


Nitrogen Containing Heterocycles as Anticancer Agents: A Medicinal Chemistry Perspective

 , ,
, ,  , , , , , and
, , , , , and
Abstract
:1. Introduction
2. Nitrogen-Containing FDA-Approved Anti-Cancer Drugs

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| S. No. | Name | Structure | Mode of Action | Drug Target | Approval Year | References |
|---|---|---|---|---|---|---|
| 1. | Alectinib Hydrochloride |  | It prevents the multiplication of cancer cells by blocking the action of abnormal proteins. | Anaplastic lymphoma kinase (ALK) Inhibitor | 2015 | [24] |
| 2. | Brigatinib |  | It inhibits the ability of the phosphate group to bind ALK and inhibits the activity of proteins STAT3, A.K.T., ERK1/2, and S6 both in vitro and in vivo. | Anaplastic lymphoma kinase (ALK) Inhibitor | 2017 | [25] |
| 3. | Lorlatinib |  | Inhibits will disrupt the ALK and ROS1- mediated signaling, further inhibiting the growing tumor cells in ALK and ROS1 cells. | Anaplastic lymphoma kinase (ALK) Inhibitor | 2018 | [26] |
| 4. | Entrectinib |  | It interferes with the growth of cancer cells, which are eventually destroyed. | Anaplastic lymphoma kinase (ALK) Inhibitor | 2019 | [27] |
| 5. | Midostaurin |  | It Prevents the multiplication of cancer cells by blocking the action of abnormal proteins. | Fms-like tyrosine kinase (FLT3) inhibitor | 2017 | [28,29] |
| 6. | Gilteritinib fumarate |  | Blocks the action of naturally occurring substances that promote cancer cell growth. | Fms-like tyrosine kinase (FLT3) inhibitor | 2018 | [30] |
| 7. | Quizartinib |  | It inhibits cancer cell proliferation which leads to the death of the cells. | Fms-like tyrosine kinase (FLT3) inhibitor | 2018 | [31] |
| 8. | Pexidarinib |  | Inhibits the colony-stimulating factor (CSF1)/CSF1 receptor pathway | Fms-like tyrosine kinase (FLT3) inhibitor | 2019 | [32] |
| 9. | Osimertinib mesylate |  | The abnormal protein’s ability to cause cancer cells to grow is blocked, which may also help tumors get smaller and slow down the spread of cancer cells. | Epidermal growth factor (EGF) receptor inhibitors | 2018 | [33] |
| 10. | Olmutinib |  | Restricting the mutant form of E.G.F.R. causes the death of E.G.F.R. expressing tumor cells. | Epidermal growth factor (EGF) receptor inhibitors | 2015 | [34] |
| 11. | Neratinib maleate |  | The autophosphorylation is prevented on tyrosine residues receptor, and the Oncogenic signaling was reduced through mitogen-activated protein kinase and Akt pathways. | Epidermal growth factor (EGF) receptor inhibitors | 2017 | [35] |
| 12. | Dacomitinib |  | The E.G.F.R. activating mutations like exon 19 deletion or the exon 21 L858R substitution mutation and E.G.F.R. proteins like EGFR/HER1, HER2, and HER4 activities are blocked. | Epidermal growth factor (EGF) receptor inhibitors | 2018 | [36] |
| 13. | Pyrotinib maleate |  | Covalently binds to the intracellular kinase domain of HER1, HER2, and HER4 to block downstream signaling pathways, inhibits autophosphorylation, and prevents the creation of homologous/heterodimers of the HER family. | Epidermal growth factor (EGF) receptor inhibitors | 2018 | [37] |
| 14. | Almonertinib mesylate |  | It inhibits E.G.F.R. tyrosine kinase targeting EGFR-sensitizing and T790M resistance mutations. | Epidermal growth factor (EGF) receptor inhibitors | 2020 | [38] |
| 15. | Lenvatinib mesylate |  | The kinase activities of vascular endothelial growth factor (VEGF) receptors VEGFR1 (FLT1), VEGFR2 (K.D.R.), and VEGFR3 (FLT4) are inhibited. | Vascular endothelial growth factor (VEGF) inhibitors | 2015 | [39] |
| 16. | Tivozanib |  | The phosphorylation of V.E.G.F.R. (vascular endothelial growth factor) receptors (V.E.G.F.R.)-1, VEGFR-2, and VEGFR-3, as well as kinases including c-kit and platelet-derived growth factor beta (P.D.G.F.R.), are suppressed. | Vascular endothelial growth factor (VEGF) inhibitors | 2021 | [40] |
| 17. | Fruquintinib |  | It prevents the VEGF from causing the phosphorylation of V.E.G.F.R.s 1, 2, and 3, which may reduce the proliferation, migration, and survival of endothelial cells, the development of microvessels, and the proliferation and death of tumor cells. | Vascular endothelial growth factor (VEGF) inhibitors | 2020 | [41] |
| 18. | Anlotinib dihydrochloride |  | It inhibits the development of tumor cells by inhibiting the pathways like PI3K/AKT, RAS/MAPK, and PLCy/PKC by using the receptors: V.E.G.F.R., F.G.F.R., and P.D.G.F.R. | Vascular endothelial growth factor (VEGF) inhibitors | 2020 | [42] |
| 19. | Acalabrutinib |  | Bruton Tyrosine Kinase inhibitor that stops B cells from chemotactic, proliferating, moving, and adhesion of B cells | Tyrosine kinase inhibitor(tki) | 2017 | [43] |
| 20. | Zanubrutinib |  | Inhibits the growth and survival of cancerous B cells to shrink the size of the tumor in mantle cell lymphoma | Tyrosine kinase inhibitor(tki) | 2021 | [44] |
| 21. | Fedratinib |  | Inhibits cell division and induces apoptosis | Janus kinase 2 (JAK2) inhibitors | 2019 | [45] |
| 22. | Copanlisib |  | Apoptosis-induced tumor cell death and primary malignant B cell line growth suppression. | Phosphatidylinositol 3-hydroxy kinase inhibitors | 2014 | [46,47] |
| 23. | Mocetinostat |  | Acts by turning on tumor suppressor genes that have been inappropriately turned off | Hydroxamic acid-based hdac Inhibitors (hdaci) | 2014 | [48,49] |
| 24. | Duvelisib |  | Inhibit isoform gamma, essential for cytokine signaling and the pro-inflammatory response, and inhibit the isoform delta of PI3K(phosphoinositide3-kinase), which is required for cell proliferation and survival. | Phosphatidylinositol 3-hydroxy kinase inhibitors | 2018 | [50] |
| 25. | Oxaliplatin |  | Inhibits D.N.A. synthesis and transcription by binding preferentially to the guanine and cytosine moieties of D.N.A. | Platinum derivative | 2004 | [51,52] |
| 26. | Irinotecan |  | Binds to the topoisomerase I-DNA complex and prevents the D.N.A. strand from relegating, which then forms a ternary complex disrupting the replication fork and moving slowly, leading to breakage of lethal double-stranded DNA. | Topoisomerase I inhibitor | 2000 | [53,54] |
| 27. | Cytarabine |  | It prevents cancer cells from generating and repairing the D.N.A., which they require to thrive and proliferate. | Antineoplastic anti-metabolite | 1999 | [55,56] |
| 28. | Gemcitabine |  | Inhibits D.N.A. synthesis | Pyrimidine nucleoside antimetabolite | 2011 | [57,58] |
| 29. | Trabectedin |  | Inhibition of trans-activated transcription and the interaction with D.N.A. repair proteins | Alkylating agent | 2015 | [59,60] |
| 30. | Kahalalide F |  | Induces oncosis | Lysosomes | 2004 | [61,62] |
| 31. | Salinosporamide A |  | Covalently modifies the threonine residues in the 20S proteasome’s active site to decrease proteasome activity. | Inhibitor of the 20S proteasomes | 2004 | [63] |
| 32. | Osimertinib |  | Binds to specific E.G.F.R. mutants (T790M, L858R, and exon 19 deletions), which are more prevalent in non-small cell lung cancer (N.S.C.L.C.) following the first-line EGFR-TKI treatment. | Epidermal growth factor receptor (E.G.F.R.) tyrosine kinase inhibitor (T.K.I.) | 2015 | [64] |
| 33. | TZT1027 (Soblidotin) |  | Inhibits tubulin polymerization | Inhibit microtubule polymerization | 2011 | [65] |
| 34. | Selinexor |  | XPO1 is selectively inhibited, and tumor-suppressing proteins are activated, causing tumor cell death | Inhibiting exportin 1 (XPO1) | 2019 | [66,67] |
| 35. | Bendamustine |  | Eradicates current cancer cells and restricts the development of new cancer cells. | Alkylation | 2008 | [68] |
| 36. | Gefitinib |  | It prevents a variety of tyrosine kinases linked to transmembrane cell surface receptors from becoming phosphorylated intracellularly, including those linked to the epidermal growth factor receptor (EGFR-TK). | Epidermal growth factor receptor tyrosine kinase inhibitors (EGFR-tkis) | 2003 | [69] |
| 37. | Abemaciclib |  | The action of an abnormal protein is blocked, which causes the cancer cell’s signals to multiply | Blocks the activity of CDK4 and CDK6 proteins (CDK4/6) | 2017 | [70,71] |
| 38. | Ribociclib |  | Cell-cycle progression is blocked by acting directly on the cyclin D–CDK4/6–p16–Rb pathway | Cyclin-dependent kinase 4 and cyclin-dependent 6 (CDK 4 and CDK 6) Inhibitor | 2018 | [72] |
| 39. | Mitomycin C |  | Causes cross-linking and inhibition of D.N.A. synthesis and function | Double-stranded D.N.A. alkylating agent | 1974 | [73] |
| 40. | Porfimer |  | When activated by laser light, a cytotoxic activity that depends on oxygen and light causes the release of thromboxane A2, which in turn causes vascular necrosis and oxygen-free radicals. | Photodynamic therapy agent High-affinity immunoglobulin gamma Fc receptor I antagonist | 2003 | [74] |
| 41. | Topotecan |  | D.N.A. damage occurs by prevention of topoisomerase from relegating cleaved D.N.A. strand | Novel topoisomerase-1 inhibitor | 1996 | [75] |
| 42. | Idelalisib |  | Prevents proliferation and induces apoptosis in cell lines derived from malignant B-cells and in primary tumor cells | PI3K inhibitor | 2014 | [76,77] |
| 43. | Acalabrutinib |  | It inhibits B.C.R. signaling by reduced phosphorylation of PLCγ2. | Bruton tyrosine kinase (btk) inhibitor | 2017 | [78] |
| 44. | Cabozantinib |  | Resistance of V.E.G.F.R. inhibitor via the c-MET axis is decreased by inhibiting V.E.G.F.R. and c-MET | Tyrosine kinase inhibitor | 2016 | [79,80] |
| 45. | Lenvatinib |  | The kinase activities of vascular endothelial growth factor (VEGF) receptors VEGFR1 (FLT1), VEGFR2 (K.D.R.), and VEGFR3 (FLT4) are inhibited. | Multi-receptor Tyrosine Kinase Inhibitor. | 2018 | [81,82] |
| 46. | Palbociclib |  | Blocking cell cycle progression from G1 into S phase and inhibits the phosphorylation of retinoblastoma (Rb) protein, | Proteins cyclin-dependent kinase 4 and 6 (CDK4 and CDK6) Inhibitor | 2016 | [83,84] |
| 47. | Trametinib |  | Decreases cell growth, causes G1 cell-cycle arrest and causes apoptosis. | M.E.K. 1 and M.E.K. 2 protein (kinase) Inhibitor | 2013 | [85,86,87] |
| 48. | Afatinib |  | Suppresses mTORC1, which initiates apoptosis of the cancer cells. | Tyrosine kinase inhibitor | 2016 | [88,89] |
| 49. | Dabrafenib |  | The major inhibitors of the B.R.A.F., R.A.F. proteins and C.R.A.F., are through the competitive binding of ATP and the active conformation of B.R.A.F. kinase. | Mutated B.R.A.F. proteins (kinase) Inhibitor | 2013 | [90,91] |
| 50. | Gilteritinib |  | Inhibits FLT3 receptor signaling and proliferation | Tyrosine kinase inhibitor | 2018 | [92,93] |
| 51. | Alpelisib |  | Blocks the catalytic subunit of PI3K, class I PI3K p110, a lipid kinase involved in several biological processes, including proliferation, survival, differentiation, and metabolism | Phosphatidylinositol 3-kinase (PI3K) inhibitor | 2019 | [94] |
| 52. | Binimetinib |  | The activation of MEK1/2-dependent effector proteins and transcription factors is prevented by inhibiting MEK1/2 | Mitogen-activated protein kinase kinase 1, Dual specificity mitogen-activated protein kinase kinase 2 Inhibitor | 2018 | [95,96] |
| 53. | Crizotinib |  | Blocks certain chemical messengers that tell cells to grow. | Small-molecule kinase inhibitor ALK, c-MET, and ROS1 | 2022 | [97,98] |
| 54. | Dactinomycin |  | D.N.A. intercalation and the stabilization of DNA-topoisomerase I and II cleavable complexes | Potent transcription inhibitor | 2009 | [99] |
| 55. | Erlotinib |  | The intracellular phosphorylation of tyrosine kinase associated with the epidermal growth factor receptor (E.G.F.R.) is inhibited | Inhibition of epidermal growth factor receptor (E.G.F.R.) | 2004 | [100] |
| 56. | Ibrutinib |  | It inhibits Btk’s enzyme activity by forming a covalent bond with a cysteine residue (CYS-481) at the active site. | Bruton tyrosine kinase inhibitor | 2022 | [101,102] |
| 57. | Lapatinib |  | It works by competitively binding to the intracellular ATP-binding site of the receptor, inhibiting the tyrosine kinase domains of the human epidermal growth factor receptor (HER)-2 and the epidermal growth factor receptor. | HER2 and E.G.F.R. antagonist | 2007 | [103] |
| 58. | Larotrectinib |  | The activity of T.R.K. proteins is disrupted, which is caused by fusion in a family of genes known as N.T.R.K. | T.R.K. (Tropomyosin Receptor Kinase) inhibitor | 2018 | [104,105] |
| 59. | Mercaptopurine |  | De novo purine synthesis is inhibited and acts as an antiproliferative agent by interfering with protein, R.N.A. and D.N.A. synthesis and induces apoptosis. | Hypoxanthine-guanine phospho ribosyl transferase, Amido phospho ribosyl transferase, Inosine-5′-monophosphate dehydrogenase Inhibitor | 2014 | [106] |
| 60. | Methotrexate |  | The enzymes responsible for nucleotide synthesis are aminoimidazole carboxamide ribonucleotide transformylase (A.I.C.A.R.T.), dihydrofolate reductase, thymidylate synthase, and amido phosphor ribosyl transferase are inhibited. | Thymidylate synthase and Dihydrofolate reductase Inhibitors | 1999 | [107,108] |
| 61. | Nilotinib |  | Stabilizes the Abl protein’s kinase domain’s inactive conformation by binding to it. | Tyrosine kinase inhibitor | 2007 | [109] |
| 62. | Nilutamide |  | The action of androgens of testicular and adrenal origin that stimulate the growth of normal and malignant prostatic tissue is blocked | Androgen receptor antagonist | 2016 | [110] |
| 63. | Olaparib |  | Blocks the repair of single-strand D.N.A. breaks by inhibiting poly(ADP-ribose) polymerase. | Poly (ADP-ribose) polymerase (PARP) inhibitor | 2014 | [111,112] |
3. Current Advances in Nitrogen Containing Heterocycles as Anticancer Agents
3.1. Pyrimidine Derivatives as Anticancer Agents
3.2. Quinoline Derivatives as Anticancer Agents
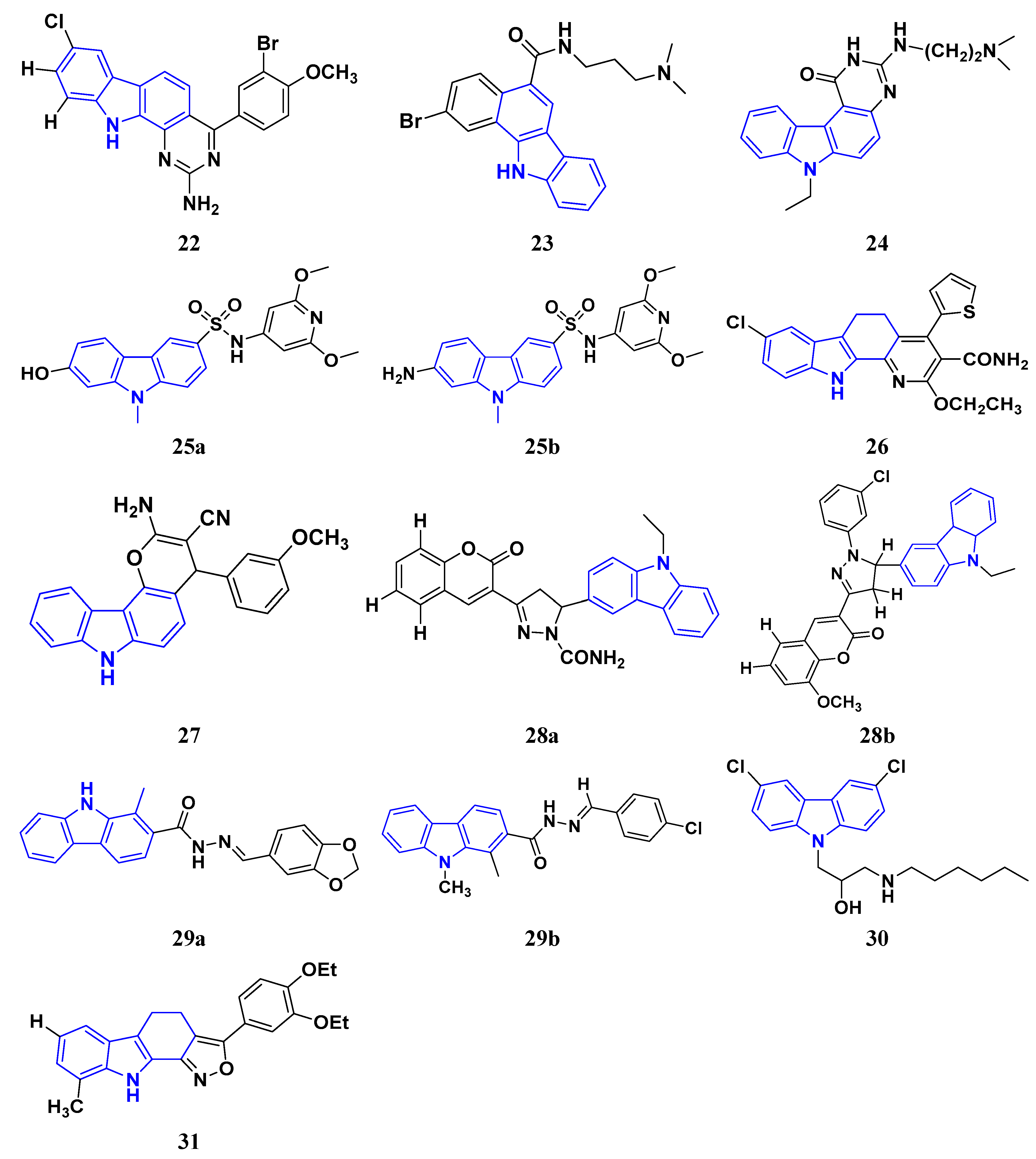
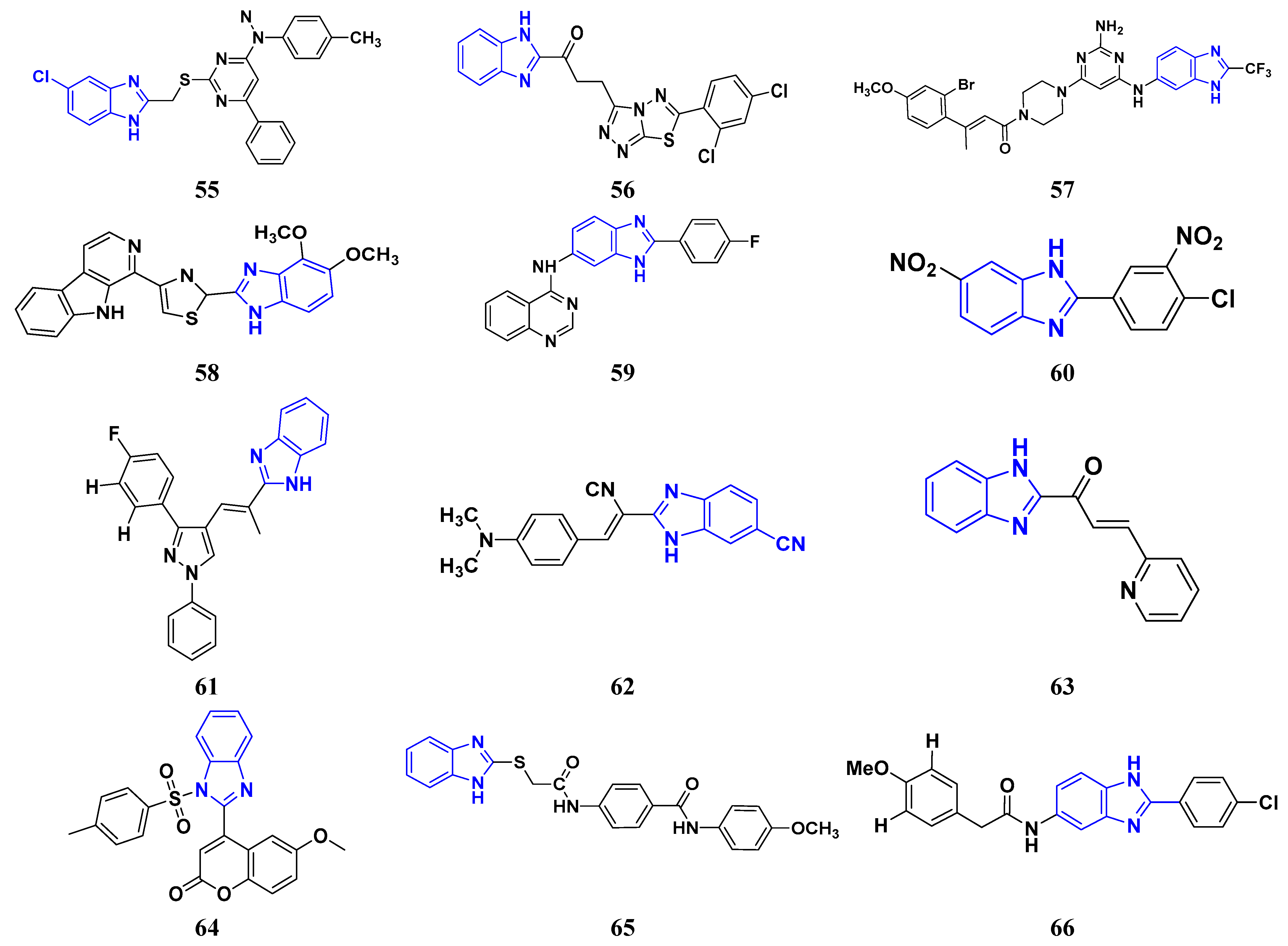
3.3. Carbazole Derivatives as Anticancer Agents
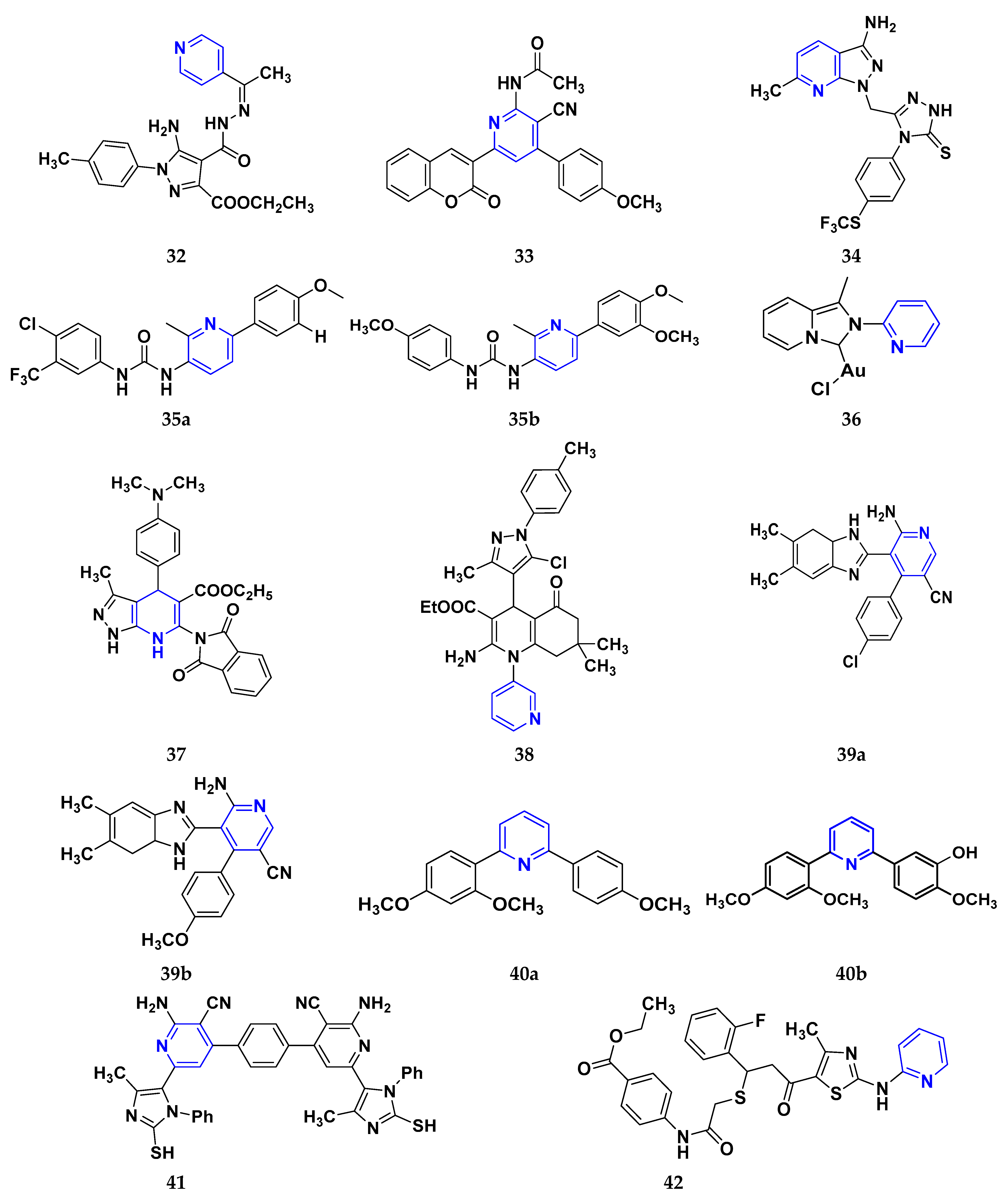
3.4. Pyridine Derivatives as Anti-Cancer Agents
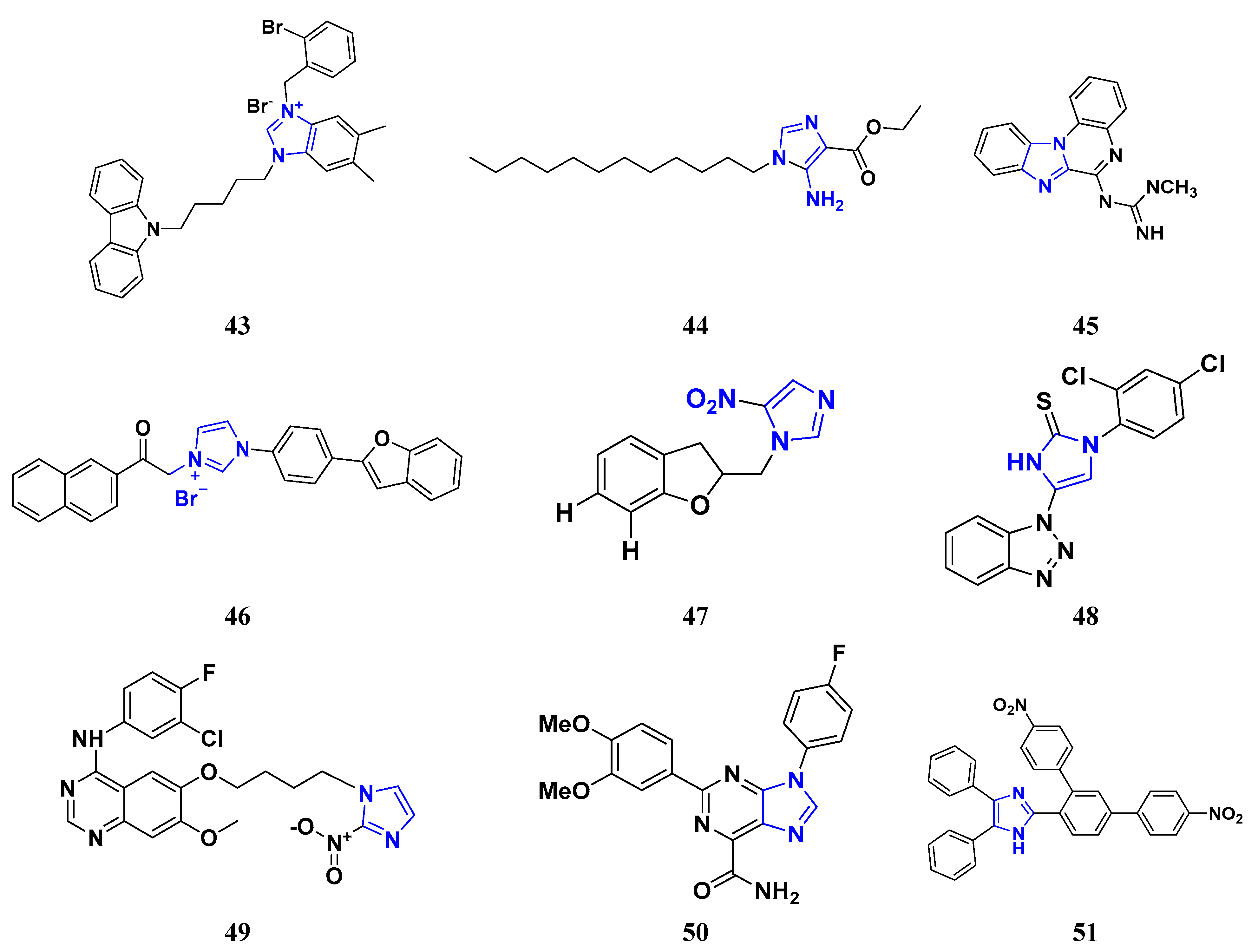
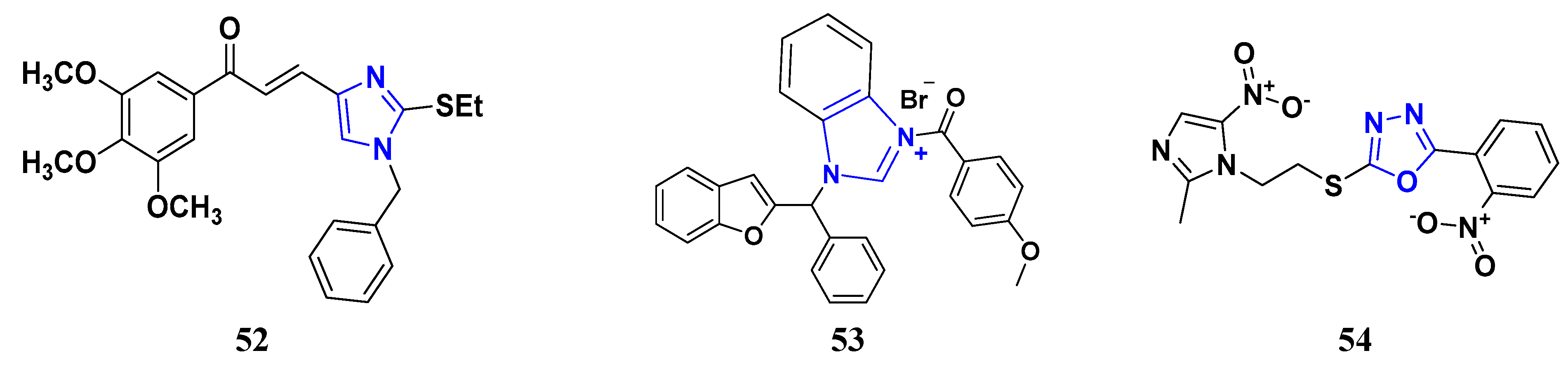
3.5. Imidazole Derivatives as Anticancer Agents
3.6. Benzimidazole Derivatives as Anticancer Agents
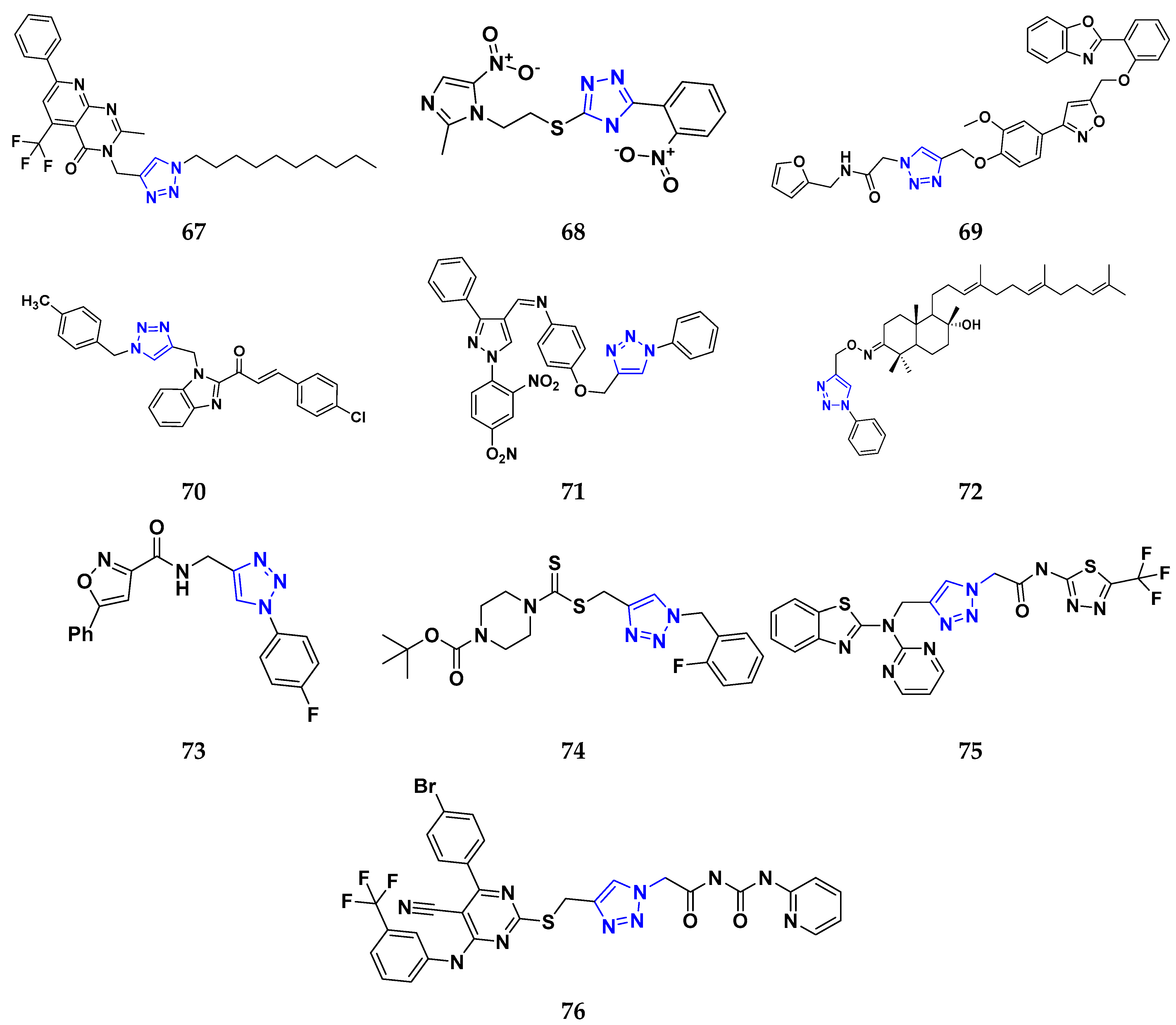
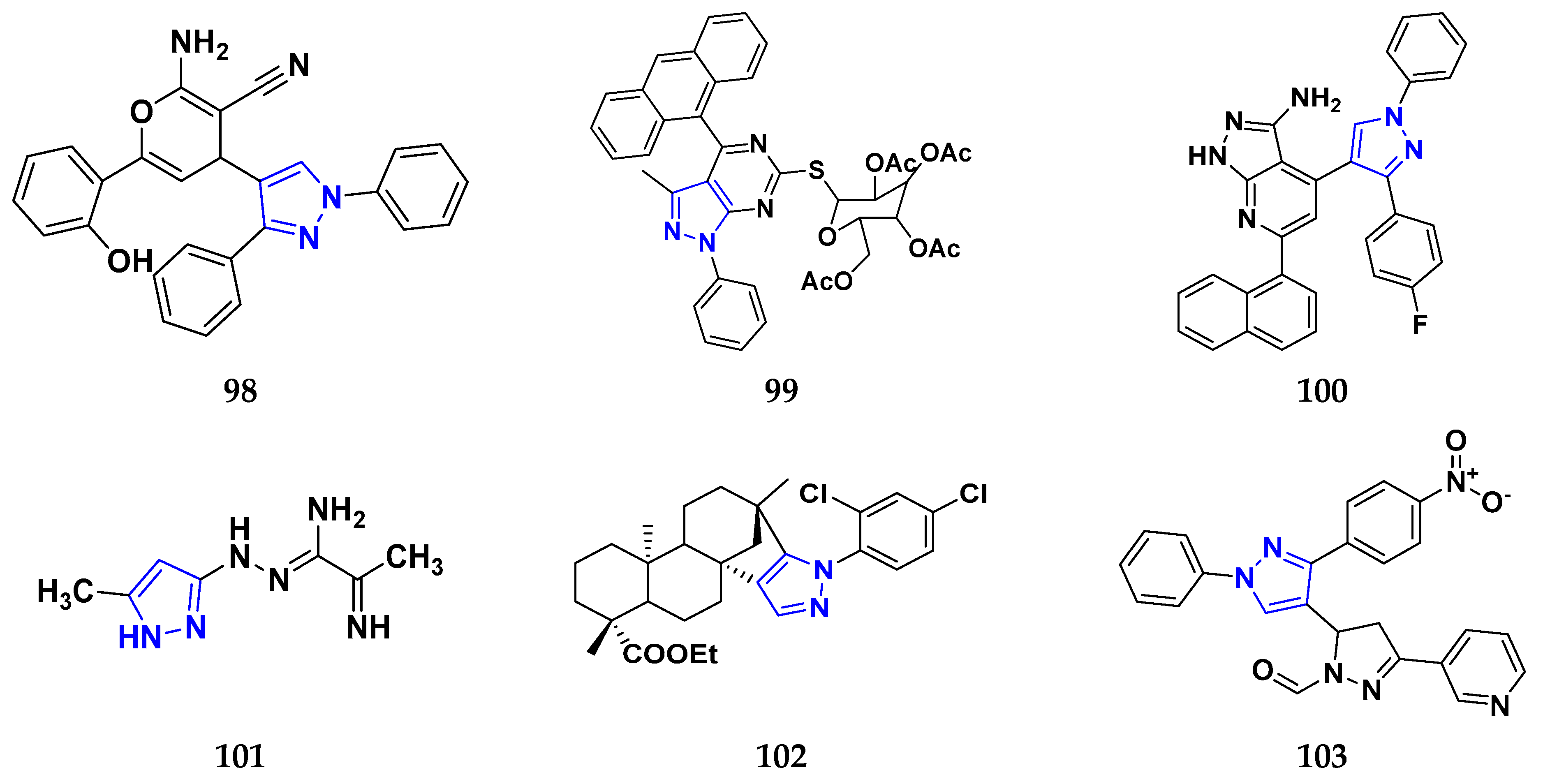
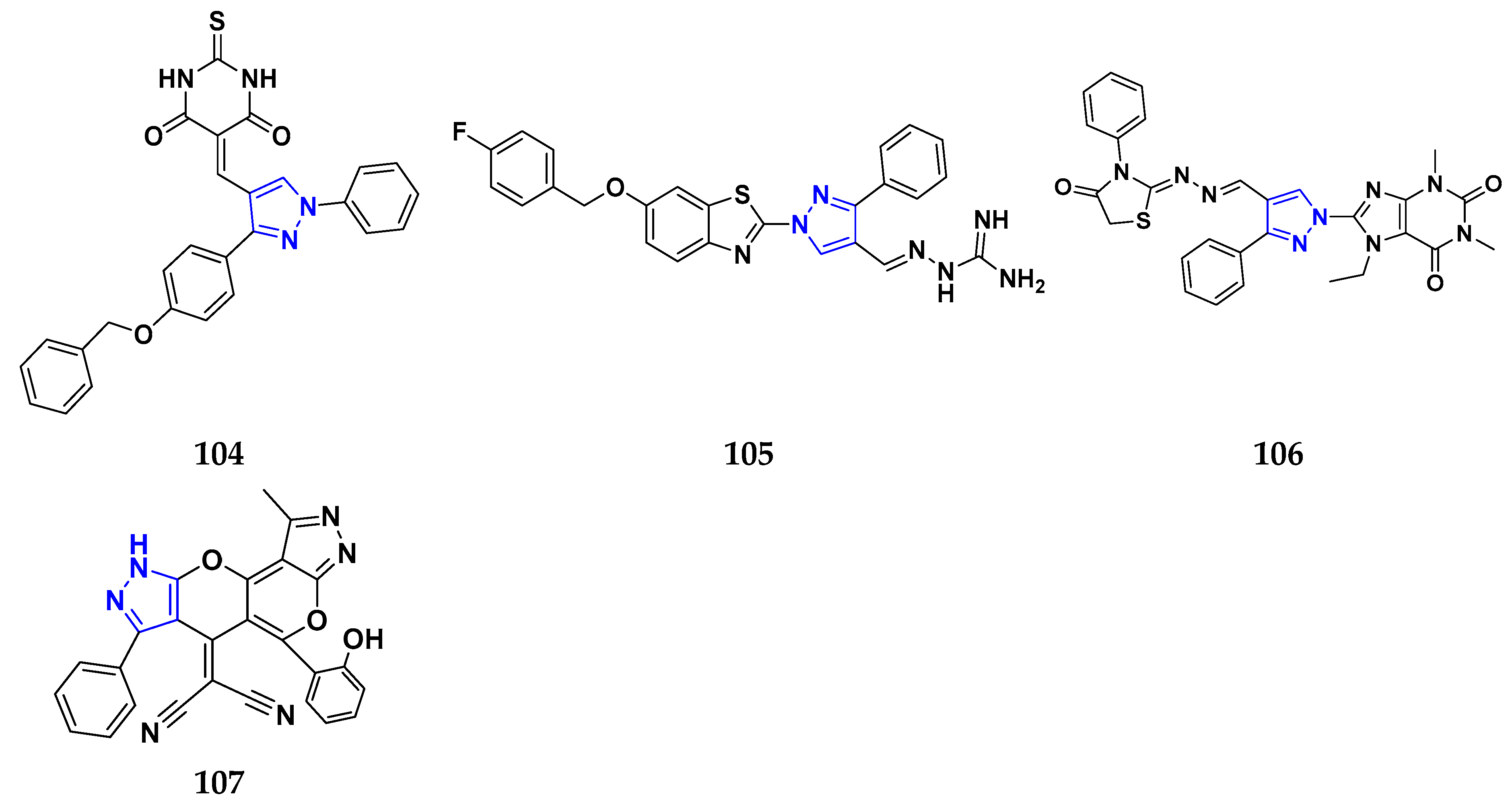
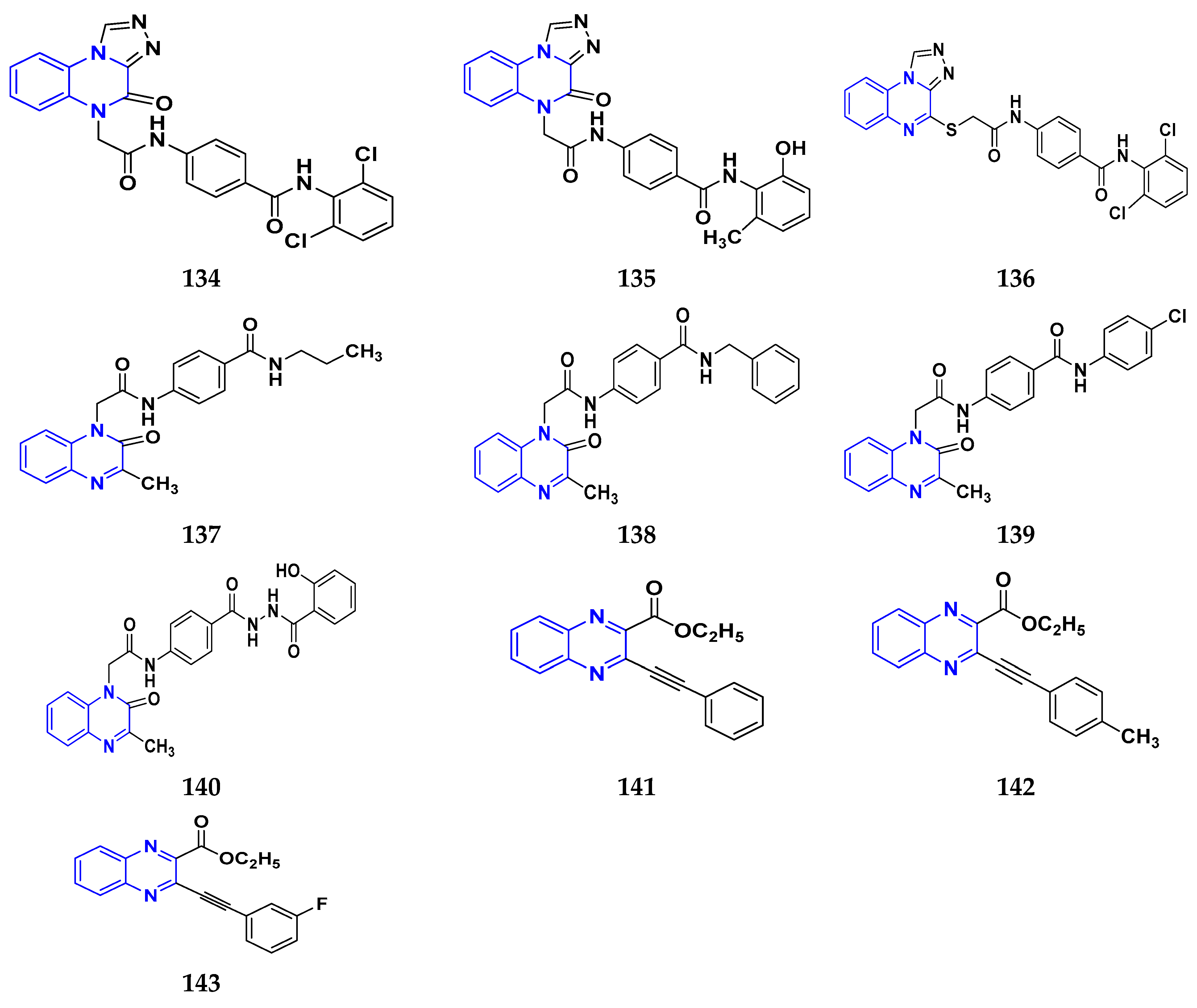
3.7. Triazole Derivatives as Anticancer Agents
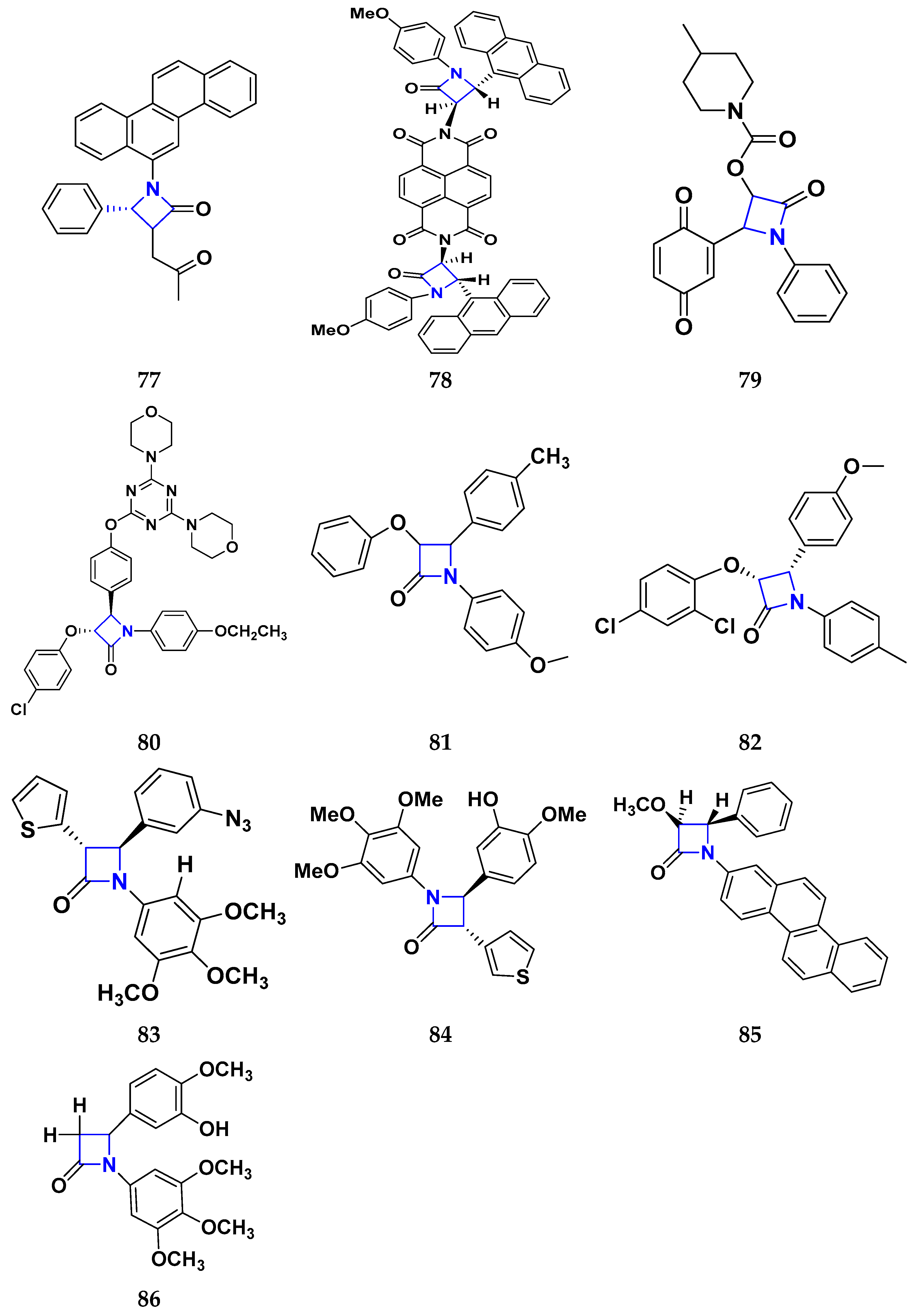
3.8. β-Lactam Derivatives as Anticancer Agents
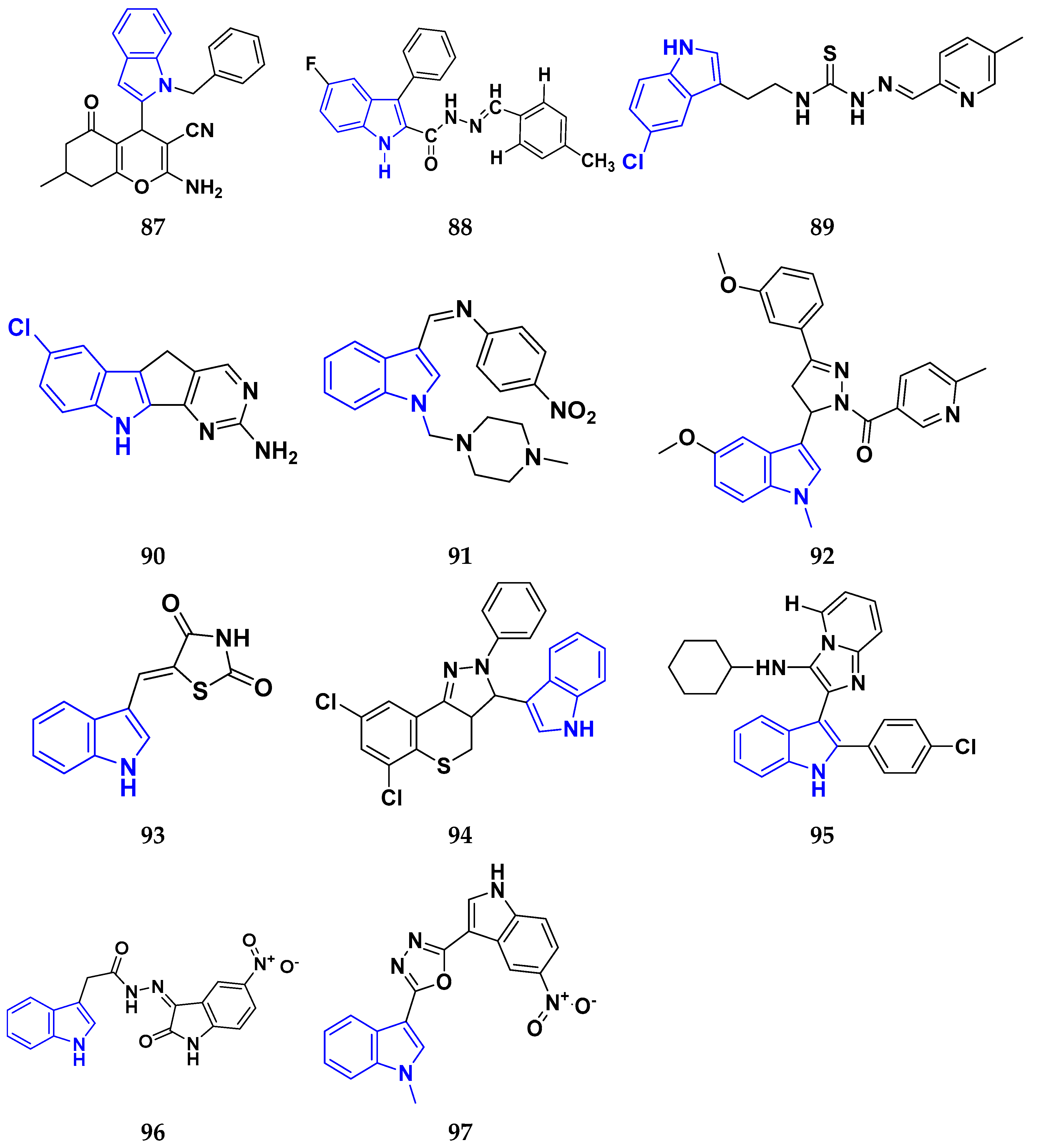
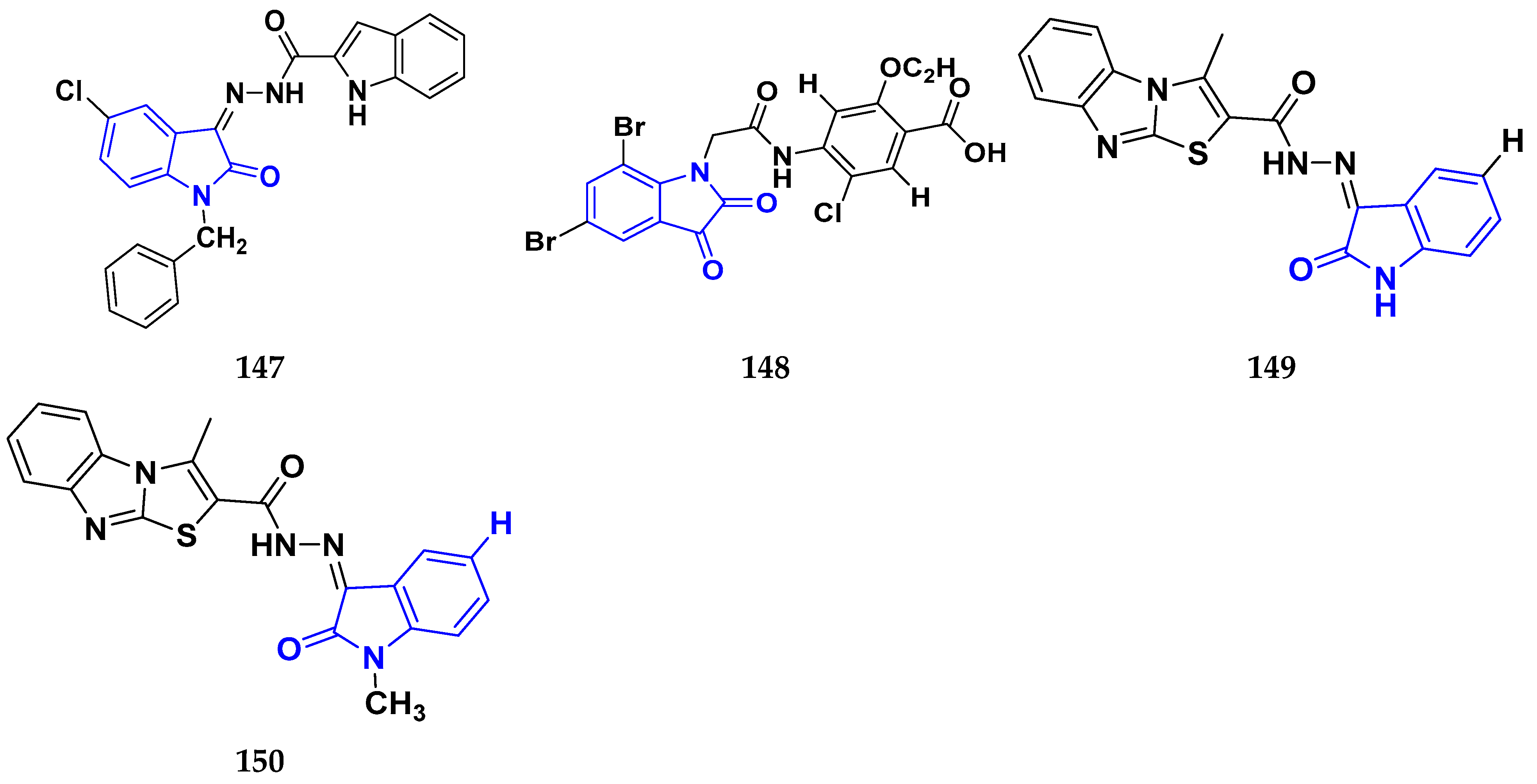
3.9. Indole Derivatives as Anticancer Ahents
3.10. Pyrazole Derivatives as Anticancer Agents
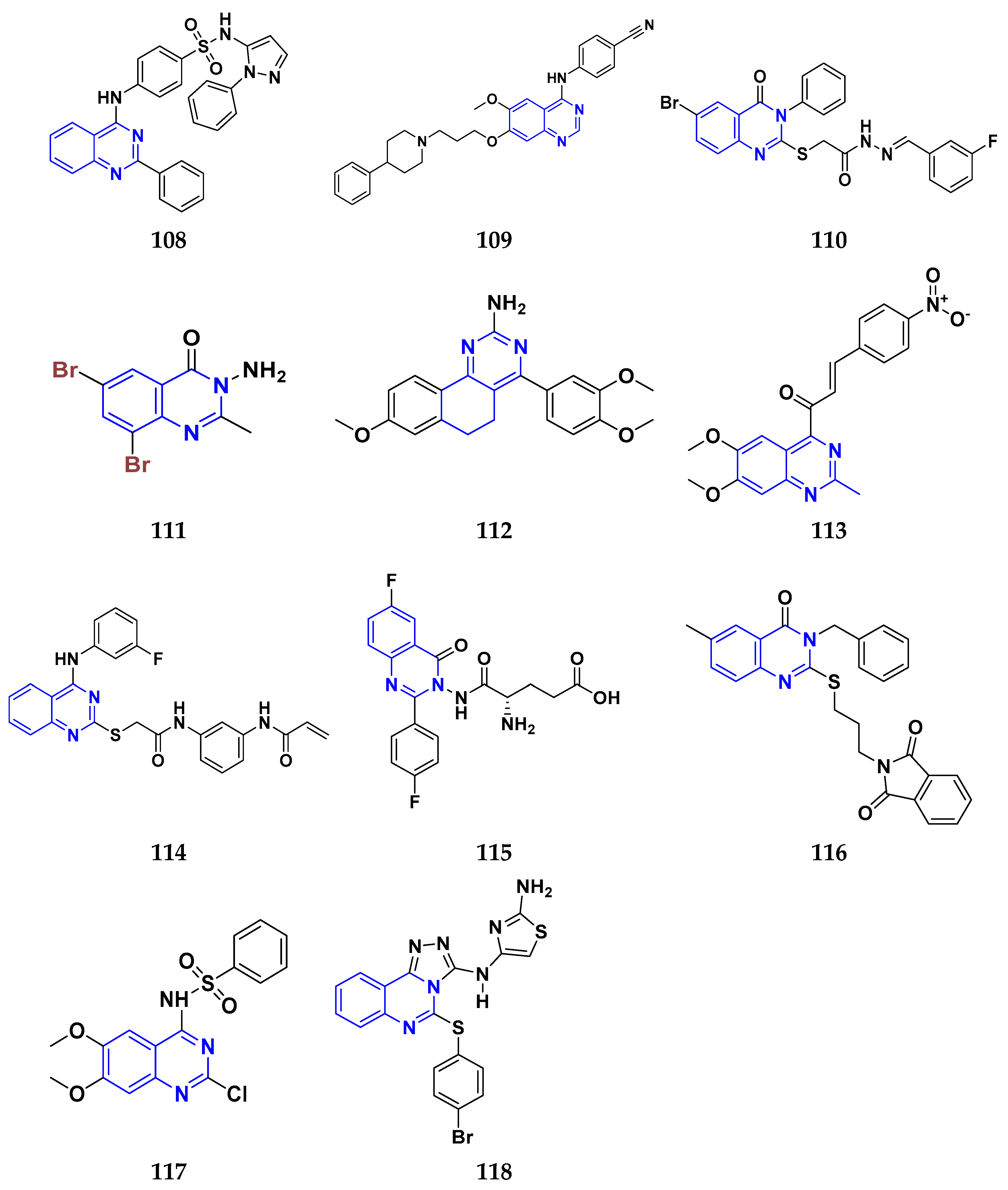
3.11. Quinazoline Derivatives as Anticancer Agents
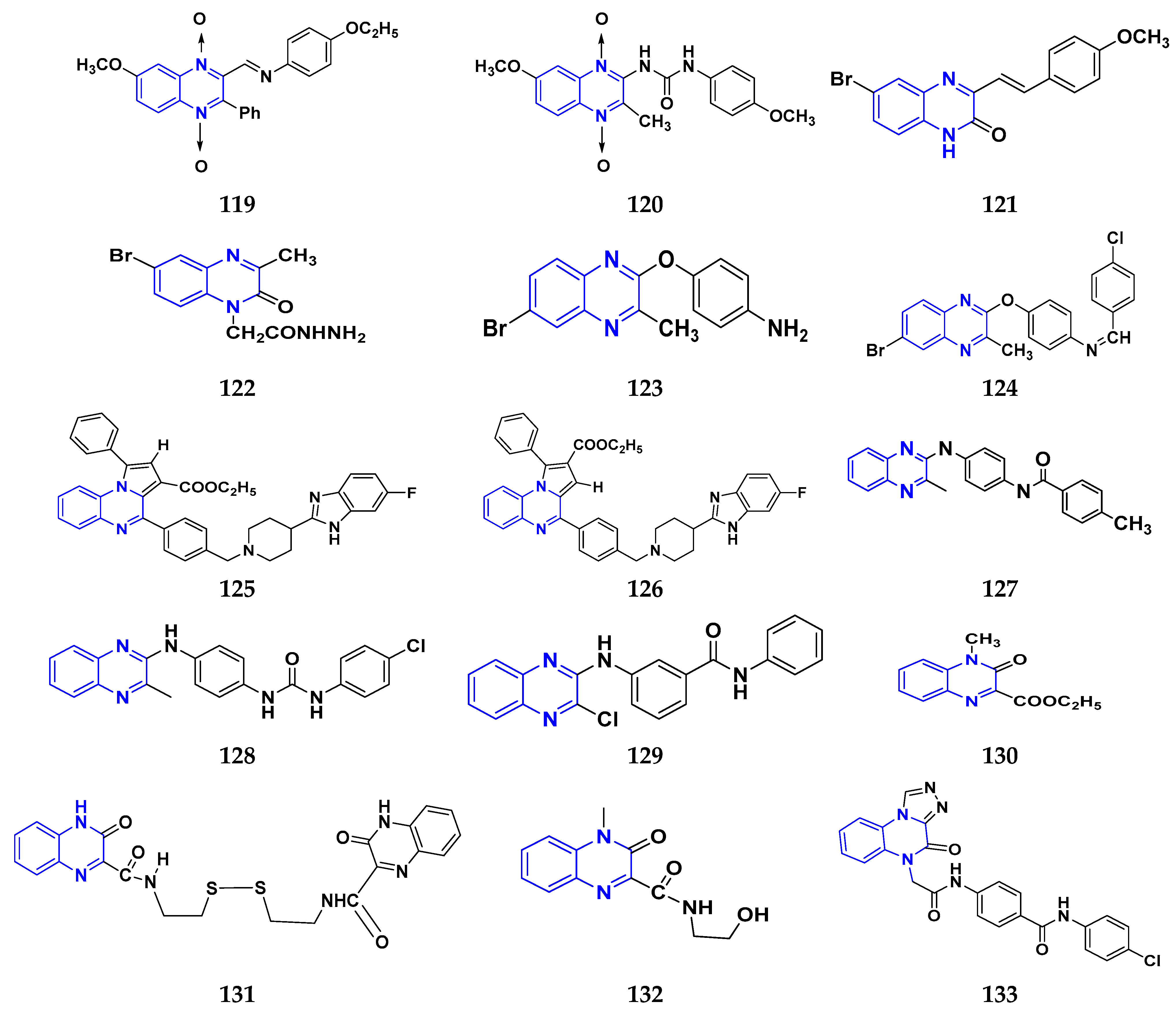
3.12. Quinoxaline Derivatives as Anticancer Agents
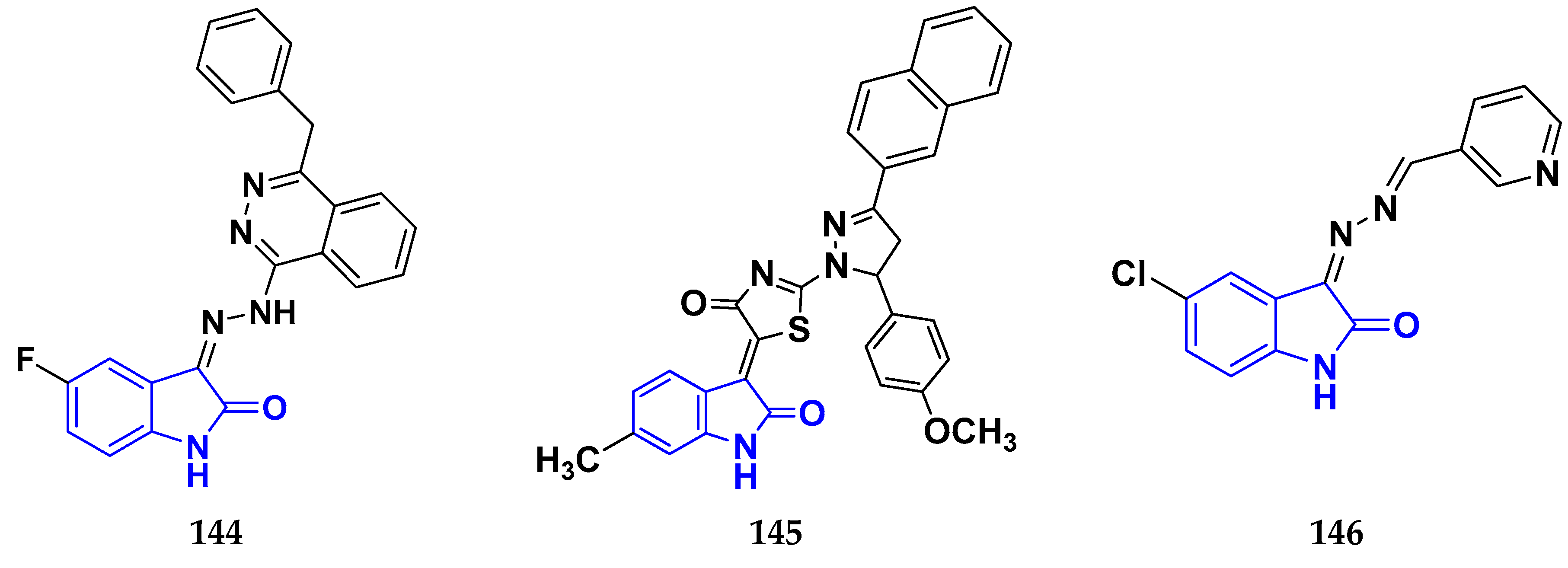
3.13. Isatin Derivatives as Anticancer Agents
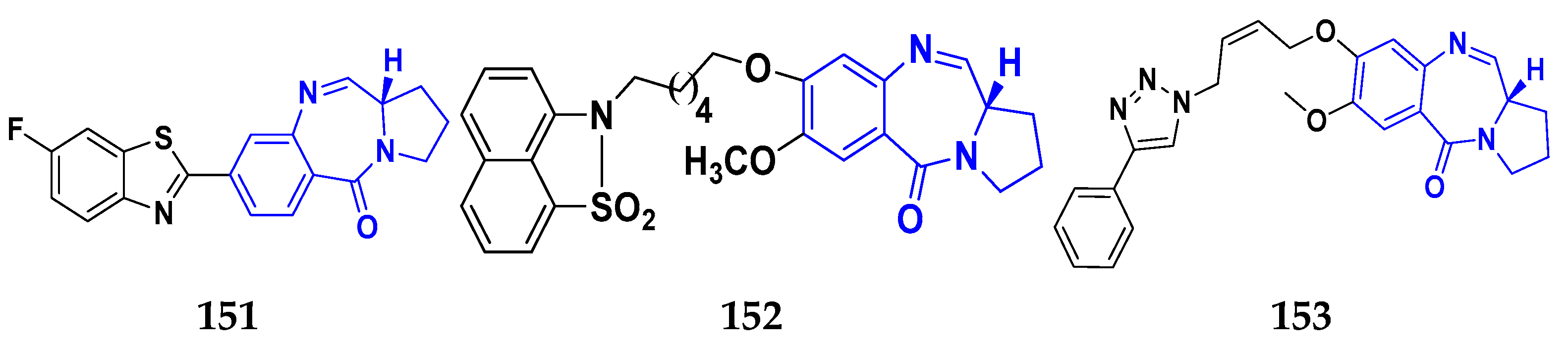
3.14. Pyrrolo-Benzodiazepines Derivatives as Anticancer Agents
3.15. Pyrido[2,3-d] Pyrimidine Derivatives as Anticancer Agents
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AICART | Aminoimidazole carboxamide ribonucleotide transformylase |
| ALK | Anaplastic lymphoma kinase |
| AQ | anthraquinone |
| ARS | Artesunate |
| BCR | Breakpoint cluster region |
| BRAF | v-raf murine sarcoma viral oncogene homolog B1 |
| CA-4 | Combretasin A-4 |
| CDK | Cyclin dependent kinase |
| CNS | Central Nervous System |
| CSF-1 | Colony-stimulating factor |
| DENV-2 | Dengue virus serotype 2 |
| DHA | Dihydroartemisinin |
| DLC | Drug loading content |
| DLE | Drug-loading efficiency |
| DMEM | Dulbecco’s Modified Eagle Medium |
| DOX | Doxorubicin |
| DPPH | 2,2-diphenylpicrylhydrazyl |
| EAC | Ehrlich Ascites Carcinoma |
| EC50 | Half maximal effective concentration |
| EGFR | Epidermal growth factor receptor |
| EMT | Epithelial Mesenchymal transition |
| ERK | Extracellular signal-regulated kinase |
| FLT3 | Fms-like tyrosine kinase |
| GI50 | Half maximal Growth Inhibition |
| HDAC | Histone deacetylases |
| HER | Human epidermal growth factor receptor |
| HetQ | heterocycle-fused quinone |
| IC50 | Half maximal Inhibitory concentration |
| JAK2 | Janus kinase 2 |
| KDR | Kinase insert domain receptor |
| LC50 | Half maximal Lethal concentration |
| MEK | Mitogen-activated protein kinase |
| MG-MID | Mean graph midpoint |
| MTT | 3-(4,5-dimethylthiazolyl-2)-2,5-diphenyltetrazolium bromide |
| NCI | National Cancer Institute |
| ND | Not Defined |
| NQ | Naphthoquinone |
| NSCLC | Non-small cell lung cancer |
| P13K | Phosphoinositide3-kinase |
| PBD | Pyrrolo-benzodiazepines |
| PGDFR | Platelet-derived growth factor |
| Raf | Rapidly accelerated fibrosarcoma |
| Ras | Rat Sarcoma |
| ROS | Reactive Oxygen Species |
| SAHA | standard hydroxamic acid |
| SRB | Sulforhodamine B |
| TKi | Tyrosine kinase inhibitor |
| TRK | Tropomyosin Receptor Kinase |
| VEGFR | vascular endothelial growth factor receptor |
| XPO1 | Exportin 1 |
References
- Angre, T.; Kumar, A.; Singh, A.K.; Thareja, S.; Kumar, P. Role of Collagen Regulators in Cancer Treatment: A Comprehensive Review. Anti-Cancer Agents Med. Chem. 2022, 22, 2956–2984. [Google Scholar]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Gu, L.; Shan, T.; Ma, Y.-X.; Tay, F.R.; Niu, L. Novel biomedical applications of crosslinked collagen. Trends Biotechnol. 2019, 37, 464–491. [Google Scholar] [CrossRef] [PubMed]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.K.; Kumar, A.; Thareja, S.; Kumar, P. Current insights into the role of BRAF inhibitors in treatment of melanoma. Anti-Cancer Agents Med. Chem. 2022, 1–7. [Google Scholar] [CrossRef]
- Rang, H.; Dale, M.; Ritter, J.; Moore, P. Edinburgh, Scotland, Pharmacology, 5th ed.; Churchill Livingstone: London, UK, 2003. [Google Scholar]
- Williams David, A.; Lemke Thomson, A. Foyes Principle of Medicinal Chemistry; Lippincott, Williams & Wilkins: Philadelphia, FL, USA, 2002. [Google Scholar]
- Stefanowicz, P.; Jaremko, Ł.; Jaremko, M.; Lis, T. Crystal-state studies on p-toluenesulfonates of N-oxyimides—A possible structural basis of serine proteases inhibition. New J. Chem. 2006, 30, 258–265. [Google Scholar] [CrossRef]
- Alvárez-Builla, J.; Barluenga, J. Heterocyclic compounds: An introduction. Mod Heterocycl Chem 2011, 1, 1–9. [Google Scholar]
- Ferreira, P.M.; Maia, H.L.; Monteiro, L.s.S. Synthesis of 2, 3, 5-substituted pyrrole derivatives. Tetrahedron. Lett. 2002, 43, 4491–4493. [Google Scholar] [CrossRef]
- Kijewska, M.; Sharfalddin, A.A.; Jaremko, Ł.; Cal, M.; Setner, B.; Siczek, M.; Stefanowicz, P.; Hussien, M.A.; Emwas, A.-H.; Jaremko, M. Lossen Rearrangement of p-Toluenesulfonates of N-Oxyimides in Basic Condition, Theoretical Study, and Molecular Docking. Front. Chem. 2021, 9, 662533. [Google Scholar] [CrossRef]
- Shukla, P.K.; Verma, A.; Mishra, P. Significance of nitrogen heterocyclic nuclei in the search of pharmacological active compounds. In New Perspective in Agricultural and Human Health; Shukla, R.P., Mishra, R.S., Tripathi, A.D., Yadav, A.K., Tiwari, M., Mishra, R.R., Eds.; Bharti Publications: Delhi, India, 2017; pp. 100–126. [Google Scholar]
- Martins, P.; Jesus, J.; Santos, S.; Raposo, L.R.; Roma-Rodrigues, C.; Baptista, P.V.; Fernandes, A.R. Heterocyclic anticancer compounds: Recent advances and the paradigm shift towards the use of nanomedicine’s tool box. Molecules 2015, 20, 16852–16891. [Google Scholar] [CrossRef]
- Ajani, O.; Audu, O.; Aderohunmu, D.; Owolabi, F.; Olomieja, A. Undeniable pharmacological potentials of quinazoline motifs in therapeutic medicine. Am. J. Drug Discov. Dev. 2017, 7, 1–24. [Google Scholar] [CrossRef]
- Özkay, Y.; Işıkdağ, İ.; İncesu, Z.; Akalın, G. Synthesis of 2-substituted-N-[4-(1-methyl-4, 5-diphenyl-1H-imidazole-2-yl) phenyl] acetamide derivatives and evaluation of their anticancer activity. Eur. J. Med. Chem. 2010, 45, 3320–3328. [Google Scholar] [CrossRef] [PubMed]
- Jangale, A.D.; Dalal, D.S. Green synthetic approaches for biologically relevant organic compounds. Synth. Commun. 2017, 47, 2139–2173. [Google Scholar] [CrossRef]
- Yang, D.; An, B.; Wei, W.; Tian, L.; Huang, B.; Wang, H. Copper-catalyzed domino synthesis of nitrogen heterocycle-fused benzoimidazole and 1, 2, 4-benzothiadiazine 1, 1-dioxide derivatives. ACS Comb. Sci. 2015, 17, 113–119. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, M.; Singh, J.; Singh, S.B.; Tiwari, K.; Pathak, V.K.; Singh, J. Synthesis of novel fused heterocycle-oxa-aza-phenanthrene and anthracene derivatives via sequential one-pot synthesis in aqueous micellar system. Green Chem. 2012, 14, 901–905. [Google Scholar] [CrossRef]
- Czarnik, A.W. Peer Reviewed: Combinatorial Chemistry. Anal. Chem. 1998, 70, 378–386. [Google Scholar] [CrossRef]
- García-Valverde, M.; Torroba, T. Sulfur-Nitrogen Heterocycles. Guest editorial; Molecules 2005, 10, 318–320. [Google Scholar] [CrossRef]
- Al-Ghorbani, M.; Bushra, B.A.; Mamatha, S.; Khanum, S.A. Piperazine and morpholine: Synthetic preview and pharmaceutical applications. Res. J. Pharm. Technol. 2015, 8, 611–628. [Google Scholar] [CrossRef]
- Heravi, M.M.; Zadsirjan, V. Prescribed drugs containing nitrogen heterocycles: An overview. RSC Adv. 2020, 10, 44247–44311. [Google Scholar] [CrossRef]
- Mendez, D.; Gaulton, A.; Bento, A.P.; Chambers, J.; De Veij, M.; Félix, E.; Magariños, M.P.; Mosquera, J.F.; Mutowo, P.; Nowotka, M. ChEMBL: Towards direct deposition of bioassay data. Nucleic Acids Res. 2019, 47, 930–940. [Google Scholar] [CrossRef]
- Larkins, E.; Blumenthal, G.M.; Chen, H.; He, K.; Agarwal, R.; Gieser, G.; Stephens, O.; Zahalka, E.; Ringgold, K.; Helms, W. FDA Approval: Alectinib for the Treatment of Metastatic, ALK-Positive Non–Small Cell Lung Cancer Following CrizotinibAlectinib for ALK-Positive Non–Small Cell Lung Cancer. Clin. Cancer Res. 2016, 22, 5171–5176. [Google Scholar] [CrossRef] [PubMed]
- Sun, N.; Ren, C.; Kong, Y.; Zhong, H.; Chen, J.; Li, Y.; Zhang, J.; Zhou, Y.; Qiu, X.; Lin, H. Development of a Brigatinib degrader (SIAIS117) as a potential treatment for ALK positive cancer resistance. Eur. J. Med. Chem. 2020, 193, 112190. [Google Scholar] [CrossRef] [PubMed]
- Collier, T.L.; Normandin, M.D.; Stephenson, N.A.; Livni, E.; Liang, S.H.; Wooten, D.W.; Esfahani, S.A.; Stabin, M.G.; Mahmood, U.; Chen, J. Synthesis and preliminary PET imaging of 11C and 18F isotopologues of the ROS1/ALK inhibitor lorlatinib. Nat. Commun. 2017, 8, 1–12. [Google Scholar] [CrossRef]
- Sartore-Bianchi, A.; Pizzutilo, E.G.; Marrapese, G.; Tosi, F.; Cerea, G.; Siena, S. Entrectinib for the treatment of metastatic NSCLC: Safety and efficacy. Expert Rev. Anticancer. Ther. 2020, 20, 333–341. [Google Scholar] [CrossRef] [PubMed]
- Kasamon, Y.L.; Ko, C.W.; Subramaniam, S.; Ma, L.; Yang, Y.; Nie, L.; Shord, S.; Przepiorka, D.; Farrell, A.T.; McKee, A.E. FDA approval summary: Midostaurin for the treatment of advanced systemic mastocytosis. Oncologist 2018, 23, 1511–1519. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.; Wu, P.; Yang, Q.; Xie, Y.; He, C.; Yin, L.; Yin, Z.; Yue, G.; Zou, Y.; Li, L. An update of new small-molecule anticancer drugs approved from 2015 to 2020. Eur. J. Med. Chem. 2021, 220, 113473. [Google Scholar] [CrossRef]
- Tzogani, K.; Røshol, H.; Olsen, H.H.; Aas, I.B.; Dalhus, M.L.; Håkonsen, G.D.; Nilssen, L.S.; Lindberg, V.; Økvist, M.; Bolstad, B. The European Medicines Agency review of Gilteritinib (Xospata) for the treatment of adult patients with relapsed or refractory acute myeloid leukemia with an FLT3 mutation. Oncologist 2020, 25, e1070–e1076. [Google Scholar] [CrossRef] [PubMed]
- Gunawardane, R.N.; Nepomuceno, R.R.; Rooks, A.M.; Hunt, J.P.; Ricono, J.M.; Belli, B.; Armstrong, R.C. Transient exposure to quizartinib mediates sustained inhibition of FLT3 signaling while specifically inducing apoptosis in FLT3-activated leukemia cells. Mol. Cancer Ther. 2013, 12, 438–447. [Google Scholar] [CrossRef]
- Benner, B.; Good, L.; Quiroga, D.; Schultz, T.E.; Kassem, M.; Carson, W.E.; Cherian, M.A.; Sardesai, S.; Wesolowski, R. Pexidartinib, a novel small molecule CSF-1R inhibitor in use for tenosynovial giant cell tumor: A systematic review of pre-clinical and clinical development. Drug Des. Dev. Ther. 2020, 14, 1693. [Google Scholar] [CrossRef]
- Butterworth, S.; Cross, D.A.; Finlay, M.R.V.; Ward, R.A.; Waring, M.J. The structure-guided discovery of osimertinib: The first US FDA approved mutant selective inhibitor of EGFR T790M. Medchemcomm 2017, 8, 820–822. [Google Scholar] [CrossRef]
- Zhang, W.; Fan, Y.-F.; Cai, C.-Y.; Wang, J.-Q.; Teng, Q.-X.; Lei, Z.-N.; Zeng, L.; Gupta, P.; Chen, Z.-S. Olmutinib (BI1482694/HM61713), a novel epidermal growth factor receptor tyrosine kinase inhibitor, reverses ABCG2-mediated multidrug resistance in cancer cells. Front. Pharmacol. 2018, 9, 1097. [Google Scholar] [CrossRef] [PubMed]
- Singh, H.; Walker, A.J.; Amiri-Kordestani, L.; Cheng, J.; Tang, S.; Balcazar, P.; Barnett-Ringgold, K.; Palmby, T.R.; Cao, X.; Zheng, N. US Food and Drug Administration Approval: Neratinib for the Extended Adjuvant Treatment of Early-Stage HER2-Positive Breast CancerFDA Approval Summary: Neratinib. Clin. Cancer Res. 2018, 24, 3486–3491. [Google Scholar] [CrossRef] [Green Version]
- Lavacchi, D.; Mazzoni, F.; Giaccone, G. Clinical evaluation of dacomitinib for the treatment of metastatic non-small cell lung cancer (NSCLC): Current perspectives. Drug Des. Dev. Ther. 2019, 13, 3187. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wang, B. Pharmaceutically Acceptable Salt of (e)-n-[4-[[3-chloro-4-(2-Pyridylmethoxy) phenyl] amino]-3-cyano-7-ethoxy-6-quinolyl]-3-[(2r)-1-methylpyrrolidin-2-yl] prop-2-enamide, Preparation Method Thereof, and Medical Use Thereof. US Patent Application No. 14/001,778, 2013. [Google Scholar]
- Zhou, C.; Xie, L.; Liu, W.; Zhang, L.; Zhou, S.; Wang, L.; Chen, J.; Li, H.; Zhao, Y.; Zhu, B. Absorption, metabolism, excretion, and safety of [14C] almonertinib in healthy Chinese subjects. Ann. Transl. Med. 2021, 9, 867. [Google Scholar] [CrossRef] [PubMed]
- Hao, Z.; Wang, P. Lenvatinib in management of solid tumors. Oncologist 2020, 25, e302–e310. [Google Scholar] [CrossRef]
- Sochacka-Ćwikła, A.; Mączyński, M.; Regiec, A. FDA-Approved Small Molecule Compounds as Drugs for Solid Cancers from Early 2011 to the End of 2021. Molecules 2022, 27, 2259. [Google Scholar] [CrossRef]
- Zhang, Y.; Zou, J.-Y.; Wang, Z.; Wang, Y. Fruquintinib: A novel antivascular endothelial growth factor receptor tyrosine kinase inhibitor for the treatment of metastatic colorectal cancer. Cancer Manag. Res. 2019, 11, 7787. [Google Scholar] [CrossRef]
- Shen, G.; Zheng, F.; Ren, D.; Du, F.; Dong, Q.; Wang, Z.; Zhao, F.; Ahmad, R.; Zhao, J. Anlotinib: A novel multi-targeting tyrosine kinase inhibitor in clinical development. J. Hematol. Oncol. 2018, 11, 1–11. [Google Scholar] [CrossRef]
- Isaac, K.; Mato, A.R. Acalabrutinib and its therapeutic potential in the treatment of chronic lymphocytic leukemia: A short review on emerging data. Cancer Manag. Res. 2020, 12, 2079. [Google Scholar] [CrossRef]
- Guo, Y.; Liu, Y.; Hu, N.; Yu, D.; Zhou, C.; Shi, G.; Zhang, B.; Wei, M.; Liu, J.; Luo, L. Discovery of zanubrutinib (BGB-3111), a novel, potent, and selective covalent inhibitor of Bruton’s tyrosine kinase. J. Med. Chem. 2019, 62, 7923–7940. [Google Scholar] [CrossRef]
- Palmer, J.; Mesa, R. The role of fedratinib for the treatment of patients with primary or secondary myelofibrosis. Ther. Adv. Hematol. 2020, 11, 2040620720925201. [Google Scholar] [CrossRef] [PubMed]
- Markham, A. Copanlisib: First global approval. Drugs 2017, 77, 2057–2062. [Google Scholar] [CrossRef]
- Dreyling, M.; Morschhauser, F.; Bouabdallah, K.; Bron, D.; Cunningham, D.; Assouline, S.; Verhoef, G.; Linton, K.; Thieblemont, C.; Vitolo, U. Phase II study of copanlisib, a PI3K inhibitor, in relapsed or refractory, indolent or aggressive lymphoma. Ann. Oncol. 2017, 28, 2169–2178. [Google Scholar] [CrossRef] [PubMed]
- Boumber, Y.; Younes, A.; Garcia-Manero, G. Mocetinostat (MGCD0103): A review of an isotype-specific histone deacetylase inhibitor. Expert Opin. Investig. Drugs 2011, 20, 823–829. [Google Scholar] [CrossRef] [PubMed]
- Karandish, F.; Haldar, M.K.; You, S.; Brooks, A.E.; Brooks, B.D.; Guo, B.; Choi, Y.; Mallik, S. Prostate-specific membrane antigen targeted polymersomes for delivering mocetinostat and docetaxel to prostate cancer cell spheroids. ACS Omega 2016, 1, 952–962. [Google Scholar] [CrossRef]
- Rodrigues, D.A.; Sagrillo, F.S.; Fraga, C.A. Duvelisib: A 2018 novel FDA-approved small molecule inhibiting phosphoinositide 3-kinases. Pharmaceuticals 2019, 12, 69. [Google Scholar] [CrossRef]
- Ibrahim, A.; Hirschfeld, S.; Cohen, M.H.; Griebel, D.J.; Williams, G.A.; Pazdur, R. FDA drug approval summaries: Oxaliplatin. Oncologist 2004, 9, 8–12. [Google Scholar] [CrossRef]
- Monneret, C. Platinum anticancer drugs. From serendipity to rational design. In Annales Pharmaceutiques Francaises; Elsevier: Amsterdam, The Netherlands, 2011; pp. 286–295. [Google Scholar]
- Dean, L. Irinotecan therapy and UGT1A1 genotype. In Medical Genetics Summaries; National Center for Biotechnology Information (US): Bethesda, MD, USA, 2012; updated 2018. [Google Scholar]
- Bailly, C. Irinotecan: 25 years of cancer treatment. Pharmacol. Res. 2019, 148, 104398. [Google Scholar] [CrossRef]
- El-Subbagh, H.I.; Al-Badr, A.A. Cytarabine. In Profiles of Drug Substances, Excipients and Related Methodology; Elsevier: Amsterdam, The Netherlands, 2009; Volume 34, pp. 37–113. [Google Scholar]
- Baker, W.J.; Royer Jr, G.L.; Weiss, R.B. Cytarabine and neurologic toxicity. Oncology 1991, 9, 679–693. [Google Scholar] [CrossRef]
- Noble, S.; Goa, K.L. Gemcitabine. Drugs 1997, 54, 447–472. [Google Scholar] [CrossRef]
- Paroha, S.; Verma, J.; Dubey, R.D.; Dewangan, R.P.; Molugulu, N.; Bapat, R.A.; Sahoo, P.K.; Kesharwani, P. Recent advances and prospects in gemcitabine drug delivery systems. Int. J. Pharm. 2021, 592, 120043. [Google Scholar] [CrossRef]
- Ganjoo, K.N.; Patel, S.J. Trabectedin: An anticancer drug from the sea. Expert Opin. Pharmacother. 2009, 10, 2735–2743. [Google Scholar] [CrossRef]
- Barone, A.; Chi, D.-C.; Theoret, M.R.; Chen, H.; He, K.; Kufrin, D.; Helms, W.S.; Subramaniam, S.; Zhao, H.; Patel, A. FDA Approval Summary: Trabectedin for Unresectable or Metastatic Liposarcoma or Leiomyosarcoma Following an Anthracycline-Containing Regimen. Clin. Cancer Res. 2017, 23, 7448–7453. [Google Scholar] [CrossRef]
- Brown, A.P.; Morrissey, R.L.; Faircloth, G.T.; Levine, B.S. Preclinical toxicity studies of kahalalide F, a new anticancer agent: Single and multiple dosing regimens in the rat. Cancer Chemother. Pharmacol. 2002, 50, 333–340. [Google Scholar] [CrossRef] [PubMed]
- Nastrucci, C.; Cesario, A.; Russo, P. Anticancer drug discovery from the marine environment. Recent Pat. Anti-Cancer Drug Discov. 2012, 7, 218–232. [Google Scholar] [CrossRef]
- Fenical, W.; Jensen, P.R.; Palladino, M.A.; Lam, K.S.; Lloyd, G.K.; Potts, B.C. Discovery and development of the anticancer agent salinosporamide A (NPI-0052). Bioorg. Med. Chem. 2009, 17, 2175–2180. [Google Scholar] [CrossRef]
- Greig, S.L. Osimertinib: First global approval. Drugs 2016, 76, 263–273. [Google Scholar] [CrossRef]
- Watanabe, J.; Minami, M.; Kobayashi, M. Antitumor activity of TZT-1027 (Soblidotin). Anticancer Res. 2006, 26, 1973–1981. [Google Scholar] [PubMed]
- Syed, Y.Y. Selinexor: First global approval. Drugs 2019, 79, 1485–1494. [Google Scholar] [CrossRef] [PubMed]
- Podar, K.; Shah, J.; Chari, A.; Richardson, P.G.; Jagannath, S. Selinexor for the treatment of multiple myeloma. Expert Opin. Pharmacother. 2020, 21, 399–408. [Google Scholar] [CrossRef]
- Leong, H.; Bonk, M.E. Therapeutics: Bendamustine (Treanda) for chronic lymphocytic leukemia: A brief overview. Pharm. Ther. 2009, 34, 73. [Google Scholar]
- Cohen, M.H.; Williams, G.A.; Sridhara, R.; Chen, G.; Pazdur, R. FDA drug approval summary: Gefitinib (ZD1839)(Iressa®) tablets. Oncologist 2003, 8, 303–306. [Google Scholar] [CrossRef]
- Kim, E.S. Abemaciclib: First global approval. Drugs 2017, 77, 2063–2070. [Google Scholar] [CrossRef] [PubMed]
- Voli, L.A.; Mamyrbékova, J.A.; Bazureau, J.-P. Abemaciclib, a recent novel FDA-Approved small molecule inhibiting cyclin-dependant kinase 4/6 for the treatment of metastatic breast cancer: A mini-review. Open J. Med. Chem. 2020, 10, 128–138. [Google Scholar] [CrossRef]
- Shah, A.; Bloomquist, E.; Tang, S.; Fu, W.; Bi, Y.; Liu, Q.; Yu, J.; Zhao, P.; Palmby, T.R.; Goldberg, K.B. FDA Approval: Ribociclib for the Treatment of Postmenopausal Women with Hormone Receptor–Positive HER2-Negative Advanced or Metastatic Breast CancerRibociclib for HR+ HER2− Metastatic Breast, Cancer. Clin. Cancer Res. 2018, 24, 2999–3004. [Google Scholar] [CrossRef] [PubMed]
- Dimou, A.; Syrigos, K.N.; Saif, M.W. Is there a role for mitomycin C in metastatic colorectal, cancer? Expert opinion on investigational drugs. Expert Opin. Investig. Drugs 2010, 19, 723–735. [Google Scholar] [CrossRef]
- Benayoun, L.; Schaffer, M.; Bril, R.; Gingis-Velitski, S.; Segal, E.; Nevelsky, A.; Satchi-Fainaro, R.; Shaked, Y. Porfimer-sodium (Photofrin-II) in combination with ionizing radiation inhibits tumor-initiating cell proliferation improves glioblastoma treatment efficacy. Cancer Biol. Ther. 2013, 14, 64–74. [Google Scholar] [CrossRef]
- Hörmann, V.; Kumi-Diaka, J.; Durity, M.; Rathinavelu, A. Anticancer activities of genistein-topotecan combination in prostate cancer, cells. J. Cell. Mol. Med. 2012, 16, 2631–2636. [Google Scholar] [CrossRef]
- Shah, A.; Mangaonkar, A. Idelalisib: A novel PI3Kδ inhibitor for chronic lymphocytic, leukemia. Ann. Pharmacother. 2015, 49, 1162–1170. [Google Scholar] [CrossRef]
- Zirlik, K.; Veelken, H. Idelalisib. Small Mol. Hematol. 2018, 243–264. [Google Scholar] [CrossRef]
- Markham, A.; Dhillon, S. Acalabrutinib: First global, approval. Drugs 2018, 78, 139–145. [Google Scholar] [CrossRef] [PubMed]
- Singh, H.; Brave, M.; Beaver, J.A.; Cheng, J.; Tang, S.; Zahalka, E.; Palmby, T.R.; Venugopal, R.; Song, P.; Liu, Q. Food Drug Administration approval: Cabozantinib for the treatment of advanced renal cell, carcinoma. Ther. Adv. Urol. 2017, 23, 330–335. [Google Scholar] [CrossRef] [PubMed]
- Duke, E.S.; Barone, A.K.; Chatterjee, S.; Mishra-Kalyani, P.S.; Shen, Y.L.; Isikwei, E.; Zhao, H.; Bi, Y.; Liu, J.; Rahman, N.A. FDA Approval Summary: Cabozantinib for Differentiated Thyroid, Cancer. Clin. Cancer Res. 2022, 28, 4173–4177. [Google Scholar] [CrossRef] [PubMed]
- Arora, S.; Balasubramaniam, S.; Zhang, W.; Zhang, L.; Sridhara, R.; Spillman, D.; Mathai, J.P.; Scott, B.; Golding, S.J.; Coory, M. FDA Approval Summary: Pembrolizumab plus Lenvatinib for Endometrial Carcinoma a Collaborative International Review under Project Orbis. FDAApproval: Pembrolizumab plus, lenvatinib; Project Orbis. Clin. Cancer Res. 2020, 26, 5062–5067. [Google Scholar] [CrossRef]
- Nair, A.; Reece, K.; Donoghue, M.B.; Yuan, W.; Rodriguez, L.; Keegan, P.; Pazdur, R. FDA supplemental approval summary: Lenvatinib for the treatment of unresectable hepatocellular, carcinoma. Oncologist 2021, 26, e484–e491. [Google Scholar] [CrossRef] [PubMed]
- Lu, J. Oncology: Palbociclib: A first-in-class CDK4/CDK6 inhibitor for the treatment of hormone-receptor positive advanced breast cancer. J. Hematol. Oncol. 2015, 8, 1–3. [Google Scholar] [CrossRef]
- Roskoski, R. Cyclin-dependent protein kinase inhibitors including palbociclib as anticancer drugs. Pharmacol. Res. 2016, 107, 249–275. [Google Scholar] [CrossRef]
- Wright, C.J.; McCormack, P.L. Trametinib: First global approval. Drugs 2013, 73, 1245–1254. [Google Scholar] [CrossRef]
- Zeiser, R.; Andrlová, H.; Meiss, F. Trametinib (GSK1120212). Small Mol. Oncol. 2018, 91–100. [Google Scholar] [CrossRef]
- Odogwu, L.; Mathieu, L.; Blumenthal, G.; Larkins, E.; Goldberg, K.B.; Griffin, N.; Bijwaard, K.; Lee, E.Y.; Philip, R.; Jiang, X. FDA approval summary: Dabrafenib and trametinib for the treatment of metastatic non-small cell lung cancers harboring BRAF V600E mutations. Oncologist 2018, 23, 740–745. [Google Scholar] [CrossRef]
- Dungo, R.T.; Keating, G.M. Afatinib: First global approval. Drugs 2013, 73, 1503–1515. [Google Scholar] [CrossRef] [PubMed]
- Helena, A.Y.; Pao, W. Targeted therapies: Afatinib—New therapy option for EGFR-mutant lung cancer. Nat. Rev. Clin. Oncol. 2013, 10, 551. [Google Scholar]
- Ballantyne, A.D.; Garnock-Jones, K.P. Dabrafenib: First global approval. Drugs 2013, 73, 1367–1376. [Google Scholar] [CrossRef]
- Menzies, A.M.; Long, G.V.; Murali, R. Dabrafenib and its potential for the treatment of metastatic melanoma. Drug Des. Dev. Ther. 2012, 6, 391. [Google Scholar]
- Dhillon, S. Gilteritinib: First global approval. Drugs 2019, 79, 331–339. [Google Scholar] [CrossRef]
- Perl, A.E.; Martinelli, G.; Cortes, J.E.; Neubauer, A.; Berman, E.; Paolini, S.; Montesinos, P.; Baer, M.R.; Larson, R.A.; Ustun, C. Gilteritinib or chemotherapy for relapsed or refractory FLT3-mutated AML. N. Engl. J. Med. 2019, 18, 1728–1740. [Google Scholar] [CrossRef]
- Markham, A. Alpelisib: First global approval. Drugs 2019, 79, 1249–1253. [Google Scholar] [CrossRef]
- Shirley, M. Encorafenib and binimetinib: First global approvals. Drugs 2018, 78, 1277–1284. [Google Scholar] [CrossRef]
- Tran, B.; Cohen, M.S. The discovery and development of binimetinib for the treatment of melanoma. Expert Opin. Drug Discov. 2020, 15, 745–754. [Google Scholar] [CrossRef]
- Kazandjian, D.; Blumenthal, G.M.; Chen, H.-Y.; He, K.; Patel, M.; Justice, R.; Keegan, P.; Pazdur, R. FDA approval summary: Crizotinib for the treatment of metastatic non-small cell lung cancer with anaplastic lymphoma kinase rearrangements. Oncologist 2014, 19, e5–e11. [Google Scholar] [CrossRef]
- O’Bryant, C.L.; Wenger, S.D.; Kim, M.; Thompson, L.A. Crizotinib: A new treatment option for ALK-positive non-small cell lung cancer. Ann. Pharmacother. 2013, 47, 189–197. [Google Scholar] [CrossRef]
- Slayton, R.E.; Park, R.C.; Silverberg, S.G.; Shingleton, H.; Creasman, W.T.; Blessing, J.A. Vincristine, dactinomycin, and cyclophosphamide in the treatment of malignant germ cell tumors of the ovary. A Gynecologic Oncology Group Study (a final report). Cancer 1985, 56, 243–248. [Google Scholar] [CrossRef] [PubMed]
- Cohen, M.H.; Johnson, J.R.; Chen, Y.-F.; Sridhara, R.; Pazdur, R. FDA drug approval summary: Erlotinib (Tarceva®) tablets. Oncologist 2005, 10, 461–466. [Google Scholar] [CrossRef] [PubMed]
- Cameron, F.; Sanford, M. Ibrutinib: First global approval. Drugs 2014, 74, 263–271. [Google Scholar] [CrossRef] [PubMed]
- de Claro, R.A.; McGinn, K.M.; Verdun, N.; Lee, S.-L.; Chiu, H.-J.; Saber, H.; Brower, M.E.; Chang, C.G.; Pfuma, E.; Habtemariam, B. FDA approval: Ibrutinib for patients with previously treated mantle cell lymphoma and previously treated chronic lymphocytic leukemia. Clin. Cancer Res. 2015, 21, 3586–3590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryan, Q.; Ibrahim, A.; Cohen, M.H.; Johnson, J.; Ko, C.-W.; Sridhara, R.; Justice, R.; Pazdur, R. FDA drug approval summary: Lapatinib in combination with capecitabine for previously treated metastatic breast cancer that overexpresses HER-2. Oncologist 2008, 13, 1114–1119. [Google Scholar] [CrossRef] [PubMed]
- Scott, L. Larotrectinib: First global approval. Drugs 2019, 79, 201–206. [Google Scholar] [CrossRef]
- Federman, N.; McDermott, R. Larotrectinib, a highly selective tropomyosin receptor kinase (TRK) inhibitor for the treatment of TRK fusion cancer. Expert Rev. Clin. Pharmacol. 2019, 12, 931–939. [Google Scholar] [CrossRef]
- Neerman, M.F.; Chen, H.-T.; Parrish, A.R.; Simanek, E.E. Reduction of drug toxicity using dendrimers based on melamine. Mol. Pharm. 2004, 1, 390–393. [Google Scholar] [CrossRef]
- Weinstein, G.D. Drugs five years later: Methotrexate. Ann. Intern. Med. 1977, 86, 199–204. [Google Scholar] [CrossRef]
- Moshikur, R.M.; Chowdhury, M.R.; Wakabayashi, R.; Tahara, Y.; Moniruzzaman, M.; Goto, M.J. Ionic liquids with methotrexate moieties as a potential anticancer prodrug: Synthesis, characterization and solubility evaluation. J. Mol. Liq. 2019, 278, 226–233. [Google Scholar] [CrossRef]
- DeRemer, D.L.; Ustun, C.; Natarajan, K. Nilotinib: A second-generation tyrosine kinase inhibitor for the treatment of chronic myelogenous leukemia. Clin. Ther. 2008, 30, 1956–1975. [Google Scholar] [CrossRef] [PubMed]
- Harris, M.G.; Coleman, S.G.; Faulds, D.; Chrisp, P. Nilutamide: A review of its pharmacodynamic and pharmacokinetic properties, and therapeutic efficacy in prostate cancer. Drugs Aging 1993, 3, 9–25. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.; Ison, G.; McKee, A.E.; Zhang, H.; Tang, S.; Gwise, T.; Sridhara, R.; Lee, E.; Tzou, A.; Philip, R. FDA Approval Summary: Olaparib Monotherapy in Patients with Deleterious Germline BRCA-Mutated Advanced Ovarian Cancer Treated with Three or More Lines of ChemotherapyOlaparib for Advanced Ovarian Cancer with BRCA Mutation. Clin. Cancer Res. 2015, 21, 4257–4261. [Google Scholar] [CrossRef] [PubMed]
- Caulfield, S.E.; Davis, C.C.; Byers, K.F. Olaparib: A novel therapy for metastatic breast cancer in patients with a BRCA1/2 mutation. J. Adv. Pract. Oncol. 2019, 10, 167. [Google Scholar] [PubMed]
- Lagoja, I.M. Pyrimidine as constituent of natural biologically active compounds. Chem. Biodivers. 2005, 2, 1–50. [Google Scholar] [CrossRef]
- Albratty, M.; Alhazmi, H.A. Novel pyridine and pyrimidine derivatives as promising anticancer agents: A review. Arab. J. Chem. 2022, 15, 103846. [Google Scholar] [CrossRef]
- Fathalla, O.A.; Mohamed, N.A.; El-Serwy, W.S.; AbdelHamid, H.F.; El-Moez, A.; Sherein, I.; Soliman, A.-m.M. Synthesis of some new pyrimidine derivatives and evaluation of their anticancer and antibacterial activities. Res. Chem. Intermed. 2013, 39, 821–841. [Google Scholar] [CrossRef]
- Ahmed, M.H.; El-Hashash, M.A.; Marzouk, M.I.; El-Naggar, A.M. Synthesis and antitumor activity of some nitrogen heterocycles bearing pyrimidine moiety. J. Heterocycl. Chem. 2020, 57, 3412–3427. [Google Scholar] [CrossRef]
- Gupta, S.; Bartwal, G.; Singh, A.; Tanwar, J.; Khurana, J. Design, synthesis and biological evaluation of spiro-isoquinoline-pyrimidine derivatives as anticancer agents against MCF-7 cancer cell lines. Results Chem. 2022, 4, 100386. [Google Scholar] [CrossRef]
- Al-Issa, S. Synthesis and anticancer activity of some fused pyrimidines and related heterocycles. Saudi Pharm. J. 2013, 21, 305–316. [Google Scholar] [CrossRef] [PubMed]
- Osmaniye, D.; Hıdır, A.; Sağlık, B.N.; Levent, S.; Özkay, Y.; Kaplancıklı, Z.A. Synthesis of New Pyrimidine-Triazole Derivatives and Investigation of Their Anticancer Activities. Chem. Biodivers. 2022, 19, e202200216. [Google Scholar] [CrossRef]
- Qin, W.-W.; Sang, C.-Y.; Zhang, L.-L.; Wei, W.; Tian, H.-Z.; Liu, H.-X.; Chen, S.-W.; Hui, L. Synthesis and biological evaluation of 2, 4-diaminopyrimidines as selective Aurora A kinase inhibitors. Eur. J. Med. Chem. 2015, 95, 174–184. [Google Scholar] [CrossRef] [PubMed]
- Venturini Filho, E.; Pina, J.W.; Antoniazi, M.K.; Loureiro, L.B.; Ribeiro, M.A.; Pinheiro, C.B.; Guimarães, C.J.; de Oliveira, F.C.; Pessoa, C.; Taranto, A.G. Synthesis, docking, machine learning and antiproliferative activity of the 6-ferrocene/heterocycle-2-aminopyrimidine and 5-ferrocene-1H-Pyrazole derivatives obtained by microwave-assisted Atwal reaction as potential anticancer agents. Bioorg. Med. Chem. Lett. 2021, 48, 128240. [Google Scholar] [CrossRef]
- Mohi El-Deen, E.M.; Anwar, M.M.; El-Gwaad, A.A.A.; Karam, E.A.; El-Ashrey, M.K.; Kassab, R.R. Novel Pyridothienopyrimidine Derivatives: Design, Synthesis and Biological Evaluation as Antimicrobial and Anticancer Agents. Molecules 2022, 27, 803. [Google Scholar] [CrossRef] [PubMed]
- Al-Anazi, M.; Khairuddean, M.; Al-Najjar, B.O.; Alidmat, M.M.; Kamal, N.N.S.N.M.; Muhamad, M. Synthesis, anticancer activity and docking studies of pyrazoline and pyrimidine derivatives as potential epidermal growth factor receptor (EGFR) inhibitors. Arab. J. Chem. 2022, 15, 103864. [Google Scholar] [CrossRef]
- El-Metwally, S.A.; Abou-El-Regal, M.M.; Eissa, I.H.; Mehany, A.B.; Mahdy, H.A.; Elkady, H.; Elwan, A.; Elkaeed, E.B. Discovery of thieno [2, 3-d] pyrimidine-based derivatives as potent VEGFR-2 kinase inhibitors and anti-cancer agents. Bioorg. Chem. 2021, 112, 104947. [Google Scholar] [CrossRef]
- Madia, V.N.; Nicolai, A.; Messore, A.; De Leo, A.; Ialongo, D.; Tudino, V.; Saccoliti, F.; De Vita, D.; Scipione, L.; Artico, M.; et al. Design, synthesis and biological evaluation of new pyrimidine derivatives as anticancer agents. Molecules 2021, 26, 771. [Google Scholar] [CrossRef]
- Marella, A.; Tanwar, O.P.; Saha, R.; Ali, M.R.; Srivastava, S.; Akhter, M.; Shaquiquzzaman, M.; Alam, M.M. Quinoline: A versatile heterocyclic. Saudi Pharm. J. 2013, 21, 1–12. [Google Scholar] [CrossRef]
- Hamdy, R.; Elseginy, S.A.; Ziedan, N.I.; Jones, A.T.; Westwell, A.D. New quinoline-based heterocycles as anticancer agents targeting bcl-2. Molecules 2019, 24, 1274. [Google Scholar] [CrossRef]
- Jin, X.-Y.; Chen, H.; Li, D.-D.; Li, A.-L.; Wang, W.-Y.; Gu, W. Design, synthesis, and anticancer evaluation of novel quinoline derivatives of ursolic acid with hydrazide, oxadiazole, and thiadiazole moieties as potent MEK inhibitors. J. Enzym. Inhib. Med. Chem. 2019, 34, 955–972. [Google Scholar] [CrossRef] [PubMed]
- Katariya, K.D.; Shah, S.R.; Reddy, D. Anticancer, antimicrobial activities of quinoline based hydrazone analogues: Synthesis, characterization and molecular docking. Bioorg. Chem. 2020, 94, 103406. [Google Scholar] [CrossRef] [PubMed]
- George, R.F.; Samir, E.M.; Abdelhamed, M.N.; Abdel-Aziz, H.A.; Abbas, S.E. Synthesis and anti-proliferative activity of some new quinoline based 4, 5-dihydropyrazoles and their thiazole hybrids as EGFR inhibitors. Bioorg. Chem. 2019, 83, 186–197. [Google Scholar] [CrossRef] [PubMed]
- Köprülü, T.K.; Ökten, S.; Atalay, V.E.; Tekin, Ş.; Çakmak, O. Biological activity and molecular docking studies of some new quinolines as potent anticancer agents. Med. Oncol. 2021, 38, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Ramya, P.S.; Guntuku, L.; Angapelly, S.; Karri, S.; Digwal, C.S.; Babu, B.N.; Naidu, V.; Kamal, A. Curcumin inspired 2-chloro/phenoxy quinoline analogues: Synthesis and biological evaluation as potential anticancer agents. Bioorg. Med. Chem. Lett. 2018, 28, 892–898. [Google Scholar] [CrossRef]
- Upadhyay, K.D.; Dodia, N.M.; Khunt, R.C.; Chaniara, R.S.; Shah, A.K. Synthesis and biological screening of pyrano [3, 2-c] quinoline analogues as anti-inflammatory and anticancer agents. ACS Med. Chem. Lett. 2018, 9, 283–288. [Google Scholar] [CrossRef] [PubMed]
- Hagras, M.; El Deeb, M.A.; Elzahabi, H.S.; Elkaeed, E.B.; Mehany, A.B.; Eissa, I.H. Discovery of new quinolines as potent colchicine binding site inhibitors: Design, synthesis, docking studies, and anti-proliferative evaluation. J. Enzym. Inhib. Med. Chem. 2021, 36, 640–658. [Google Scholar] [CrossRef]
- Mirzaei, S.; Hadizadeh, F.; Eisvand, F.; Mosaffa, F.; Ghodsi, R. Synthesis, structure-activity relationship and molecular docking studies of novel quinoline-chalcone hybrids as potential anticancer agents and tubulin inhibitors. J. Mol. Struct. 2020, 1202, 127310. [Google Scholar] [CrossRef]
- Chate, A.V.; Kamdi, S.P.; Bhagat, A.N.; Jadhav, C.K.; Nipte, A.; Sarkate, A.P.; Tiwari, S.V.; Gill, C.H. Design, Synthesis and SAR Study of Novel Spiro [Pyrimido[5,4-b]Quinoline-10,5′-Pyrrolo[2,3-d]Pyrimidine] Derivatives as Promising Anticancer Agents. J. Heterocycl. Chem. 2018, 55, 2297–2302. [Google Scholar] [CrossRef]
- Ziarani, G.; Moradi, R.; Lashgari, N.; Kruger, H.G. Metal-Free Synthetic Organic Dyes; Elsevier: Amsterdam, The Netherlands, 2018. [Google Scholar]
- Murali, K.; Sparkes, H.A.; Prasad, K.J.R. Synthesis of hetero annulated isoxazolo-, pyrido-and pyrimido carbazoles: Screened for in vitro antitumor activity and structure activity relationships, a novel 2-amino-4-(3′-bromo-4′-methoxyphenyl)-8-chloro-11H-pyrimido [4, 5-a] carbazole as an antitumor agent. Eur. J. Med. Chem. 2017, 128, 319–331. [Google Scholar]
- Wang, Y.-Q.; Li, X.-H.; He, Q.; Chen, Y.; Xie, Y.-Y.; Ding, J.; Miao, Z.-H.; Yang, C.-H. Design, synthesis and biological evaluation of substituted 11H-benzo [a] carbazole-5-carboxamides as novel antitumor agents. Eur. J. Med. Chem. 2011, 46, 5878–5884. [Google Scholar] [CrossRef]
- Debray, J.; Zeghida, W.; Baldeyrou, B.; Mahieu, C.; Lansiaux, A.; Demeunynck, M. Montmorillonite K-10 catalyzed cyclization of N-ethoxycarbonyl-N′-arylguanidines: Access to pyrimido [4,5-c] carbazole and pyrimido [5,4-b] indole derivatives. Bioorg. Med. Chem. Lett. 2010, 20, 4244–4247. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Wu, Y.; Liu, Y.; Chen, X.; Hu, L. Novel carbazole sulfonamide derivatives of antitumor agent: Synthesis, antiproliferative activity and aqueous solubility. Bioorg. Med. Chem. Lett. 2017, 27, 261–265. [Google Scholar] [CrossRef] [PubMed]
- Arya, K.R.; Rajendra Prasad, K.J. Rational eco-compatible synthesis and biological screening of new highly functionalized pyrido [2, 3-a] carbazole derivatives: A novel class of antioxidant and anticancer agents. Synth. Commun. 2018, 48, 1465–1481. [Google Scholar] [CrossRef]
- Padmaja, P.; Rao, G.K.; Indrasena, A.; Reddy, B.V.S.; Patel, N.; Shaik, A.B.; Reddy, N.; Dubey, P.K.; Bhadra, M.P. Synthesis and biological evaluation of novel pyrano [3,2-c] carbazole derivatives as anti-tumor agents inducing apoptosis via tubulin polymerization inhibition. Org. Biomol. Chem. 2015, 13, 1404–1414. [Google Scholar] [CrossRef]
- Patel, M.; Pandey, N.; Timaniya, J.; Parikh, P.; Chauhan, A.; Jain, N.; Patel, K. Coumarin–carbazole based functionalized pyrazolines: Synthesis, characterization, anticancer investigation and molecular docking. RSC Adv. 2021, 11, 27627–27644. [Google Scholar] [CrossRef]
- Huang, W.; Gao, Z.; Zhang, Z.; Fang, W.; Wang, Z.; Wan, Z.; Shi, L.; Wang, K.; Ke, S. Selective and effective anticancer agents: Synthesis, biological evaluation and structure–activity relationships of novel carbazole derivatives. Bioorg. Chem. 2021, 113, 104991. [Google Scholar] [CrossRef]
- Vairavelu, L.; Zeller, M.; Prasad, K.R. Solvent-free synthesis of heteroannulated carbazoles: A novel class of anti-tumor agents. Bioorg. Chem. 2014, 54, 12–20. [Google Scholar] [CrossRef]
- Altaf, A.A.; Shahzad, A.; Gul, Z.; Rasool, N.; Badshah, A.; Lal, B.; Khan, E. A review on the medicinal importance of pyridine derivatives. J. Drug Des. Med. Chem. 2015, 1, 1–11. [Google Scholar]
- Gomha, S.M.; Abdelrazek, F.M.; Abdelrahman, A.H.; Metz, P. Synthesis of some new pyridine-based heterocyclic compounds with anticipated antitumor activity. J. Heterocycl. Chem. 2018, 55, 1729–1737. [Google Scholar] [CrossRef]
- Fayed, E.A.; Sabour, R.; Harras, M.F.; Mehany, A. Design, synthesis, biological evaluation and molecular modeling of new coumarin derivatives as potent anticancer agents. Med. Chem. Res. 2019, 28, 1284–1297. [Google Scholar] [CrossRef]
- Nagender, P.; Kumar, R.N.; Reddy, G.M.; Swaroop, D.K.; Poornachandra, Y.; Kumar, C.G.; Narsaiah, B. Synthesis of novel hydrazone and azole functionalized pyrazolo [3, 4-b] pyridine derivatives as promising anticancer agents. Bioorg. Med. Chem. Lett. 2016, 26, 4427–4432. [Google Scholar] [CrossRef] [PubMed]
- El-Naggar, M.; Almahli, H.; Ibrahim, H.S.; Eldehna, W.M.; Abdel-Aziz, H.A. Pyridine-ureas as potential anticancer agents: Synthesis and in vitro biological evaluation. Molecules 2018, 23, 1459. [Google Scholar] [CrossRef]
- Dinda, J.; Nandy, A.; Rana, B.K.; Bertolasi, V.; Saha, K.D.; Bielawski, C.W. Cytotoxicity of silver (I), gold (I) and gold (III) complexes of a pyridine wingtip substituted annelated N-heterocyclic carbene. RSC Adv. 2014, 4, 60776–60784. [Google Scholar] [CrossRef]
- El-Gohary, N.; Gabr, M.; Shaaban, M. Synthesis, molecular modeling and biological evaluation of new pyrazolo [3, 4-b] pyridine analogs as potential antimicrobial, antiquorum-sensing and anticancer agents. Bioorg. Chem. 2019, 89, 102976. [Google Scholar] [CrossRef] [PubMed]
- Sangani, C.B.; Makawana, J.A.; Zhang, X.; Teraiya, S.B.; Lin, L.; Zhu, H.-L. Design, synthesis and molecular modeling of pyrazole–quinoline–pyridine hybrids as a new class of antimicrobial and anticancer agents. Eur. J. Med. Chem. 2014, 76, 549–557. [Google Scholar] [CrossRef]
- Elzahabi, H.S. Synthesis, characterization of some benzazoles bearing pyridine moiety: Search for novel anticancer agents. Eur. J. Med. Chem. 2011, 46, 4025–4034. [Google Scholar] [CrossRef]
- Zheng, S.; Zhong, Q.; Mottamal, M.; Zhang, Q.; Zhang, C.; LeMelle, E.; McFerrin, H.; Wang, G. Design, synthesis, and biological evaluation of novel pyridine-bridged analogues of combretastatin-A4 as anticancer agents. J. Med. Chem. 2014, 57, 3369–3381. [Google Scholar] [CrossRef] [PubMed]
- Abbas, I.; Gomha, S.; Elaasser, M.; Bauomi, M. Synthesis and biological evaluation of new pyridines containing imidazole moiety as antimicrobial and anticancer agents. Turk. J. Chem. 2015, 39, 334–346. [Google Scholar] [CrossRef]
- Ivasechko, I.; Yushyn, I.; Roszczenko, P.; Senkiv, J.; Finiuk, N.; Lesyk, D.; Holota, S.; Czarnomysy, R.; Klyuchivska, O.; Khyluk, D. Development of Novel Pyridine-Thiazole Hybrid Molecules as Potential Anticancer Agents. Molecules 2022, 27, 6219. [Google Scholar] [CrossRef]
- Debus, H. Ueber die einwirkung des ammoniaks auf glyoxal. Ann. Chem. Pharm. 1858, 107, 199–208. [Google Scholar] [CrossRef]
- Liu, L.-X.; Wang, X.-Q.; Zhou, B.; Yang, L.-J.; Li, Y.; Zhang, H.-B.; Yang, X.-D. Synthesis and antitumor activity of novel N-substituted carbazole imidazolium salt derivatives. Sci. Rep. 2015, 5, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Ruzi, Z.; Nie, L.; Bozorov, K.; Zhao, J.; Aisa, H.A. Synthesis and anticancer activity of ethyl 5-amino-1-N-substituted-imidazole-4-carboxylate building blocks. Arch. Pharm. 2021, 354, 2000470. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Kumar, R.; Pandrala, M.; Kaur, P.; Gupta, S.; Tailor, D.; Malhotra, S.V.; Salunke, D.B. Facile synthesis of C6-substituted benz [4,5] imidazo [1,2-a] quinoxaline derivatives and their anticancer evaluation. Arch. Der Pharm. 2021, 354, 2000393. [Google Scholar] [CrossRef]
- Yang, X.-D.; Wan, W.-C.; Deng, X.-Y.; Li, Y.; Yang, L.-J.; Li, L.; Zhang, H.-B. Design, synthesis and cytotoxic activities of novel hybrid compounds between 2-phenylbenzofuran and imidazole. Bioorg. Med. Chem. Lett. 2012, 22, 2726–2729. [Google Scholar] [CrossRef]
- Song, W.-J.; Yang, X.-D.; Zeng, X.-H.; Xu, X.-L.; Zhang, G.-L.; Zhang, H.-B. Synthesis and cytotoxic activities of novel hybrid compounds of imidazole scaffold-based 2-substituted benzofurans. RSC Adv. 2012, 2, 4612–4615. [Google Scholar] [CrossRef]
- Khayyat, A.N.; Mohamed, K.O.; Malebari, A.M.; El-Malah, A. Design, Synthesis, and Antipoliferative Activities of Novel Substituted Imidazole-Thione Linked Benzotriazole Derivatives. Molecules 2021, 26, 5983. [Google Scholar] [CrossRef]
- Singh, A.K.; Kumar, A.; Singh, H.; Sonawane, P.; Paliwal, H.; Thareja, S.; Pathak, P.; Grishina, M.; Jaremko, M.; Emwas, A.-H. Concept of hybrid drugs and recent advancements in anticancer hybrids. Pharmaceuticals 2022, 15, 1071. [Google Scholar] [CrossRef]
- Kalra, S.; Joshi, G.; Kumar, M.; Arora, S.; Kaur, H.; Singh, S.; Munshi, A.; Kumar, R. Anticancer potential of some imidazole and fused imidazole derivatives: Exploring the mechanism via epidermal growth factor receptor (EGFR) inhibition. RSC Med. Chem. 2020, 11, 923–939. [Google Scholar] [CrossRef]
- Taheri, B.; Taghavi, M.; Zarei, M.; Chamkouri, N.; Mojaddami, A. Imidazole and carbazole derivatives as potential anticancer agents: Molecular docking studies and cytotoxic activity evaluation. Bul. Chem. Soc. Ethiop. 2020, 34, 377–384. [Google Scholar] [CrossRef]
- Oskuei, S.R.; Mirzaei, S.; Jafari-Nik, M.R.; Hadizadeh, F.; Eisvand, F.; Mosaffa, F.; Ghodsi, R. Design, synthesis and biological evaluation of novel imidazole-chalcone derivatives as potential anticancer agents and tubulin polymerization inhibitors. Bioorg. Chem. 2021, 112, 104904. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.-Q.; Liu, L.-X.; Li, Y.; Sun, C.-J.; Chen, W.; Li, L.; Zhang, H.-B.; Yang, X.-D. Design, synthesis and biological evaluation of novel hybrid compounds of imidazole scaffold-based 2-benzylbenzofuran as potent anticancer agents. Eur. J. Med. Chem. 2013, 62, 111–121. [Google Scholar] [CrossRef]
- Du, Q.R.; Li, D.D.; Pi, Y.Z.; Li, J.R.; Sun, J.; Fang, F.; Zhong, W.Q.; Gong, H.B.; Zhu, H.L. Novel 1,3,4-oxadiazole thioether derivatives targeting thymidylate synthase as dual anticancer/antimicrobial agents. Bioorg. Med. Chem. 2013, 21, 2286–2297. [Google Scholar] [CrossRef] [PubMed]
- Shrivastava, N.; Naim, M.J.; Alam, M.J.; Nawaz, F.; Ahmed, S.; Alam, O. Benzimidazole scaffold as anticancer agent: Synthetic approaches and structure–activity relationship. Arch. Pharm. 2017, 350, e201700040. [Google Scholar] [CrossRef] [PubMed]
- Rawat, S.; Ghate, M. Potential Anticancer Agents From Benzimidazole Derivatives. Nveo-Nat. Volatiles Essent. Oils J. NVEO 2021, 8, 4109–4120. [Google Scholar]
- Nicolai, A.; Madia, V.N.; Messore, A.; De Vita, D.; De Leo, A.; Ialongo, D.; Tudino, V.; Tortorella, E.; Scipione, L.; Taurone, S.; et al. Anti-Tumoral Effects of a (1 H-Pyrrol-1-yl) Methyl-1 H-Benzoimidazole Carbamate Ester Derivative on Head and Neck Squamous Carcinoma Cell Lines. Pharmaceuticals 2021, 14, 564. [Google Scholar] [CrossRef]
- Shao, K.P.; Zhang, X.Y.; Chen, P.J.; Xue, D.Q.; He, P.; Ma, L.Y.; Zheng, J.X.; Zhang, Q.R.; Liu, H.M. Synthesis and biological evaluation of novel pyrimidine-benzimidazol hybrids as potential anticancer agents. Bioorg. Med. Chem. Lett. 2014, 24, 3877–3881. [Google Scholar] [CrossRef] [PubMed]
- Husain, A.; Rashid, M.; Shaharyar, M.; Siddiqui, A.A.; Mishra, R. Benzimidazole clubbed with triazolo-thiadiazoles and triazolo-thiadiazines: New anticancer agents. Eur. J. Med. Chem. 2013, 62, 785–798. [Google Scholar] [CrossRef]
- Sana, S.; Reddy, V.G.; Reddy, T.S.; Tokala, R.; Kumar, R.; Bhargava, S.K.; Shankaraiah, N. Cinnamide derived pyrimidine-benzimidazole hybrids as tubulin inhibitors: Synthesis, in silico and cell growth inhibition studies. Bioorg. Chem. 2021, 110, 104765. [Google Scholar] [CrossRef]
- Sireesha, R.; Sreenivasulu, R.; Chandrasekhar, C.; Jadav, S.S.; Pavani, Y.; Rao, M.V.B.; Subbarao, M. Design, synthesis, anti-cancer evaluation and binding mode studies of benzimidazole/benzoxazole linked β-carboline derivatives. J. Mol. Struct. 2021, 1226, 129351. [Google Scholar] [CrossRef]
- Shi, L.; Wu, T.-T.; Wang, Z.; Xue, J.-Y.; Xu, Y.-G. Discovery of quinazolin-4-amines bearing benzimidazole fragments as dual inhibitors of c-Met and VEGFR-2. Bioorg. Med. Chem. 2014, 22, 4735–4744. [Google Scholar] [CrossRef] [PubMed]
- Romero-Castro, A.; León-Rivera, I.; Avila-Rojas, L.C.; Navarrete-Vázquez, G.; Nieto-Rodríguez, A. Synthesis and preliminary evaluation of selected 2-aryl-5(6)-nitro- 1H-benzimidazole derivatives as potential anticancer agents. Arch. Pharm. Chemi. 2011, 34, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Sivaramakarthikeyan, R.; Iniyaval, S.; Saravanan, V.; Lim, W.-M.; Mai, C.-W.; Ramalingan, C. Molecular hybrids integrated with benzimidazole and pyrazole structural motifs: Design, synthesis, biological evaluation, and molecular docking studies. ACS Omega 2020, 5, 10089–10098. [Google Scholar] [CrossRef] [PubMed]
- Perin, N.; Hok, L.; Beč, A.; Persoons, L.; Vanstreels, E.; Daelemans, D.; Vianello, R.; Hranjec, M. N-substituted benzimidazole acrylonitriles as in vitro tubulin polymerization inhibitors: Synthesis, biological activity and computational analysis. Eur. J. Med. Chem. 2021, 211, 113003. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.-T.; Jiang, Z.; Shen, J.-J.; Yi, H.; Zhan, Y.-C.; Sha, M.-Q.; Wang, Z.; Xue, S.-T.; Li, Z.-R. Design, synthesis and biological evaluation of novel benzimidazole-2-substituted phenyl or pyridine propyl ketene derivatives as antitumour agents. Eur. J. Med. Chem. 2016, 114, 328–336. [Google Scholar] [CrossRef]
- Holiyachi, M.; Shastri, S.L.; Chougala, B.M.; Shastri, L.A.; Joshi, S.D.; Dixit, S.R.; Nagarajaiah, H.; Sunagar, V.A. Design, Synthesis and Structure-Activity Relationship Study of Coumarin Benzimidazole Hybrid as Potent Antibacterial and Anticancer Agents. Chem. Sel. 2016, 1, 4638–4644. [Google Scholar] [CrossRef]
- Tahlan, S.; Ramasamy, K.; Lim, S.M.; Shah, S.A.A.; Mani, V.; Narasimhan, B. Design, synthesis and therapeutic potential of 3-(2-(1H-benzo [d] imidazol-2-ylthio) acetamido)-N-(substituted phenyl) benzamide analogues. Chem. Cent. J. 2018, 12, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.; Yang, Q.; Liu, T.; Li, K.; Liu, Y.; Zhu, C.; Zhang, Z.; Li, L.; Zhang, C.; Xie, M. Design, synthesis and in vitro evaluation of 6-amide-2-aryl benzoxazole/benzimidazole derivatives against tumor cells by inhibiting VEGFR-2 kinase. Eur. J. Med. Chem. 2019, 179, 147–165. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Gan, L.; Zhang, Y.; Zhang, F.; Wang, G.; Jin, L.; Geng, R. Review on supermolecules as chemical drugs. Sci. China Ser. B Chem. 2009, 52, 415–458. [Google Scholar] [CrossRef]
- Juan-Juan, C.; Yan, W.; Hui-Zhen, Z.; Cheng-He, Z.; Rong-Xia, G.; Qing-Gang, J. Recent advances in researches of triazole-based supramolecular chemistry and medicinal drugs. Chem. J. Chin. Univ. Chin. 2011, 32, 1970–1985. [Google Scholar]
- Kurumurthy, C.; Rao, P.S.; Rao, P.S.; Narsaiah, B.; Velatooru, L.; Pamanji, R.; Rao, J.V. Synthesis of novel alkyltriazole tagged pyrido [2, 3-d] pyrimidine derivatives and their anticancer activity. Eur. J. Med. Chem. 2011, 46, 3462–3468. [Google Scholar] [CrossRef]
- Al-Blewi, F.; Shaikh, S.A.; Naqvi, A.; Aljohani, F.; Aouad, M.R.; Ihmaid, S.; Rezki, N. Design and synthesis of novel imidazole derivatives possessing triazole pharmacophore with potent anticancer activity, and in silico ADMET with GSK-3β molecular docking investigations. Int. J. Mol. Sci. 2021, 22, 1162. [Google Scholar] [CrossRef] [PubMed]
- Bozorov, K.; Zhao, J.; Aisa, H.A. 1, 2, 3-Triazole-containing hybrids as leads in medicinal chemistry: A recent overview. Bioorg. Med. Chem. 2019, 27, 3511–3531. [Google Scholar] [CrossRef] [PubMed]
- Djemoui, A.; Naouri, A.; Ouahrani, M.R.; Djemoui, D.; Lahcene, S.; Lahrech, M.B.; Boukenna, L.; Albuquerque, H.M.; Saher, L.; Rocha, D.H. A step-by-step synthesis of triazole-benzimidazole-chalcone hybrids: Anticancer activity in human cells+. J. Mol. Struct. 2020, 1204, 127487. [Google Scholar] [CrossRef]
- Suryanarayana, K.; Robert, A.R.; Kerru, N.; Pooventhiran, T.; Thomas, R.; Maddila, S.; Jonnalagadda, S.B. Design, synthesis, anticancer activity and molecular docking analysis of novel dinitrophenylpyrazole bearing 1, 2, 3-triazoles. J. Mol. Struct. 2021, 1243, 130865. [Google Scholar] [CrossRef]
- Chandrashekhar, M.; Nayak, V.L.; Ramakrishna, S.; Mallavadhani, U.V. Novel triazole hybrids of myrrhanone C, a natural polypodane triterpene: Synthesis, cytotoxic activity and cell based studies. Eur. J. Med. Chem. 2016, 114, 293–307. [Google Scholar] [CrossRef] [PubMed]
- Najafi, Z.; Mahdavi, M.; Safavi, M.; Saeedi, M.; Alinezhad, H.; Pordeli, M.; Kabudanian Ardestani, S.; Shafiee, A.; Foroumadi, A.; Akbarzadeh, T. Synthesis and In Vitro Cytotoxic Activity of Novel Triazole-Isoxazole Derivatives. J. Heterocycl. Chem. 2015, 52, 1743–1747. [Google Scholar] [CrossRef]
- Duan, Y.C.; Ma, Y.C.; Zhang, E.; Shi, X.J.; Wang, M.M.; Ye, X.W.; Liu, H.M. Design and synthesis of novel 1,2,3-triazole-dithiocarbamate hybrids as potential anticancer agents. Eur. J. Med. Chem. 2013, 62, 11–19. [Google Scholar] [CrossRef]
- Kumbhare, R.M.; Dadmal, T.L.; Ramaiah, M.J.; Kishore, K.S.; Pushpa Valli, S.N.; Tiwari, S.K.; Appalanaidu, K.; Rao, Y.K.; Bhadra, M.P. Synthesis and anticancer evaluation of novel triazole linked N-(pyrimidin-2-yl)benzo[d]thiazol-2-amine derivatives as inhibitors of cell survival proteins and inducers of apoptosis in MCF-7 breast cancer cells. Bioorg. Med. Chem. Lett. 2015, 25, 654–658. [Google Scholar] [CrossRef]
- Ma, L.Y.; Wang, B.; Pang, L.P.; Zhang, M.; Wang, S.Q.; Zheng, Y.C.; Shao, K.P.; Xue, D.Q.; Liu, H.M. Design and synthesis of novel 1,2,3-triazole-pyrimidine-urea hybrids as potential anticancer agents. Sci. Rep. 2015, 25, 1124–1128. [Google Scholar] [CrossRef]
- Bush, K.; Bradford, P.A. β-Lactams and β-Lactamase Inhibitors: An Overview. Cold Spring Harb. Perspect. Med. 2016, 6, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Banik, I.; Becker, F.F.; Banik, B.K. Stereoselective synthesis of β-lactams with polyaromatic imines: Entry to new and novel anticancer agents. J. Med. Chem. 2003, 46, 12–15. [Google Scholar] [CrossRef] [PubMed]
- Borazjani, N.; Behzadi, M.; Dadkhah Aseman, M.; Jarrahpour, A.; Rad, J.A.; Kianpour, S.; Iraji, A.; Nabavizadeh, S.M.; Ghanbari, M.M.; Batta, G. Cytotoxicity, anticancer, and antioxidant properties of mono and bis-naphthalimido β-lactam conjugates. Med. Chem. Res. 2020, 29, 1355–1375. [Google Scholar] [CrossRef]
- Payili, N.; Yennam, S.; Rekula, S.R.; Naidu, C.G.; Bobde, Y.; Ghosh, B. Design, Synthesis, and Evaluation of the Anticancer Properties of Novel Quinone Bearing Carbamyl β-Lactam Hybrids. J. Heterocycl. Chem. 2018, 55, 1358–1365. [Google Scholar] [CrossRef]
- Ranjbari, S.; Behzadi, M.; Sepehri, S.; Dadkhah Aseman, M.; Jarrahpour, A.; Mohkam, M.; Ghasemi, Y.; Reza Akbarizadeh, A.; Kianpour, S.; Atioğlu, Z.; et al. Investigations of antiproliferative and antioxidant activity of β-lactam morpholino-1,3,5-triazine hybrids. Bioorg. Med. Chem. 2020, 28, 115408. [Google Scholar] [CrossRef] [PubMed]
- Olazaran, F.n.E.; Rivera, G.; Pérez-Vázquez, A.M.; Morales-Reyes, C.M.; Segura-Cabrera, A.; Balderas-Rentería, I. Biological Evaluation in vitro and in silico of Azetidin-2-one Derivatives as Potential Anticancer Agents. ACS Med. Chem. Lett. 2017, 8, 32–37. [Google Scholar] [CrossRef]
- Rashidi, M.; Islami, M.R.; Esmaeili-Mahani, S. Design and stereoselective synthesis of novel β-lactone and β-lactams as potent anticancer agents on breast cancer cells. Tetrahedron 2018, 74, 835–841. [Google Scholar] [CrossRef]
- Fu, D.-J.; Fu, L.; Liu, Y.-C.; Wang, J.-W.; Wang, Y.-Q.; Han, B.-K.; Li, X.-R.; Zhang, C.; Li, F.; Song, J. Structure-activity relationship studies of β-lactam-azide analogues as orally active antitumor agents targeting the tubulin colchicine site. Sci. Rep. 2017, 7, 1–12. [Google Scholar] [CrossRef]
- Gupta, A.; Halve, A. [Beta]-lactams: A mini review of their biological activity. Int. J. Pharm. Sci. Res. 2015, 6, 978. [Google Scholar]
- Banik, B.K.; Banik, I.; Becker, F.F. Asymmetric synthesis of anticancer β-lactams via Staudinger reaction: Utilization of chiral ketene from carbohydrate. Eur. J. Med. Chem. 2010, 45, 846–848. [Google Scholar] [CrossRef]
- Carr, M.; Greene, L.M.; Knox, A.J.; Lloyd, D.G.; Zisterer, D.M.; Meegan, M.J. Lead identification of conformationally restricted β-lactam type combretastatin analogues: Synthesis, antiproliferative activity and tubulin targeting effects. Eur. J. Med. Chem. 2010, 45, 5752–5766. [Google Scholar] [CrossRef]
- Higashio, Y.; Shoji, T. Heterocyclic compounds such as pyrrole, pyridines, pyrrolidine, piperidine, indole, imidazol and pyrazines. Appl. Catal. A Gen. 2004, 260, 251–259. [Google Scholar] [CrossRef]
- Singla, R.; Prakash, K.; Gupta, K.B.; Upadhyay, S.; Dhiman, M.; Jaitak, V. Identification of novel indole based heterocycles as selective estrogen receptor modulator. Bioorg. Chem. 2018, 79, 72–88. [Google Scholar] [CrossRef]
- Cihan-Üstündağ, G.; Şatana, D.; Özhan, G.; Çapan, G. Indole-based hydrazide-hydrazones and 4-thiazolidinones: Synthesis and evaluation as antitubercular and anticancer agents. J. Enzym. Inhib. Med. Chem. 2016, 31, 369–380. [Google Scholar] [CrossRef] [PubMed]
- He, Z.; Qiao, H.; Yang, F.; Zhou, W.; Gong, Y.; Zhang, X.; Wang, H.; Zhao, B.; Ma, L.; Liu, H.-m. Novel thiosemicarbazone derivatives containing indole fragment as potent and selective anticancer agent. Eur. J. Med. Chem. 2019, 184, 111764. [Google Scholar] [CrossRef] [PubMed]
- Prakash, B.; Amuthavalli, A.; Edison, D.; Sivaramkumar, M.; Velmurugan, R. Novel indole derivatives as potential anticancer agents: Design, synthesis and biological screening. Med. Chem. Res. 2018, 27, 321–331. [Google Scholar] [CrossRef]
- Ali, I.; Mukhtar, S.D.; Hsieh, M.F.; Alothman, Z.A.; Alwarthan, A. Facile synthesis of indole heterocyclic compounds based micellar nano anti-cancer drugs. RSC Adv. 2018, 8, 37905–37914. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Zhang, Y.L.; Fan, J.; Ma, X.; Qin, Y.J.; Zhu, H.L. Novel nicotinoyl pyrazoline derivates bearing N-methyl indole moiety as antitumor agents: Design, synthesis and evaluation. Eur. J. Med. Chem. 2018, 156, 722–737. [Google Scholar] [CrossRef]
- Lafayette, E.A.; de Almeida, S.M.V.; Cavalcanti Santos, R.V.; de Oliveira, J.F.; Amorim, C.; da Silva, R.M.F.; Pitta, M.; Pitta, I.D.R.; de Moura, R.O.; de Carvalho Júnior, L.B.; et al. Synthesis of novel indole derivatives as promising DNA-binding agents and evaluation of antitumor and antitopoisomerase I activities. Eur. J. Med. Chem. 2017, 136, 511–522. [Google Scholar] [CrossRef]
- Song, Y.; Feng, S.; Feng, J.; Dong, J.; Yang, K.; Liu, Z.; Qiao, X. Synthesis and biological evaluation of novel pyrazoline derivatives containing indole skeleton as anti-cancer agents targeting topoisomerase II. Eur. J. Med. Chem. 2020, 200, 112459. [Google Scholar] [CrossRef]
- Bakherad, Z.; Safavi, M.; Sepehri, S.; Fassihi, A.; Sadeghi-Aliabadi, H.; Bakherad, M.; Rastegar, H.; Larijani, B.; Saghaie, L.; Mahdavi, M. Preparation of some novel imidazopyridine derivatives of indole as anticancer agents: One-pot multicomponent synthesis, biological evaluation and docking studies. Res. Chem. Intermed. 2019, 45, 5261–5290. [Google Scholar] [CrossRef]
- Eldehna, W.M.; Hassan, G.S.; Al-Rashood, S.T.; Alkahtani, H.M.; Almehizia, A.A.; Al-Ansary, G.H. Marine-Inspired Bis-indoles Possessing Antiproliferative Activity against Breast Cancer; Design, Synthesis, and Biological Evaluation. Mar. Drugs 2020, 18, 190. [Google Scholar] [CrossRef] [PubMed]
- Sreenivasulu, R.; Tej, M.B.; Jadav, S.S.; Sujitha, P.; Kumar, C.G.; Raju, R.R. Synthesis, anticancer evaluation and molecular docking studies of 2,5-bis(indolyl)-1,3,4-oxadiazoles, Nortopsentin analogues. J. Mol. Struct. 2020, 1208, 127875. [Google Scholar] [CrossRef]
- Naim, M.J.; Alam, O.; Nawaz, F.; Alam, M.J.; Alam, P. Current status of pyrazole and its biological activities. J. Pharm. Bioallied Sci. 2016, 8, 2–17. [Google Scholar] [PubMed]
- Abdelgawad, N.; Ismail, M.F.; Hekal, M.H.; Marzouk, M.I. Design, synthesis, and evaluation of some novel heterocycles bearing pyrazole moiety as potential anticancer agents. J. Heterocycl. Chem. 2019, 56, 1771–1779. [Google Scholar] [CrossRef]
- Nassar, I.F.; El Farargy, A.F.; Abdelrazek, F.M.; Ismail, N.S. Design, synthesis and anticancer evaluation of novel pyrazole, pyrazolo [3, 4-d] pyrimidine and their glycoside derivatives. Nucleosides Nucleotides Nucleic Acids 2017, 36, 275–291. [Google Scholar] [CrossRef] [PubMed]
- El-Sayed, A.A.; Amr, A.E.E.; El-Ziaty, A.K.; Elsayed, E.A. Cytotoxic Effects of Newly Synthesized Heterocyclic Candidates Containing Nicotinonitrile and Pyrazole Moieties on Hepatocellular and Cervical Carcinomas. Molecules 2019, 24, 1965. [Google Scholar] [CrossRef]
- Omran, D.M.; Ghaly, M.A.; El-Messery, S.M.; Badria, F.A.; Abdel-Latif, E.; Shehata, I.A. Targeting hepatocellular carcinoma: Synthesis of new pyrazole-based derivatives, biological evaluation, DNA binding, and molecular modeling studies. Bioorg. Chem. 2019, 88, 102917. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.-L.; Wu, Y.; Liu, C.-J.; Wei, C.-Y.; Tao, J.-C.; Liu, H.-M. Design and stereoselective synthesis of novel isosteviol-fused pyrazolines and pyrazoles as potential anticancer agents. Eur. J. Med. Chem. 2013, 65, 70–82. [Google Scholar] [CrossRef] [PubMed]
- Alam, R.; Alam, M.A.; Panda, A.K. Design, Synthesis, and Cytotoxicity Evaluation of 3-(5-(3-(aryl)-1-phenyl-1H-pyrazol-4-yl)-1-phenyl-4, 5-dihydro-1H-pyrazol-3-yl) pyridine and 5-(3-(aryl)-1-phenyl-1H-pyrazol-4-yl)-3-(pyridin-3-yl)-4, 5-dihydropyrazole-1-carbaldehyde Derivatives as Potential Anticancer Agents. J. Heterocycl. Chem. 2017, 54, 1812–1821. [Google Scholar]
- Harras, M.F.; Sabour, R. Design, synthesis and biological evaluation of novel 1,3,4-trisubstituted pyrazole derivatives as potential chemotherapeutic agents for hepatocellular carcinoma. Bioorg. Chem. 2018, 78, 149–157. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.C.; Gao, M.J.; Huo, Q.; Ma, T.; Wang, Y.; Wu, C.Z. Design, synthesis, and apoptosis-promoting effect evaluation of novel pyrazole with benzo[d]thiazole derivatives containing aminoguanidine units. J. Enzym. Inhib. Med. Chem. 2019, 34, 829–837. [Google Scholar] [CrossRef] [PubMed]
- Afifi, O.S.; Shaaban, O.G.; Abd El Razik, H.A.; Shams El-Dine, S.E.-D.A.; Ashour, F.A.; El-Tombary, A.A.; Abu-Serie, M.M. Synthesis and biological evaluation of purine-pyrazole hybrids incorporating thiazole, thiazolidinone or rhodanine moiety as 15-LOX inhibitors endowed with anticancer and antioxidant potential. Bioorg. Chem. 2019, 87, 821–837. [Google Scholar] [CrossRef] [PubMed]
- Bakhotmah, D.A.; Ali, T.E.; Assiri, M.A.; Yahia, I.S. Synthesis of Some Novel 2-{Pyrano[2,3-c]Pyrazoles-4-Ylidene}Malononitrile Fused with Pyrazole, Pyridine, Pyrimidine, Diazepine, Chromone, Pyrano[2,3-c]Pyrazole and Pyrano[2,3-d]Pyrimidine Systems as Anticancer Agents. Polycycl. Aromat. Compd. 2022, 42, 2136–2150. [Google Scholar] [CrossRef]
- Hameed, A.; Al-Rashida, M.; Uroos, M.; Ali, S.A.; Arshia Ishtiaq, M.; Khan, K.M. Quinazoline and quinazolinone as important medicinal scaffolds: A comparative patent review (2011–2016). Expert Opin. Ther. Pat. 2018, 28, 281–297. [Google Scholar] [CrossRef]
- Ghorab, M.M.; Alsaid, M.S.; Al-Dosari, M.S.; El-Gazzar, M.G.; Parvez, M.K. Design, Synthesis and Anticancer Evaluation of Novel Quinazoline-Sulfonamide Hybrids. Molecules 2016, 21, 189. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.; Ren, H.; Zhao, M.; Chong, Y.; Zhao, W.; He, Y.; Zhao, Y.; Zhang, H.; Qi, C. Development of a series of novel 4-anlinoquinazoline derivatives possessing quinazoline skeleton: Design, synthesis, EGFR kinase inhibitory efficacy, and evaluation of anticancer activities in vitro. Eur. J. Med. Chem. 2017, 138, 669–688. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, M.F.; Khalifa, A.S.; Eed, E.M. Discovery of New Quinazoline-Based Anticancer Agents as VEGFR-2 Inhibitors and Apoptosis Inducers. Russ. J. Bioorg. Chem. 2022, 48, 739–748. [Google Scholar] [CrossRef]
- ElZahabi, H.S.A.; Nafie, M.S.; Osman, D.; Elghazawy, N.H.; Soliman, D.H.; El-Helby, A.A.H.; Arafa, R.K. Design, synthesis and evaluation of new quinazolin-4-one derivatives as apoptotic enhancers and autophagy inhibitors with potent antitumor activity. Eur. J. Med. Chem. 2021, 222, 113609. [Google Scholar] [CrossRef]
- Abdelsalam, E.A.; Zaghary, W.A.; Amin, K.M.; Abou Taleb, N.A.; Mekawey, A.A.I.; Eldehna, W.M.; Abdel-Aziz, H.A.; Hammad, S.F. Synthesis and in vitro anticancer evaluation of some fused indazoles, quinazolines and quinolines as potential EGFR inhibitors. Bioorg. Chem. 2019, 89, 102985. [Google Scholar] [CrossRef]
- Hoan, D.Q.; Hoa, L.T.; Huan, T.T.; Dinh, N.H. Synthesis and Transformation of 4-(1-Chloro-1-nitroethyl)-6, 7-dimethoxy-2-methylquinazoline: Spectral Characterization and Anti-cancer Properties of some Novel Quinazoline Derivatives. J. Heterocycl. Chem. 2020, 57, 1720–1728. [Google Scholar] [CrossRef]
- Wang, Z.; Liu, L.; Dai, H.; Si, X.; Zhang, L.; Li, E.; Yang, Z.; Chao, G.; Zheng, J.; Ke, Y.; et al. Design, synthesis and biological evaluation of novel 2,4-disubstituted quinazoline derivatives targeting H1975 cells via EGFR-PI3K signaling pathway. Bioorg. Med. Chem. 2021, 43, 116265. [Google Scholar] [CrossRef]
- Zayed, M.F.; Rateb, H.S.; Ahmed, S.; Khaled, O.A.; Ibrahim, S.R.M. Quinazolinone-Amino Acid Hybrids as Dual Inhibitors of EGFR Kinase and Tubulin Polymerization. Molecules 2018, 23, 1699. [Google Scholar] [CrossRef] [PubMed]
- Abuelizz, H.A.; Marzouk, M.; Ghabbour, H.; Al-Salahi, R. Synthesis and anticancer activity of new quinazoline derivatives. Saudi Pharm. J. 2017, 25, 1047–1054. [Google Scholar] [CrossRef] [PubMed]
- Poudapally, S.; Battu, S.; Velatooru, L.R.; Bethu, M.S.; Janapala, V.R.; Sharma, S.; Sen, S.; Pottabathini, N.; Iska, V.B.R.; Katangoor, V. Synthesis and biological evaluation of novel quinazoline-sulfonamides as anti-cancer agents. Bioorg. Med. Chem. Lett. 2017, 27, 1923–1928. [Google Scholar] [CrossRef]
- Ewes, W.A.; Elmorsy, M.A.; El-Messery, S.M.; Nasr, M.N.A. Synthesis, biological evaluation and molecular modeling study of [1,2,4]-Triazolo[4,3-c]quinazolines: New class of EGFR-TK inhibitors. Bioorg. Med. Chem. 2020, 28, 115373. [Google Scholar] [CrossRef] [PubMed]
- Tariq, S.; Somakala, K.; Amir, M. Quinoxaline: An insight into the recent pharmacological advances. Eur. J. Med. Chem. 2018, 143, 542–557. [Google Scholar] [CrossRef]
- Srivastava, S.; Banerjee, J.; Srestha, N. Quinoxaline as a potent heterocyclic moiety. IOSR J. Pharm. 2014, 4, 17–27. [Google Scholar] [CrossRef]
- El Newahie, A.M.; Ismail, N.S.; Abou El Ella, D.A.; Abouzid, K.A.M. Quinoxaline-Based Scaffolds Targeting Tyrosine Kinases and Their Potential Anticancer Activity. Arch. Pharm. 2016, 349, 309–326. [Google Scholar] [CrossRef] [PubMed]
- Ismail, M.M.; Amin, K.M.; Noaman, E.; Soliman, D.H.; Ammar, Y.A. New quinoxaline 1, 4-di-N-oxides: Anticancer and hypoxia-selective therapeutic agents. Eur. J. Med. Chem. 2010, 45, 2733–2738. [Google Scholar] [CrossRef] [PubMed]
- Abbas, H.-A.S.; Al-Marhabi, A.R.; Eissa, S.I.; Ammar, Y.A. Molecular modeling studies and synthesis of novel quinoxaline derivatives with potential anticancer activity as inhibitors of c-Met kinase. Bioorg. Med. Chem. 2015, 23, 6560–6572. [Google Scholar] [CrossRef]
- Desplat, V.; Vincenzi, M.; Lucas, R.; Moreau, S.; Savrimoutou, S.; Pinaud, N.; Lesbordes, J.; Peyrilles, E.; Marchivie, M.; Routier, S. Synthesis and evaluation of the cytotoxic activity of novel ethyl 4-[4-(4-substitutedpiperidin-1-yl)] benzyl-phenylpyrrolo [1, 2-a] quinoxaline-carboxylate derivatives in myeloid and lymphoid leukemia cell lines. Eur. J. Med. Chem. 2016, 113, 214–227. [Google Scholar] [CrossRef] [PubMed]
- El Newahie, A.M.; Nissan, Y.M.; Ismail, N.S.; Abou El Ella, D.A.; Khojah, S.M.; Abouzid, K.A.M. Design and synthesis of new quinoxaline derivatives as anticancer agents and apoptotic inducers. Molecules 2019, 24, 1175. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, E.A.; Mohamed, M.F.; Omran, A.; Salah, H. Synthesis, EGFR-TK inhibition and anticancer activity of new quinoxaline derivatives. Synth. Commun. 2020, 50, 2924–2940. [Google Scholar] [CrossRef]
- Alsaif, N.A.; Dahab, M.A.; Alanazi, M.M.; Obaidullah, A.J.; Al-Mehizia, A.A.; Alanazi, M.M.; Aldawas, S.; Mahdy, H.A.; Elkady, H. New quinoxaline derivatives as VEGFR-2 inhibitors with anticancer and apoptotic activity: Design, molecular modeling, and synthesis. Bioorg. Chem. 2021, 110, 104807. [Google Scholar] [CrossRef]
- El-Adl, K.; Sakr, H.M.; Yousef, R.G.; Mehany, A.B.; Metwaly, A.M.; Elhendawy, M.A.; Radwan, M.M.; ElSohly, M.A.; Abulkhair, H.S.; Eissa, I.H. Discovery of new quinoxaline-2 (1H)-one-based anticancer agents targeting VEGFR-2 as inhibitors: Design, synthesis, and anti-proliferative evaluation. Bioorg. Chem. 2021, 114, 105105. [Google Scholar] [CrossRef]
- Hajri, M.; Esteve, M.-A.; Khoumeri, O.; Abderrahim, R.; Terme, T.; Montana, M.; Vanelle, P. Synthesis and evaluation of in vitro antiproliferative activity of new ethyl 3-(arylethynyl) quinoxaline-2-carboxylate and pyrido [4, 3-b] quinoxalin-1 (2H)-one derivatives. Eur. J. Med. Chem. 2016, 124, 959–966. [Google Scholar] [CrossRef] [PubMed]
- Karthikeyan, C.; Solomon, V.R.; Lee, H.; Trivedi, P.J.B.; Nutrition, P. Design, synthesis and biological evaluation of some isatin-linked chalcones as novel anti-breast cancer agents: A molecular hybridization approach. Biomed. Prev. Nutr. 2013, 3, 325–330. [Google Scholar] [CrossRef]
- Eldehna, W.M.; El Hassab, M.A.; Abo-Ashour, M.F.; Al-Warhi, T.; Elaasser, M.M.; Safwat, N.A.; Suliman, H.; Ahmed, M.F.; Al-Rashood, S.T.; Abdel-Aziz, H.A. Development of isatin-thiazolo [3, 2-a] benzimidazole hybrids as novel CDK2 inhibitors with potent in vitro apoptotic anti-proliferative activity: Synthesis, biological and molecular dynamics investigations. Bioorg. Chem. 2021, 110, 104748. [Google Scholar] [CrossRef]
- Abdel-Aziz, H.A.; Eldehna, W.M.; Keeton, A.B.; Piazza, G.A.; Kadi, A.A.; Attwa, M.W.; Abdelhameed, A.S.; Attia, M.I. Development, Therapy: Isatin-benzoazine molecular hybrids as potential antiproliferative agents: Synthesis and in vitro pharmacological profiling. Drug Des. Dev. Ther. 2017, 11, 2333. [Google Scholar] [CrossRef]
- Meleddu, R.; Petrikaite, V.; Distinto, S.; Arridu, A.; Angius, R.; Serusi, L.; Škarnulytė, L.; Endriulaitytė, U.; Paškevičiūtė, M.; Cottiglia, F. Investigating the anticancer activity of isatin/dihydropyrazole hybrids. ACS Med. Chem. Lett. 2018, 10, 571–576. [Google Scholar] [CrossRef] [PubMed]
- Eldehna, W.M.; Altoukhy, A.; Mahrous, H.; Abdel-Aziz, H.A. Design, synthesis and QSAR study of certain isatin-pyridine hybrids as potential anti-proliferative agents. Eur. J. Med. Chem. 2015, 90, 684–694. [Google Scholar] [CrossRef] [PubMed]
- Al-Wabli, R.I.; Almomen, A.A.; Almutairi, M.S.; Keeton, A.B.; Piazza, G.A.; Attia, M.I. Development, Therapy: New Isatin–Indole Conjugates: Synthesis, Characterization, and a Plausible Mechanism of Their in vitro Antiproliferative Activity. Drug Des. Dev. Ther. 2020, 14, 483. [Google Scholar] [CrossRef]
- Panga, S.; Podila, N.K.; Asres, K.; Ciddi, V. Synthesis and anticancer activity of new isatin-benzoic acid conjugates. Ethiop. Pharm. J. 2016, 31, 75–92. [Google Scholar] [CrossRef]
- Ferguson, L.; Bhakta, S.; Fox, K.R.; Wells, G.; Brucoli, F. Synthesis and Biological Evaluation of a Novel C8-Pyrrolobenzodiazepine (PBD) Adenosine Conjugate. A Study on the Role of the PBD Ring in the Biological Activity of PBD-Conjugates. Molecules 2020, 25, 1243. [Google Scholar] [CrossRef] [PubMed]
- Bose, D.S.; Idrees, M.; Todewale, I.K.; Jakka, N.; Rao, J.V. Hybrids of privileged structures benzothiazoles and pyrrolo [2, 1-c][1, 4] benzodiazepin-5-one, and diversity-oriented synthesis of benzothiazoles. Eur. J. Med. Chem. 2012, 50, 27–38. [Google Scholar] [CrossRef] [PubMed]
- Kamal, A.; Ramakrishna, G.; Nayak, V.L.; Raju, P.; Rao, A.S.; Viswanath, A.; Vishnuvardhan, M.; Ramakrishna, S.; Srinivas, G. Design and synthesis of benzo [c, d] indolone-pyrrolobenzodiazepine conjugates as potential anticancer agents. Bioorg. Med. Chem. 2012, 20, 789–800. [Google Scholar] [CrossRef]
- Chen, C.-Y.; Lee, P.-H.; Lin, Y.-Y.; Yu, W.-T.; Hu, W.-P.; Hsu, C.-C.; Lin, Y.-T.; Chang, L.-S.; Hsiao, C.-T.; Wang, J.; et al. Synthesis, DNA-binding abilities and anticancer activities of triazole-pyrrolo [2, 1-c][1, 4] benzodiazepines hybrid scaffolds. Bioorg. Med. Chem. Lett. 2013, 23, 6854–6859. [Google Scholar] [CrossRef]
- Abu-Zied, K.M.; Mohamed, T.K.; Al-Duiaj, O.K.; Zaki, M.E. A simple approach to fused pyrido [2, 3-d] pyrimidines incorporating khellinone and trimethoxyphenyl moieties as new scaffolds for antibacterial and antifungal agents. Heterocycl. Commun. 2014, 20, 93–102. [Google Scholar] [CrossRef]
- Robins, R.K.; Hitchings, G.H. Studies on condensed pyrimidine systems. XIX. A new synthesis of pyrido [2, 3-d] pyrimidines. The condensation of 1, 3-diketones and 3-ketoaldehydes with 4-aminopyrimidines. J. Am. Chem. Soc. 1958, 80, 3449–3457. [Google Scholar] [CrossRef]
- Kumar, R.N.; Dev, G.J.; Ravikumar, N.; Swaroop, D.K.; Debanjan, B.; Bharath, G.; Narsaiah, B.; Jain, S.N.; Rao, A.G. Synthesis of novel triazole/isoxazole functionalized 7-(trifluoromethyl) pyrido [2, 3-d] pyrimidine derivatives as promising anticancer and antibacterial agents. Bioorg. Med. Chem. Lett. 2016, 26, 2927–2930. [Google Scholar] [CrossRef]
- Hou, J.; Wan, S.; Wang, G.; Zhang, T.; Li, Z.; Tian, Y.; Yu, Y.; Wu, X.; Zhang, J. Design, synthesis, anti-tumor activity, and molecular modeling of quinazoline and pyrido [2, 3-d] pyrimidine derivatives targeting epidermal growth factor receptor. Eur. J. Med. Chem. 2016, 118, 276–289. [Google Scholar] [CrossRef]
- Banda, V.; Gaddameedi Jitender, D.; Gautham Santhosh, K.; Pillalamarri Sambasiva, R.; Chavva, K.; Rajesh, P.; Janapala Venkateswara, R.; Banda, N. Studies on Synthesis of Novel Pyrido [2, 3-d] pyrimidine Derivatives and Their Anticancer Activity. J. Heterocycl. Chem. 2018, 55, 2538–2544. [Google Scholar] [CrossRef]
- Behalo, M.S.; Mele, G.J. Synthesis and evaluation of pyrido [2, 3-d] pyrimidine and 1, 8-naphthyridine derivatives as potential antitumor agents. J. Heterocycl. Chem. 2017, 54, 295–300. [Google Scholar] [CrossRef]
- Elzahabi, H.S.; Nossier, E.S.; Khalifa, N.M.; Alasfoury, R.A.; El-Manawaty, M.A.; Chemistry, M. Anticancer evaluation and molecular modeling of multi-targeted kinase inhibitors based pyrido [2, 3-d] pyrimidine scaffold. J. Enzyme. Inhib. Med. Chem. 2018, 33, 546–557. [Google Scholar] [CrossRef]
- Faidallah, H.M.; Rostom, S.A.; Khan, K.A. Synthesis of some polysubstituted nicotinonitriles and derived pyrido [2, 3-d] pyrimidines as in vitro cytotoxic and antimicrobial candidates. J. Chem. 2016, 2016, 1–12. [Google Scholar] [CrossRef]
- Al-Warhi, T.; Sallam, A.-A.M.; Hemeda, L.R.; El Hassab, M.A.; Aljaeed, N.; Alotaibi, O.J.; Doghish, A.S.; Noshy, M.; Eldehna, W.M.; Ibrahim, M.H. Identification of Novel Cyanopyridones and Pyrido [2, 3-D] Pyrimidines as Anticancer Agents with Dual VEGFR-2/HER-2 Inhibitory Action: Synthesis, Biological Evaluation and Molecular Docking Studies. Pharmaceuticals 2022, 15, 1262. [Google Scholar] [CrossRef]
- Ding, J.; Liu, T.; Zeng, C.; Li, B.; Ai, Y.; Zhang, X.; Zhong, H. Design, synthesis, and anti-breast-cancer activity evaluation of pyrrolo (pyrido)[2, 3-d] pyrimidine derivatives. Chem. Heterocycl. Compd. 2022, 58, 438–448. [Google Scholar] [CrossRef]





















Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kumar, A.; Singh, A.K.; Singh, H.; Vijayan, V.; Kumar, D.; Naik, J.; Thareja, S.; Yadav, J.P.; Pathak, P.; Grishina, M.; et al. Nitrogen Containing Heterocycles as Anticancer Agents: A Medicinal Chemistry Perspective. Pharmaceuticals 2023, 16, 299. https://doi.org/10.3390/ph16020299
Kumar A, Singh AK, Singh H, Vijayan V, Kumar D, Naik J, Thareja S, Yadav JP, Pathak P, Grishina M, et al. Nitrogen Containing Heterocycles as Anticancer Agents: A Medicinal Chemistry Perspective. Pharmaceuticals. 2023; 16(2):299. https://doi.org/10.3390/ph16020299
Chicago/Turabian StyleKumar, Adarsh, Ankit Kumar Singh, Harshwardhan Singh, Veena Vijayan, Deepak Kumar, Jashwanth Naik, Suresh Thareja, Jagat Pal Yadav, Prateek Pathak, Maria Grishina, and et al. 2023. "Nitrogen Containing Heterocycles as Anticancer Agents: A Medicinal Chemistry Perspective" Pharmaceuticals 16, no. 2: 299. https://doi.org/10.3390/ph16020299
APA StyleKumar, A., Singh, A. K., Singh, H., Vijayan, V., Kumar, D., Naik, J., Thareja, S., Yadav, J. P., Pathak, P., Grishina, M., Verma, A., Khalilullah, H., Jaremko, M., Emwas, A. -H., & Kumar, P. (2023). Nitrogen Containing Heterocycles as Anticancer Agents: A Medicinal Chemistry Perspective. Pharmaceuticals, 16(2), 299. https://doi.org/10.3390/ph16020299