167 | Phe203 | Phe146 | |
| Phe2/Leu | Phe168 | Phe168 | Phe168 | Leu204 | Phe147 | |
| NDUFS7 | Trp/Pro | Trp56 | Trp46 | Trp46 | Trp77 | Pro38 |
| Met1 | Met69 | Met59 | Met59 | Met90 | Met50 | |
| Met2 | Met70 | Met60 | Met60 | Met91 | Met51 | |
| Val/Ile | Val75 | Val75 | Val75 | Ile106 | Val67 | |
| Phe | Phe76 | Phe76 | Phe76 | Phe107 | Phe68 | |
| Arg | Arg78 | Arg77 | Arg77 | Arg108 | Arg77 | |
| ND1 | Glu1 | Glu24 | Glu24 | Glu24 | Glu26 | Glu35 |
| Gln | Gln32 | Gln32 | Gln32 | Gln34 | Gln43 | |
| Arg1 | Arg34 | Arg34 | Arg34 | Arg36 | Arg45 | |
| Glu2 | Glu204 | Glu204 | Glu202 | Glu208 | Glu225 | |
| Phe/Gln | Phe224 | Phe224 | Phe224 | Phe228 | Gln245 | |
| Tyr | Tyr228 | Tyr228 | Tyr228 | Tyr232 | Tyr249 | |
| Arg2 | Arg274 | Arg274 | Arg274 | Arg297 | Arg294 | |
| ND4 | Arg | Arg142 | Arg142 | Arg142 | Arg162 | Arg143 |
| Lys | Lys206 | Lys206 | Lys206 | Lys221 | Lys204 | |
| Trp | Trp215 | Trp215 | Trp215 | Trp230 | Trp213 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schiller, J.; Zickermann, V. Binding of Natural Inhibitors to Respiratory Complex I. Pharmaceuticals 2022, 15, 1088. https://doi.org/10.3390/ph15091088
Schiller J, Zickermann V. Binding of Natural Inhibitors to Respiratory Complex I. Pharmaceuticals. 2022; 15(9):1088. https://doi.org/10.3390/ph15091088
Chicago/Turabian StyleSchiller, Jonathan, and Volker Zickermann. 2022. "Binding of Natural Inhibitors to Respiratory Complex I" Pharmaceuticals 15, no. 9: 1088. https://doi.org/10.3390/ph15091088
APA StyleSchiller, J., & Zickermann, V. (2022). Binding of Natural Inhibitors to Respiratory Complex I. Pharmaceuticals, 15(9), 1088. https://doi.org/10.3390/ph15091088