


Technologies for Direct Detection of Covalent Protein–Drug Adducts


Abstract
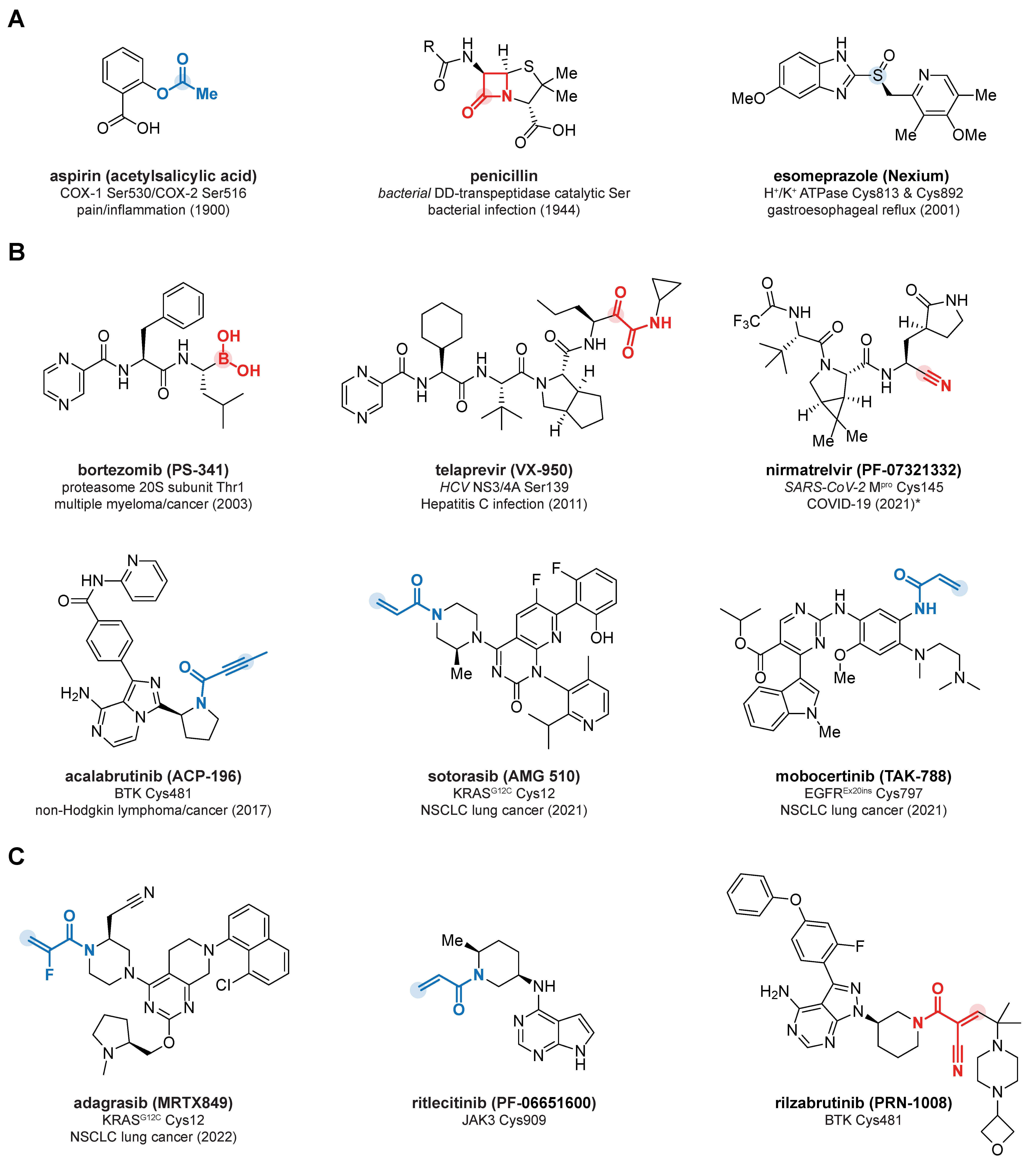
:1. Introduction
2. Mass Spectrometry (MS)
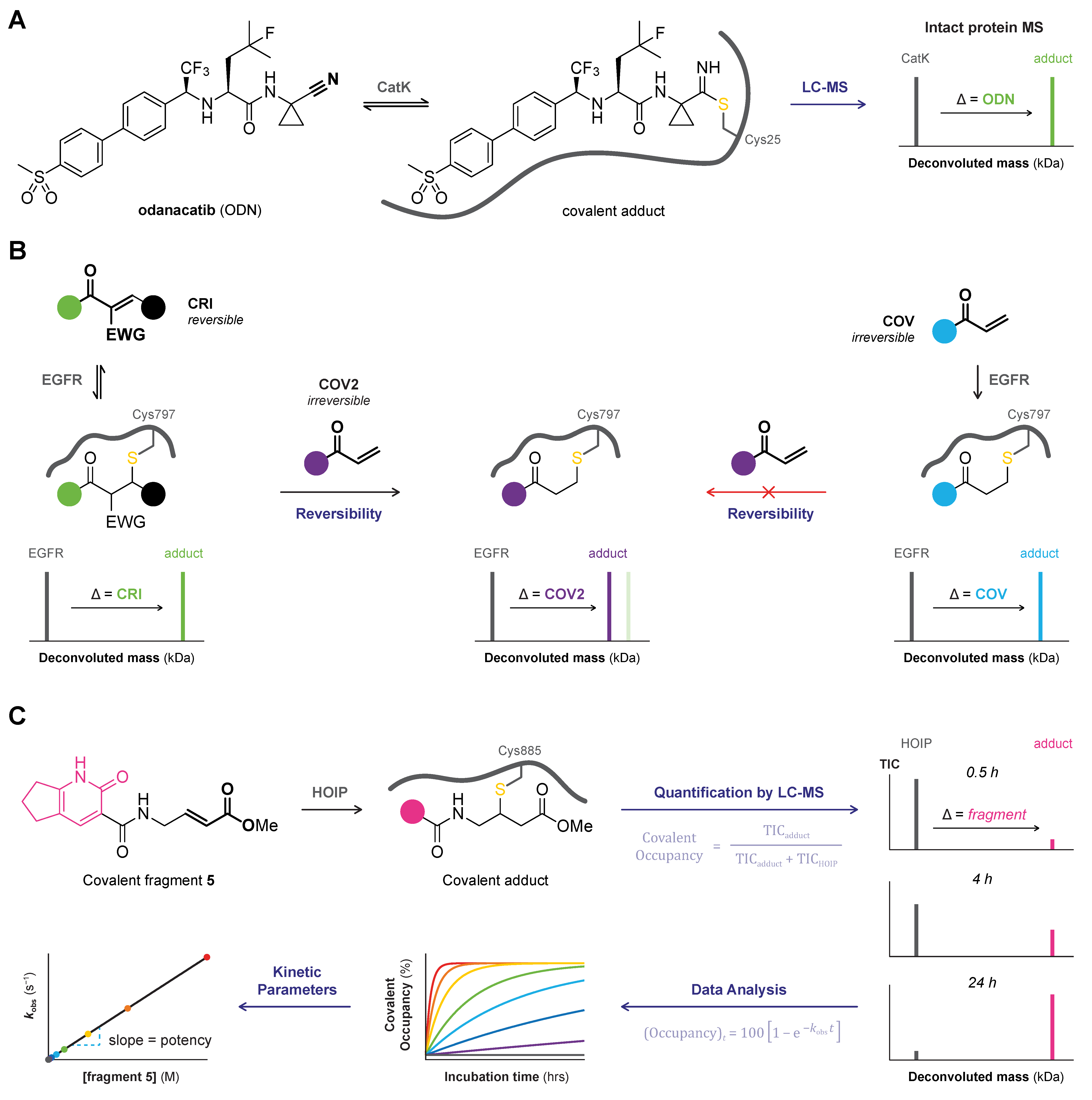
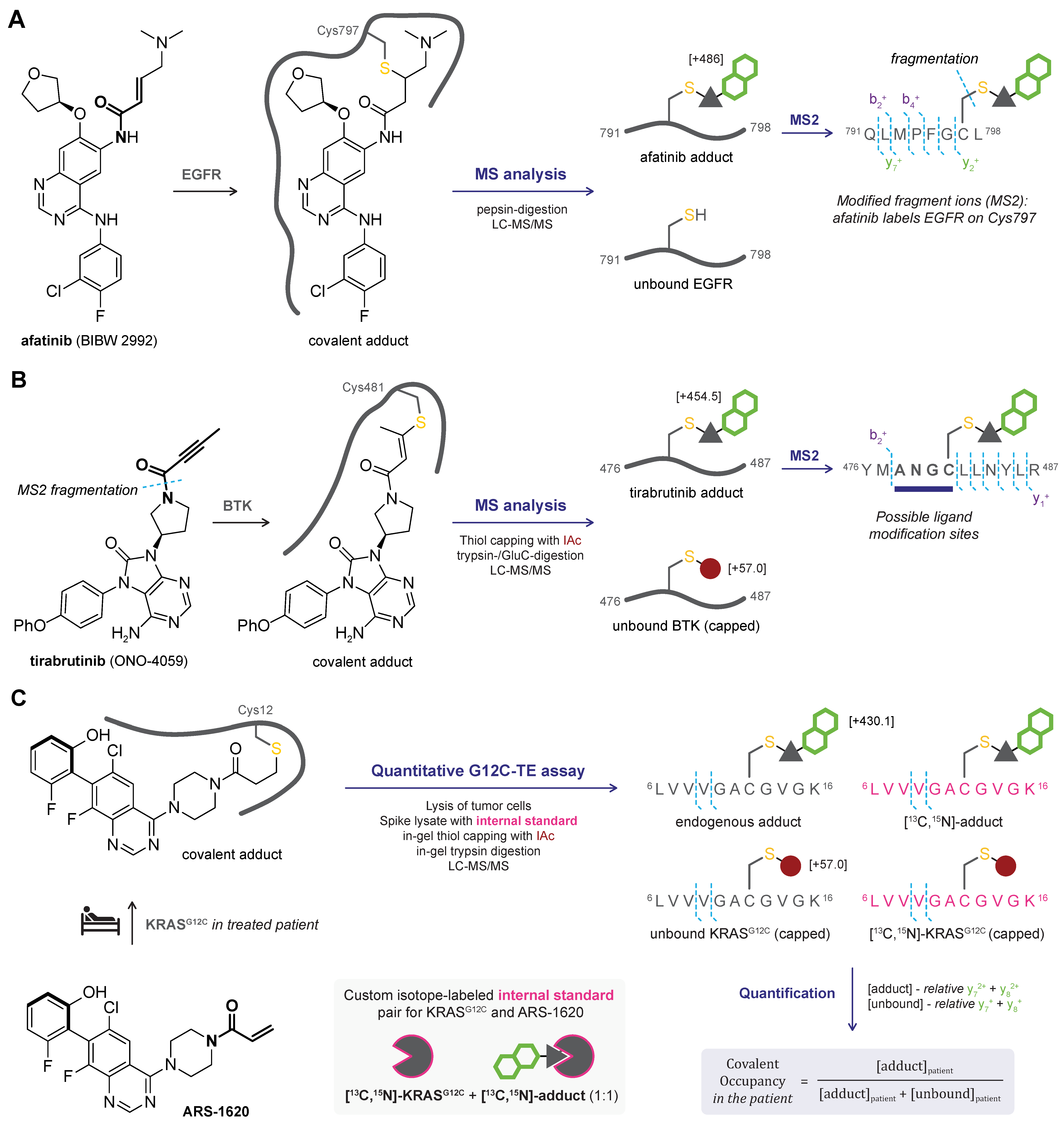
2.1. Top-Down MS
2.2. Bottom-Up MS
2.3. MS/MS or Tandem MS
3. Protein Crystallography
4. Intrinsic Fluorescence/Absorbance
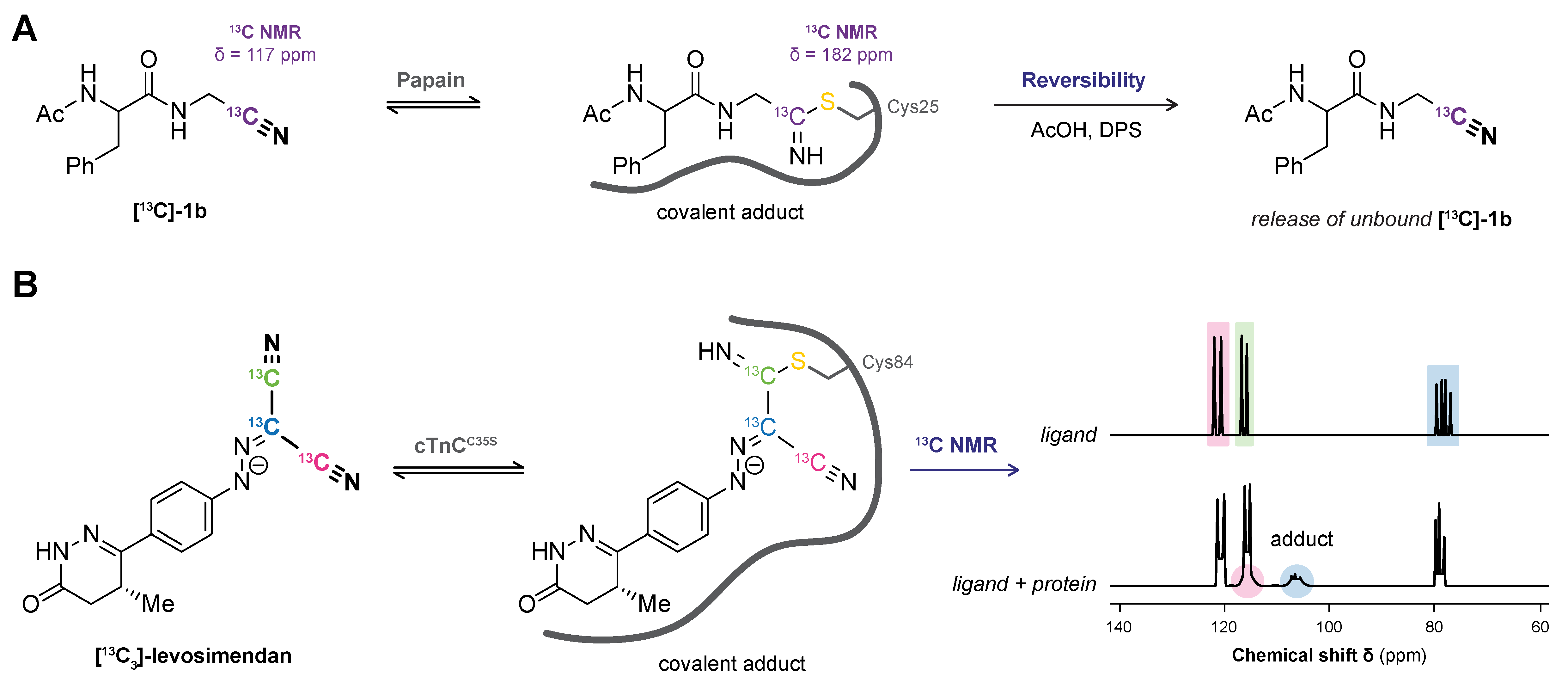
5. Nuclear Magnetic Resonance (NMR)
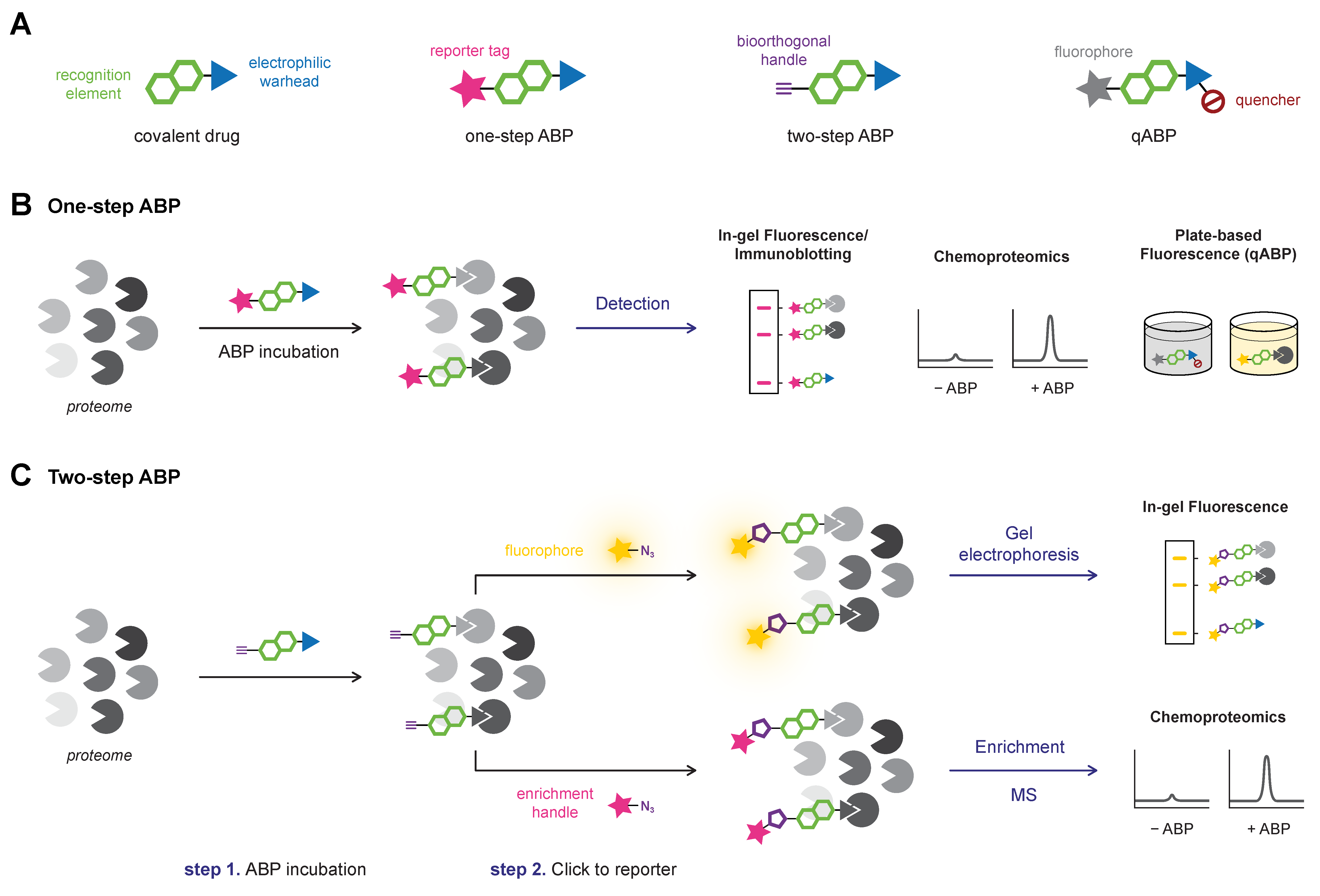
6. Activity-Based Protein Profiling (ABPP)
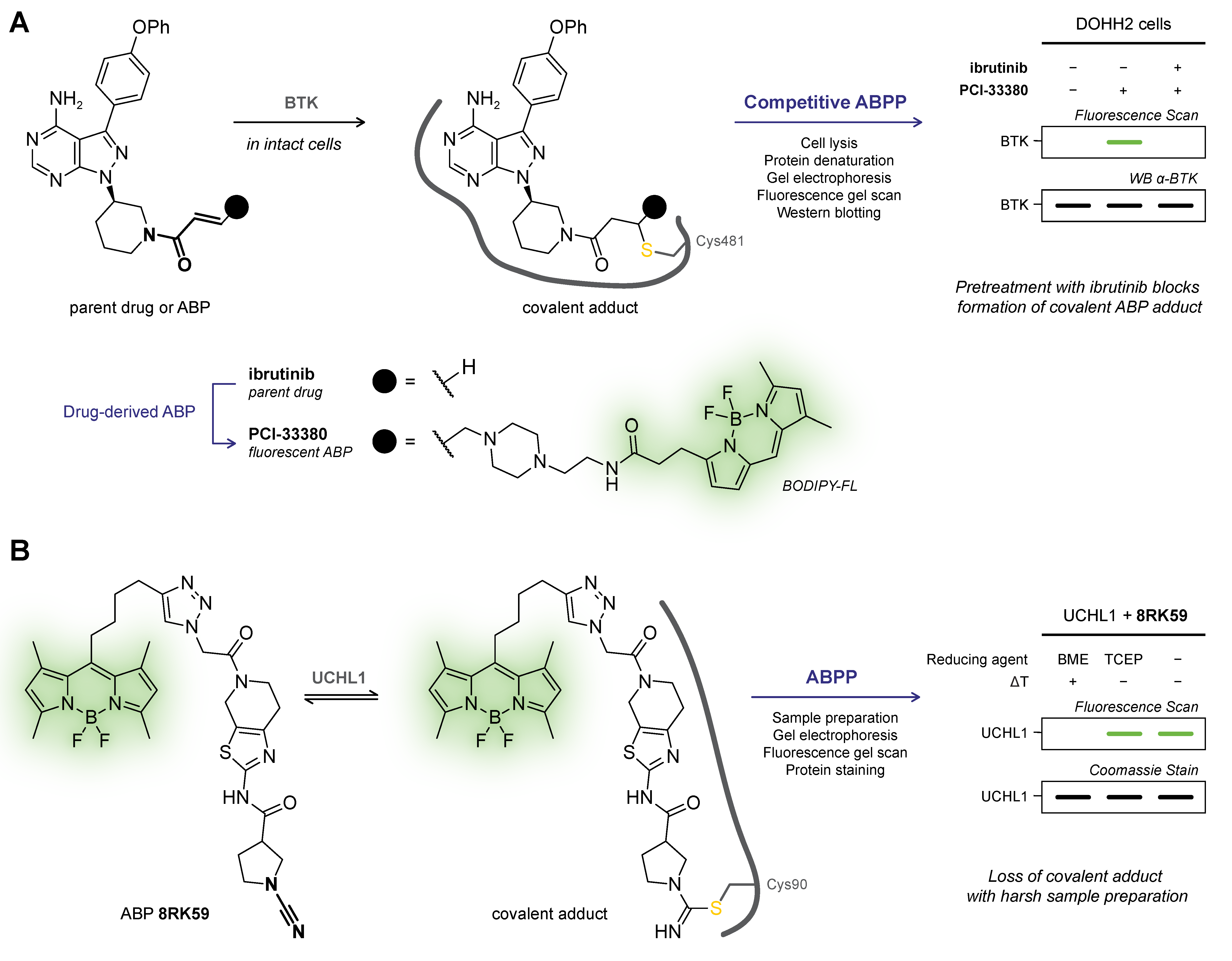
6.1. Gel Electrophoresis Platforms (In-gel Fluorescence, Immunoblotting)
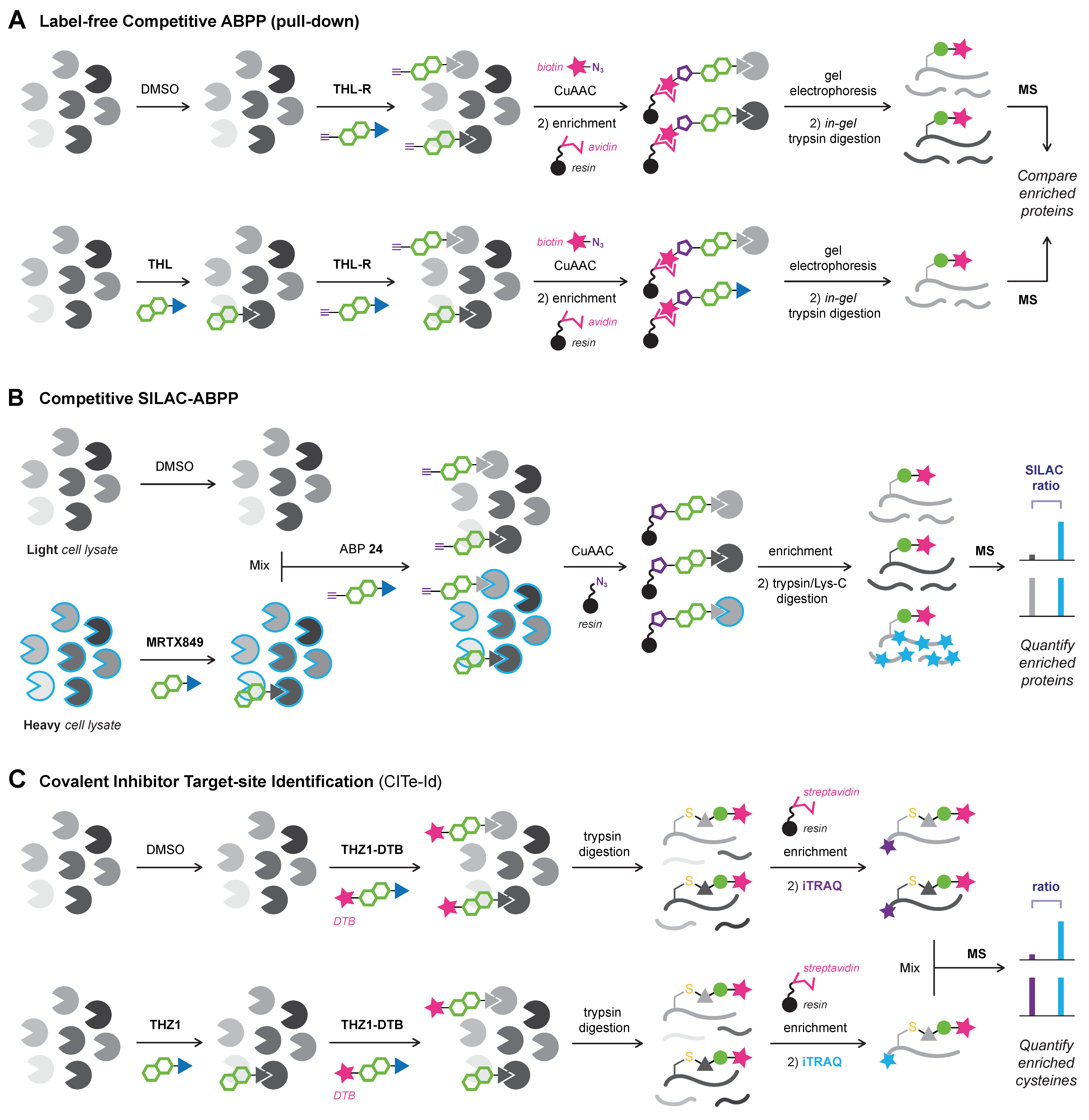
6.2. Chemoproteomic Platforms

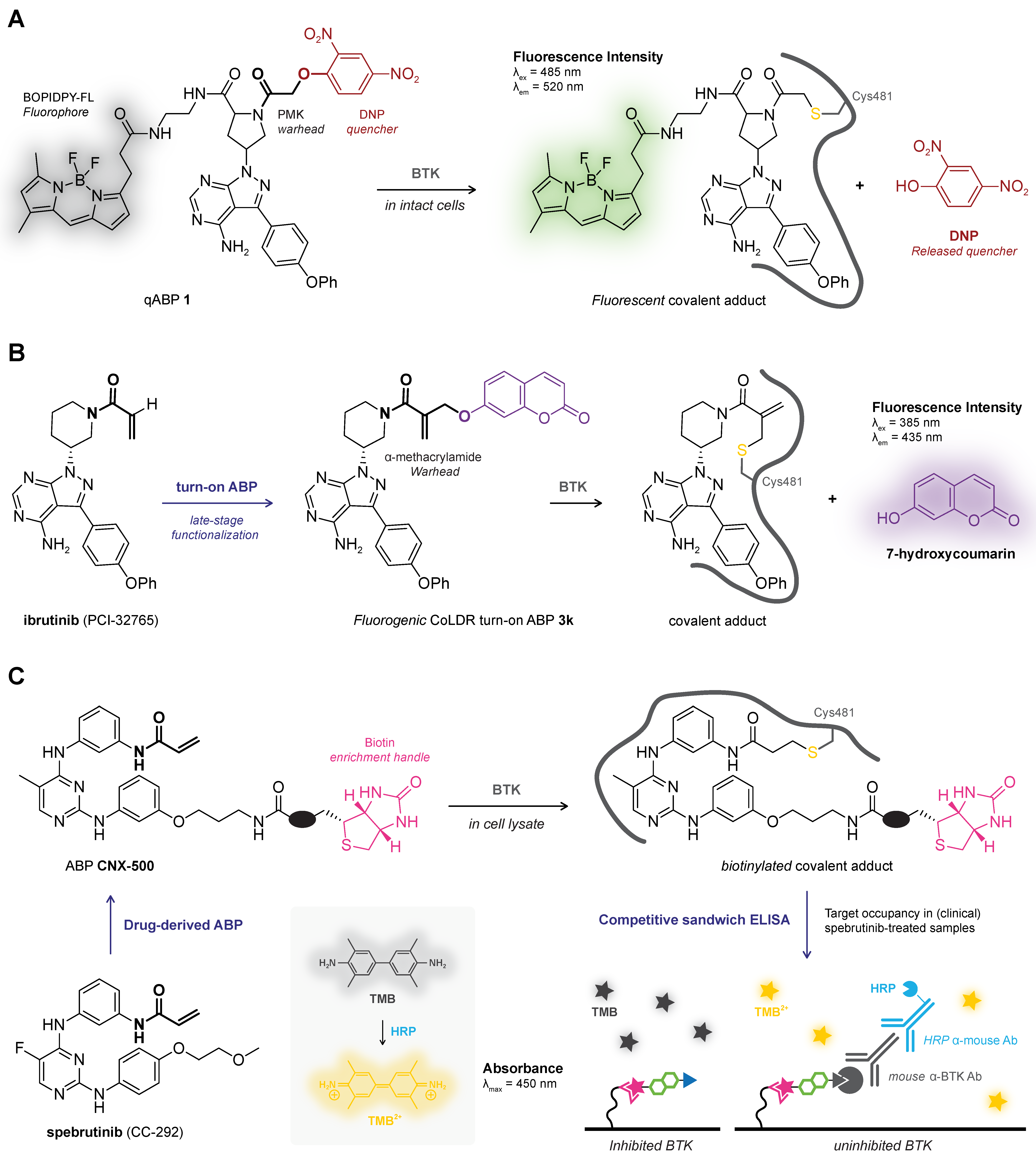
6.3. Homogeneous (Plate-Based) Platforms

7. Conclusions, Current Challenges, and Future Directions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ABP | activity-based probe |
| ABPP | activity-based protein profiling |
| ADME | absorption, distribution, metabolism, and excretion |
| ALPHA | amplified luminescent proximity homogeneous assay |
| AOMK | acyloxymethyl ketone |
| APT | attached proton test |
| BME | β-mercaptoethanol |
| BODIPY | fluorinated boron-dipyrromethene |
| CAA | chloroacetamide |
| CITe-Id | covalent inhibitor target-site identification |
| CoLDR | covalent ligand directed release |
| CRI | covalent reversible inhibitor |
| cryo-EM | cryogenic electron microscopy |
| CuAAC | Huisgen copper-catalyzed alkyne–azide cycloaddition |
| DNP | 2,4-dinitrophenyl |
| DPS | 2,2′-dipyridyldisulfide |
| DTT | dithiothreitol |
| DUB | deubiquitinating enzyme |
| ELISA | enzyme-linked immunosorbent assay |
| FBDD | fragment-based drug development |
| FBLD | fragment-based ligand discovery |
| FDA | food and drug administration (USA) |
| FFPE | formalin fixed paraffin embedded |
| G12C-TE | KRASG12C target engagement |
| GSH | glutathione |
| GST | glutathione S-transferase |
| HCV | hepatitis C virus |
| His- | polyhistidine tag |
| HTS | high-throughput screening |
| IAc | iodoacetamide |
| ICC | intrahepatic cholangiocarcinoma |
| IEDDA | inverse electron demand Diels Alder |
| isoDTB | isotopically-labeled desthiobiotin |
| isoTOP-ABPP | isotopic tandem orthogonal activity-based protein profiling |
| iTRAQ | isobaric tag for relative and absolute quantitation |
| KD | dissociation constant |
| Ki* | steady-state inhibition constant |
| LC | liquid chromatography |
| MMTS | S-methyl methanethiosulfonate |
| MS | mass spectrometry |
| NMR | nuclear magnetic resonance |
| NSCLC | non-small cell lung carcinoma |
| ODN | odanacatib |
| PD | pharmacodynamics |
| PDB | protein data bank |
| PDT | photodynamic therapy |
| PET | photo-induced electron transfer |
| PK | pharmacokinetics |
| PMK | phenoxymethyl ketone |
| Prg | propargyl |
| PROTAC | proteolysis-targeting chimera |
| qABP | quenched ABP |
| Rho | rhodamine |
| SAR | structure-activity relationship |
| SARS-CoV-2 | severe acute respiratory syndrome coronavirus 2 |
| SDS-PAGE | sodium dodecyl sulfate polyacrylamide gel electrophoresis |
| SILAC | stable-isotope labeling by amino acids in cell culture |
| SPAAC | strain-promoted alkyne–azide cycloaddition |
| SUMO | small ubiquitin-related modifier |
| TAMRA/TMR | tetramethylrhodamine |
| TCEP | tris(2-carboxyethyl)phosphine |
| TCI | targeted covalent inhibitor |
| TIC | total ion count |
| THF | tetrahydrofuran |
| THL | tetrahydrolipstatin |
| TMB | tetramethyl benzidine |
| TMT | tandem mass tag |
| Ub | ubiquitin |
| VME | vinyl methyl ester |
| XRD | X-ray diffraction |
Appendix A


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Active Ingredient b | Drug Brand Name | Sponsor c | Approval Year | Target | Indication | Treatment Area | Warhead | Reversibility | Target Residue | Ref |
| Lurbinectedin (PM01183) | Zepzelca | Pharma Mar | 2020 | DNA minor groove (alkylation) | Metastatic SCLC | Cancer | Highly reactive carbinolamine (Imine intermediate) | Reversible | Guanine N2 | [208] |
| Remdesivir (GS-5734) d | Veklury | Gilead Sciences | 2020 | SARS-CoV-2 RdRp SARS-CoV-2 Mpro | COVID-19 | Anti-microbial | Nitrile | Reversible | Ser861 (RdRp) Cys145 (Mpro) | [64,280] |
| Sotorasib (AMG 510) | Lumakras | Amgen | 2021 | KRASG12C | KRASG12C-mutated NSCLC | Cancer | Acrylamide | Irreversible | Cys12 | [18] |
| Mobocertinib (TAK788) | Exkivity | Takeda | 2021 | EGFRex20ins | Metastatic EGFRex20ins-mutated NSCLC | Cancer | Acrylamide | Irreversible | Cys797 | [19] |
| Nirmatrelvir (PF-07321332) | Paxlovid | Pfizer | 2021 e | SARS-CoV-2 Mpro | COVID-19 | Anti-microbial | Nitrile | Reversible | Cys145 | [47,281] |
| Futibatinib (TAS-120) | Lytgobi | Taiho Oncology | 2022 | FGFR1-4 | FGFR2 fusion-positive ICC | Cancer | Acrylamide | Irreversible | Cys488 (FGFR1) Cys492 (FGFR2-IIIb) | [282] |
| Adagrasib (MRTX849) | Krazati | Mirati Therapeutics | 2022 | KRASG12C | KRASG12C-mutated NSCLC | Cancer | Acrylamide | Irreversible | Cys12 | [103] |
References
- Singh, J.; Petter, R.C.; Baillie, T.A.; Whitty, A. The resurgence of covalent drugs. Nat. Rev. Drug Discov. 2011, 10, 307–317. [Google Scholar] [CrossRef]
- Johnson, D.S.; Weerapana, E.; Cravatt, B.F. Strategies for discovering and derisking covalent, irreversible enzyme inhibitors. Future Med. Chem. 2010, 2, 949–964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vane, J.R.; Botting, R.M. The mechanism of action of aspirin. Thromb. Res. 2003, 110, 255–258. [Google Scholar] [CrossRef]
- Yocum, R.R.; Rasmussen, J.R.; Strominger, J.L. The mechanism of action of penicillin. Penicillin acylates the active site of Bacillus stearothermophilus D-alanine carboxypeptidase. J. Biol. Chem. 1980, 255, 3977–3986. [Google Scholar] [CrossRef] [PubMed]
- Ding, Z.; Kim, S.; Dorsam, R.T.; Jin, J.; Kunapuli, S.P. Inactivation of the human P2Y12 receptor by thiol reagents requires interaction with both extracellular cysteine residues, Cys17 and Cys270. Blood 2003, 101, 3908–3914. [Google Scholar] [CrossRef] [Green Version]
- Shin, J.M.; Cho, Y.M.; Sachs, G. Chemistry of Covalent Inhibition of the Gastric (H+, K+)-ATPase by Proton Pump Inhibitors. J. Am. Chem. Soc. 2004, 126, 7800–7811. [Google Scholar] [CrossRef] [PubMed]
- Bauer, R.A. Covalent inhibitors in drug discovery: From accidental discoveries to avoided liabilities and designed therapies. Drug Discov. Today 2015, 20, 1061–1073. [Google Scholar] [CrossRef]
- Gehringer, M. Covalent inhibitors: Back on track? Future Med. Chem. 2020, 12, 1363–1368. [Google Scholar] [CrossRef] [PubMed]
- Barf, T.; Kaptein, A. Irreversible Protein Kinase Inhibitors: Balancing the Benefits and Risks. J. Med. Chem. 2012, 55, 6243–6262. [Google Scholar] [CrossRef]
- Chaikuad, A.; Koch, P.; Laufer, S.A.; Knapp, S. The Cysteinome of Protein Kinases as a Target in Drug Development. Angew. Chem. Int. Ed. 2018, 57, 4372–4385. [Google Scholar] [CrossRef] [PubMed]
- Honigberg, L.A.; Smith, A.M.; Sirisawad, M.; Verner, E.; Loury, D.; Chang, B.; Li, S.; Pan, Z.; Thamm, D.H.; Miller, R.A.; et al. The Bruton tyrosine kinase inhibitor PCI-32765 blocks B-cell activation and is efficacious in models of autoimmune disease and B-cell malignancy. Proc. Natl. Acad. Sci. USA 2010, 107, 13075–13080. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.; Liu, Y.; Hu, N.; Yu, D.; Zhou, C.; Shi, G.; Zhang, B.; Wei, M.; Liu, J.; Luo, L.; et al. Discovery of Zanubrutinib (BGB-3111), a Novel, Potent, and Selective Covalent Inhibitor of Bruton’s Tyrosine Kinase. J. Med. Chem. 2019, 62, 7923–7940. [Google Scholar] [CrossRef] [PubMed]
- Barf, T.; Covey, T.; Izumi, R.; van de Kar, B.; Gulrajani, M.; van Lith, B.; van Hoek, M.; de Zwart, E.; Mittag, D.; Demont, D.; et al. Acalabrutinib (ACP-196): A Covalent Bruton Tyrosine Kinase Inhibitor with a Differentiated Selectivity and In Vivo Potency Profile. J. Pharmacol. Exp. Ther. 2017, 363, 240. [Google Scholar] [CrossRef] [PubMed]
- Solca, F.; Dahl, G.; Zoephel, A.; Bader, G.; Sanderson, M.; Klein, C.; Kraemer, O.; Himmelsbach, F.; Haaksma, E.; Adolf, G.R. Target Binding Properties and Cellular Activity of Afatinib (BIBW 2992), an Irreversible ErbB Family Blocker. J. Pharmacol. Exp. Ther. 2012, 343, 342–350. [Google Scholar] [CrossRef]
- Cross, D.A.E.; Ashton, S.E.; Ghiorghiu, S.; Eberlein, C.; Nebhan, C.A.; Spitzler, P.J.; Orme, J.P.; Finlay, M.R.V.; Ward, R.A.; Mellor, M.J.; et al. AZD9291, an Irreversible EGFR TKI, Overcomes T790M-Mediated Resistance to EGFR Inhibitors in Lung Cancer. Cancer Discov. 2014, 4, 1046–1061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rabindran, S.K.; Discafani, C.M.; Rosfjord, E.C.; Baxter, M.; Floyd, M.B.; Golas, J.; Hallett, W.A.; Johnson, B.D.; Nilakantan, R.; Overbeek, E.; et al. Antitumor Activity of HKI-272, an Orally Active, Irreversible Inhibitor of the HER-2 Tyrosine Kinase. Cancer Res. 2004, 64, 3958. [Google Scholar] [CrossRef] [Green Version]
- Gonzales, A.J.; Hook, K.E.; Althaus, I.W.; Ellis, P.A.; Trachet, E.; Delaney, A.M.; Harvey, P.J.; Ellis, T.A.; Amato, D.M.; Nelson, J.M.; et al. Antitumor activity and pharmacokinetic properties of PF-00299804, a second-generation irreversible pan-erbB receptor tyrosine kinase inhibitor. Mol. Cancer Ther. 2008, 7, 1880–1889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lanman, B.A.; Allen, J.R.; Allen, J.G.; Amegadzie, A.K.; Ashton, K.S.; Booker, S.K.; Chen, J.J.; Chen, N.; Frohn, M.J.; Goodman, G.; et al. Discovery of a Covalent Inhibitor of KRASG12C (AMG 510) for the Treatment of Solid Tumors. J. Med. Chem. 2020, 63, 52–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalvez, F.; Vincent, S.; Baker, T.E.; Gould, A.E.; Li, S.; Wardwell, S.D.; Nadworny, S.; Ning, Y.; Zhang, S.; Huang, W.-S.; et al. Mobocertinib (TAK-788): A Targeted Inhibitor of EGFR Exon 20 Insertion Mutants in Non–Small Cell Lung Cancer. Cancer Discov. 2021, 11, 1672–1687. [Google Scholar] [CrossRef]
- De Vita, E. 10 years into the resurgence of covalent drugs. Future Med. Chem. 2021, 13, 193–210. [Google Scholar] [CrossRef]
- Ward, R.A.; Grimster, N.P. The Design of Covalent-Based Inhibitors. Annu. Rep. Med. Chem. 2021, 56, 2–284. [Google Scholar]
- Borsari, C.; Keles, E.; McPhail, J.A.; Schaefer, A.; Sriramaratnam, R.; Goch, W.; Schaefer, T.; De Pascale, M.; Bal, W.; Gstaiger, M.; et al. Covalent Proximity Scanning of a Distal Cysteine to Target PI3Kα. J. Am. Chem. Soc. 2022, 144, 6326–6342. [Google Scholar] [CrossRef] [PubMed]
- Gehringer, M.; Laufer, S.A. Emerging and Re-Emerging Warheads for Targeted Covalent Inhibitors: Applications in Medicinal Chemistry and Chemical Biology. J. Med. Chem. 2019, 62, 5673–5724. [Google Scholar] [CrossRef]
- Shindo, N.; Ojida, A. Recent progress in covalent warheads for in vivo targeting of endogenous proteins. Bioorg. Med. Chem. 2021, 47, 116386. [Google Scholar] [CrossRef]
- Ray, S.; Murkin, A.S. New Electrophiles and Strategies for Mechanism-Based and Targeted Covalent Inhibitor Design. Biochemistry 2019, 58, 5234–5244. [Google Scholar] [CrossRef]
- Martin, J.S.; MacKenzie, C.J.; Fletcher, D.; Gilbert, I.H. Characterising covalent warhead reactivity. Bioorg. Med. Chem. 2019, 27, 2066–2074. [Google Scholar] [CrossRef]
- Lonsdale, R.; Burgess, J.; Colclough, N.; Davies, N.L.; Lenz, E.M.; Orton, A.L.; Ward, R.A. Expanding the Armory: Predicting and Tuning Covalent Warhead Reactivity. J. Chem. Inf. Model. 2017, 57, 3124–3137. [Google Scholar] [CrossRef]
- Guan, I.; Williams, K.; Pan, J.; Liu, X. New Cysteine Covalent Modification Strategies Enable Advancement of Proteome-wide Selectivity of Kinase Modulators. Asian J. Org. Chem. 2021, 10, 949–963. [Google Scholar] [CrossRef]
- Oballa, R.M.; Truchon, J.-F.; Bayly, C.I.; Chauret, N.; Day, S.; Crane, S.; Berthelette, C. A generally applicable method for assessing the electrophilicity and reactivity of diverse nitrile-containing compounds. Bioorg. Med. Chem. Lett. 2007, 17, 998–1002. [Google Scholar] [CrossRef] [PubMed]
- Péczka, N.; Orgován, Z.; Ábrányi-Balogh, P.; Keserű, G.M. Electrophilic warheads in covalent drug discovery: An overview. Expert Opin. Drug Discov. 2022, 17, 413–422. [Google Scholar] [CrossRef] [PubMed]
- McAulay, K.; Bilsland, A.; Bon, M. Reactivity of Covalent Fragments and Their Role in Fragment Based Drug Discovery. Pharmaceuticals 2022, 15, 1366. [Google Scholar] [CrossRef]
- De Cesco, S.; Kurian, J.; Dufresne, C.; Mittermaier, A.K.; Moitessier, N. Covalent inhibitors design and discovery. Eur. J. Med. Chem. 2017, 138, 96–114. [Google Scholar] [CrossRef] [PubMed]
- Engel, J.; Richters, A.; Getlik, M.; Tomassi, S.; Keul, M.; Termathe, M.; Lategahn, J.; Becker, C.; Mayer-Wrangowski, S.; Grütter, C.; et al. Targeting Drug Resistance in EGFR with Covalent Inhibitors: A Structure-Based Design Approach. J. Med. Chem. 2015, 58, 6844–6863. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Hatcher, J.M.; Teng, M.; Gray, N.S.; Kostic, M. Recent Advances in Selective and Irreversible Covalent Ligand Development and Validation. Cell Chem. Biol. 2019, 26, 1486–1500. [Google Scholar] [CrossRef]
- Chen, S.; Lovell, S.; Lee, S.; Fellner, M.; Mace, P.D.; Bogyo, M. Identification of highly selective covalent inhibitors by phage display. Nat. Biotechnol. 2021, 39, 490–498. [Google Scholar] [CrossRef]
- Dalton, S.E.; Campos, S. Covalent Small Molecules as Enabling Platforms for Drug Discovery. ChemBioChem 2020, 21, 1080–1100. [Google Scholar] [CrossRef]
- Campuzano, I.D.G.; San Miguel, T.; Rowe, T.; Onea, D.; Cee, V.J.; Arvedson, T.; McCarter, J.D. High-Throughput Mass Spectrometric Analysis of Covalent Protein-Inhibitor Adducts for the Discovery of Irreversible Inhibitors: A Complete Workflow. J. Biomol. Screen. 2015, 21, 136–144. [Google Scholar] [CrossRef] [Green Version]
- Lu, W.; Kostic, M.; Zhang, T.; Che, J.; Patricelli, M.P.; Jones, L.H.; Chouchani, E.T.; Gray, N.S. Fragment-based covalent ligand discovery. RSC Chem. Biol. 2021, 2, 354–367. [Google Scholar] [CrossRef]
- Keeley, A.; Petri, L.; Ábrányi-Balogh, P.; Keserű, G.M. Covalent fragment libraries in drug discovery. Drug Discov. Today 2020, 25, 983–996. [Google Scholar] [CrossRef] [PubMed]
- Resnick, E.; Bradley, A.; Gan, J.; Douangamath, A.; Krojer, T.; Sethi, R.; Geurink, P.P.; Aimon, A.; Amitai, G.; Bellini, D.; et al. Rapid Covalent-Probe Discovery by Electrophile-Fragment Screening. J. Am. Chem. Soc. 2019, 141, 8951–8968. [Google Scholar] [CrossRef] [Green Version]
- Miller, R.M.; Paavilainen, V.O.; Krishnan, S.; Serafimova, I.M.; Taunton, J. Electrophilic Fragment-Based Design of Reversible Covalent Kinase Inhibitors. J. Am. Chem. Soc. 2013, 135, 5298–5301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kathman, S.G.; Statsyuk, A.V. Covalent tethering of fragments for covalent probe discovery. MedChemComm 2016, 7, 576–585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kathman, S.G.; Statsyuk, A.V. Methodology for Identification of Cysteine-Reactive Covalent Inhibitors. In Functional Disulphide Bonds: Methods and Protocols; Hogg, P., Ed.; Springer: New York, NY, USA, 2019; pp. 245–262. [Google Scholar]
- Kumalo, H.M.; Bhakat, S.; Soliman, M.E.S. Theory and Applications of Covalent Docking in Drug Discovery: Merits and Pitfalls. Molecules 2015, 20, 1984–2000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lonsdale, R.; Ward, R.A. Structure-based design of targeted covalent inhibitors. Chem. Soc. Rev. 2018, 47, 3816–3830. [Google Scholar] [CrossRef]
- Zaidman, D.; Gehrtz, P.; Filep, M.; Fearon, D.; Gabizon, R.; Douangamath, A.; Prilusky, J.; Duberstein, S.; Cohen, G.; Owen, C.D.; et al. An automatic pipeline for the design of irreversible derivatives identifies a potent SARS-CoV-2 Mpro inhibitor. Cell Chem. Biol. 2021, 28, 1795–1806.e1795. [Google Scholar] [CrossRef] [PubMed]
- Owen Dafydd, R.; Allerton Charlotte, M.N.; Anderson Annaliesa, S.; Aschenbrenner, L.; Avery, M.; Berritt, S.; Boras, B.; Cardin Rhonda, D.; Carlo, A.; Coffman Karen, J.; et al. An oral SARS-CoV-2 Mpro inhibitor clinical candidate for the treatment of COVID-19. Science 2021, 374, 1586–1593. [Google Scholar] [CrossRef] [PubMed]
- Tuley, A.; Fast, W. The Taxonomy of Covalent Inhibitors. Biochemistry 2018, 57, 3326–3337. [Google Scholar] [CrossRef]
- Langrish, C.L.; Bradshaw, J.M.; Francesco, M.R.; Owens, T.D.; Xing, Y.; Shu, J.; LaStant, J.; Bisconte, A.; Outerbridge, C.; White, S.D.; et al. Preclinical Efficacy and Anti-Inflammatory Mechanisms of Action of the Bruton Tyrosine Kinase Inhibitor Rilzabrutinib for Immune-Mediated Disease. J. Immunol. 2021, 206, 1454–1468. [Google Scholar] [CrossRef]
- Gauthier, J.Y.; Chauret, N.; Cromlish, W.; Desmarais, S.; Duong, L.T.; Falgueyret, J.-P.; Kimmel, D.B.; Lamontagne, S.; Léger, S.; LeRiche, T.; et al. The discovery of odanacatib (MK-0822), a selective inhibitor of cathepsin K. Bioorg. Med. Chem. Lett. 2008, 18, 923–928. [Google Scholar] [CrossRef]
- Groll, M.; Berkers, C.R.; Ploegh, H.L.; Ovaa, H. Crystal Structure of the Boronic Acid-Based Proteasome Inhibitor Bortezomib in Complex with the Yeast 20S Proteasome. Structure 2006, 14, 451–456. [Google Scholar] [CrossRef] [Green Version]
- Perni, R.B.; Almquist, S.J.; Byrn, R.A.; Chandorkar, G.; Chaturvedi, P.R.; Courtney, L.F.; Decker, C.J.; Dinehart, K.; Gates, C.A.; Harbeson, S.L.; et al. Preclinical Profile of VX-950, a Potent, Selective, and Orally Bioavailable Inhibitor of Hepatitis C Virus NS3-4A Serine Protease. Antimicrob. Agents Chemother. 2006, 50, 899–909. [Google Scholar] [CrossRef] [Green Version]
- Romano, K.P.; Ali, A.; Aydin, C.; Soumana, D.; Özen, A.; Deveau, L.M.; Silver, C.; Cao, H.; Newton, A.; Petropoulos, C.J.; et al. The Molecular Basis of Drug Resistance against Hepatitis C Virus NS3/4A Protease Inhibitors. PLoS Pathog. 2012, 8, e1002832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serafimova, I.M.; Pufall, M.A.; Krishnan, S.; Duda, K.; Cohen, M.S.; Maglathlin, R.L.; McFarland, J.M.; Miller, R.M.; Frödin, M.; Taunton, J. Reversible targeting of noncatalytic cysteines with chemically tuned electrophiles. Nat. Chem. Biol. 2012, 8, 471–476. [Google Scholar] [CrossRef]
- Bradshaw, J.M.; McFarland, J.M.; Paavilainen, V.O.; Bisconte, A.; Tam, D.; Phan, V.T.; Romanov, S.; Finkle, D.; Shu, J.; Patel, V.; et al. Prolonged and tunable residence time using reversible covalent kinase inhibitors. Nat. Chem. Biol. 2015, 11, 525–531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krenske, E.H.; Petter, R.C.; Houk, K.N. Kinetics and Thermodynamics of Reversible Thiol Additions to Mono- and Diactivated Michael Acceptors: Implications for the Design of Drugs That Bind Covalently to Cysteines. J. Org. Chem. 2016, 81, 11726–11733. [Google Scholar] [CrossRef]
- Basu, D.; Richters, A.; Rauh, D. Structure-based design and synthesis of covalent-reversible inhibitors to overcome drug resistance in EGFR. Bioorg. Med. Chem. 2015, 23, 2767–2780. [Google Scholar] [CrossRef] [PubMed]
- Forster, M.; Gehringer, M.; Laufer, S.A. Recent advances in JAK3 inhibition: Isoform selectivity by covalent cysteine targeting. Bioorg. Med. Chem. Lett. 2017, 27, 4229–4237. [Google Scholar] [CrossRef]
- U.S. Food & Drug Administration. Coronavirus (COVID-19) Update: FDA Authorizes First Oral Antiviral for Treatment of COVID-19 [News Release, 2021-12-22]. Available online: https://www.fda.gov/news-events/press-announcements/coronavirus-covid-19-update-fda-authorizes-first-oral-antiviral-treatment-covid-19 (accessed on 5 January 2022).
- Mons, E.; Roet, S.; Kim, R.Q.; Mulder, M.P.C. A Comprehensive Guide for Assessing Covalent Inhibition in Enzymatic Assays Illustrated with Kinetic Simulations. Curr. Protoc. 2022, 2, e419. [Google Scholar] [CrossRef]
- Strelow, J.M. A Perspective on the Kinetics of Covalent and Irreversible Inhibition. SLAS Discov. 2017, 22, 3–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McWhirter, C. Chapter One Kinetic mechanisms of covalent inhibition. In Annual Reports in Medicinal Chemistry; Ward, R.A., Grimster, N.P., Eds.; Academic Press: Cambridge, MA, USA, 2021; Volume 56, pp. 1–31. [Google Scholar]
- Harris, C.M.; Foley, S.E.; Goedken, E.R.; Michalak, M.; Murdock, S.; Wilson, N.S. Merits and Pitfalls in the Characterization of Covalent Inhibitors of Bruton’s Tyrosine Kinase. SLAS Discov. 2018, 23, 1040–1050. [Google Scholar] [CrossRef] [Green Version]
- Al-Khafaji, K.; Al-Duhaidahawi, D.; Taskin Tok, T. Using integrated computational approaches to identify safe and rapid treatment for SARS-CoV-2. J. Biomol. Struct. Dyn. 2021, 39, 3387–3395. [Google Scholar] [CrossRef]
- Yang, X.; Dilweg, M.A.; Osemwengie, D.; Burggraaff, L.; van der Es, D.; Heitman, L.H.; Ijzerman, A.P. Design and pharmacological profile of a novel covalent partial agonist for the adenosine A1 receptor. Biochem. Pharmacol. 2020, 180, 114144. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; van Veldhoven, J.P.D.; Offringa, J.; Kuiper, B.J.; Lenselink, E.B.; Heitman, L.H.; van der Es, D.; Ijzerman, A.P. Development of Covalent Ligands for G Protein-Coupled Receptors: A Case for the Human Adenosine A3 Receptor. J. Med. Chem. 2019, 62, 3539–3552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weichert, D.; Kruse, A.C.; Manglik, A.; Hiller, C.; Zhang, C.; Hübner, H.; Kobilka, B.K.; Gmeiner, P. Covalent agonists for studying G protein-coupled receptor activation. Proc. Natl. Acad. Sci. USA 2014, 111, 10744–10748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grimster, N.P. Covalent PROTACs: The best of both worlds? RSC Med. Chem. 2021, 12, 1452–1458. [Google Scholar] [CrossRef] [PubMed]
- Gabizon, R.; Shraga, A.; Gehrtz, P.; Livnah, E.; Shorer, Y.; Gurwicz, N.; Avram, L.; Unger, T.; Aharoni, H.; Albeck, S.; et al. Efficient Targeted Degradation via Reversible and Irreversible Covalent PROTACs. J. Am. Chem. Soc. 2020, 142, 11734–11742. [Google Scholar] [CrossRef]
- Li, Y.-J.; Du, L.; Wang, J.; Vega, R.; Lee, T.D.; Miao, Y.; Aldana-Masangkay, G.; Samuels, E.R.; Li, B.; Ouyang, S.X.; et al. Allosteric Inhibition of Ubiquitin-like Modifications by a Class of Inhibitor of SUMO-Activating Enzyme. Cell Chem. Biol. 2019, 26, 278–288.e276. [Google Scholar] [CrossRef]
- Pettinger, J.; Le Bihan, Y.-V.; Widya, M.; van Montfort, R.L.M.; Jones, K.; Cheeseman, M.D. An Irreversible Inhibitor of HSP72 that Unexpectedly Targets Lysine-56. Angew. Chem. Int. Ed. 2017, 56, 3536–3540. [Google Scholar] [CrossRef]
- Abdeldayem, A.; Raouf, Y.S.; Constantinescu, S.N.; Moriggl, R.; Gunning, P.T. Advances in covalent kinase inhibitors. Chem. Soc. Rev. 2020, 49, 2617–2687. [Google Scholar] [CrossRef]
- Copeland, R.A.; Basavapathruni, A.; Moyer, M.; Scott, M.P. Impact of enzyme concentration and residence time on apparent activity recovery in jump dilution analysis. Anal. Biochem. 2011, 416, 206–210. [Google Scholar] [CrossRef]
- Smith, S.; Keul, M.; Engel, J.; Basu, D.; Eppmann, S.; Rauh, D. Characterization of Covalent-Reversible EGFR Inhibitors. ACS Omega 2017, 2, 1563–1575. [Google Scholar] [CrossRef]
- Tailor, A.; Waddington, J.C.; Meng, X.; Park, B.K. Mass Spectrometric and Functional Aspects of Drug–Protein Conjugation. Chem. Res. Toxicol. 2016, 29, 1912–1935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Fonslow, B.R.; Shan, B.; Baek, M.-C.; Yates, J.R. Protein Analysis by Shotgun/Bottom-up Proteomics. Chem. Rev. 2013, 113, 2343–2394. [Google Scholar] [CrossRef] [Green Version]
- Donnelly, D.P.; Rawlins, C.M.; DeHart, C.J.; Fornelli, L.; Schachner, L.F.; Lin, Z.; Lippens, J.L.; Aluri, K.C.; Sarin, R.; Chen, B.; et al. Best practices and benchmarks for intact protein analysis for top-down mass spectrometry. Nat. Methods 2019, 16, 587–594. [Google Scholar] [CrossRef]
- Tokmina-Lukaszewska, M.; Patterson, A.; Berry, L.; Scott, L.; Balasubramanian, N.; Bothner, B. The Role of Mass Spectrometry in Structural Studies of Flavin-Based Electron Bifurcating Enzymes. Front. Microbiol. 2018, 9, 1397. [Google Scholar] [CrossRef] [PubMed]
- Hilton, G.R.; Benesch, J.L.P. Two decades of studying non-covalent biomolecular assemblies by means of electrospray ionization mass spectrometry. J. R. Soc. Interface 2012, 9, 801–816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, X.; Jin, M.; Breuker, K.; McLafferty, F.W. Extending Top-Down Mass Spectrometry to Proteins with Masses Greater Than 200 Kilodaltons. Science 2006, 314, 109–112. [Google Scholar] [CrossRef] [Green Version]
- Melby, J.A.; Roberts, D.S.; Larson, E.J.; Brown, K.A.; Bayne, E.F.; Jin, S.; Ge, Y. Novel Strategies to Address the Challenges in Top-Down Proteomics. J. Am. Soc. Mass Spectrom. 2021, 32, 1278–1294. [Google Scholar] [CrossRef]
- Mons, E.; Jansen, I.D.C.; Loboda, J.; van Doodewaerd, B.R.; Hermans, J.; Verdoes, M.; van Boeckel, C.A.A.; van Veelen, P.A.; Turk, B.; Turk, D.; et al. The Alkyne Moiety as a Latent Electrophile in Irreversible Covalent Small Molecule Inhibitors of Cathepsin K. J. Am. Chem. Soc. 2019, 141, 3507–3514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mons, E.; Kim, R.Q.; van Doodewaerd, B.R.; van Veelen, P.A.; Mulder, M.P.C.; Ovaa, H. Exploring the Versatility of the Covalent Thiol–Alkyne Reaction with Substituted Propargyl Warheads: A Deciding Role for the Cysteine Protease. J. Am. Chem. Soc. 2021, 143, 6423–6433. [Google Scholar] [CrossRef]
- Panyain, N.; Godinat, A.; Lanyon-Hogg, T.; Lachiondo-Ortega, S.; Will, E.J.; Soudy, C.; Mondal, M.; Mason, K.; Elkhalifa, S.; Smith, L.M.; et al. Discovery of a Potent and Selective Covalent Inhibitor and Activity-Based Probe for the Deubiquitylating Enzyme UCHL1, with Antifibrotic Activity. J. Am. Chem. Soc. 2020, 142, 12020–12026. [Google Scholar] [CrossRef]
- Kathman, S.G.; Xu, Z.; Statsyuk, A.V. A Fragment-Based Method to Discover Irreversible Covalent Inhibitors of Cysteine Proteases. J. Med. Chem. 2014, 57, 4969–4974. [Google Scholar] [CrossRef]
- Caldwell, R.D.; Qiu, H.; Askew, B.C.; Bender, A.T.; Brugger, N.; Camps, M.; Dhanabal, M.; Dutt, V.; Eichhorn, T.; Gardberg, A.S.; et al. Discovery of Evobrutinib: An Oral, Potent, and Highly Selective, Covalent Bruton’s Tyrosine Kinase (BTK) Inhibitor for the Treatment of Immunological Diseases. J. Med. Chem. 2019, 62, 7643–7655. [Google Scholar] [CrossRef] [Green Version]
- Angst, D.; Gessier, F.; Janser, P.; Vulpetti, A.; Wälchli, R.; Beerli, C.; Littlewood-Evans, A.; Dawson, J.; Nuesslein-Hildesheim, B.; Wieczorek, G.; et al. Discovery of LOU064 (Remibrutinib), a Potent and Highly Selective Covalent Inhibitor of Bruton’s Tyrosine Kinase. J. Med. Chem. 2020, 63, 5102–5118. [Google Scholar] [CrossRef]
- Liclican, A.; Serafini, L.; Xing, W.; Czerwieniec, G.; Steiner, B.; Wang, T.; Brendza, K.M.; Lutz, J.D.; Keegan, K.S.; Ray, A.S.; et al. Biochemical characterization of tirabrutinib and other irreversible inhibitors of Bruton’s tyrosine kinase reveals differences in on and off target inhibition. Biochim. Biophys. Acta Gen. Subj. 2020, 1864, 129531. [Google Scholar] [CrossRef] [PubMed]
- Johansson, H.; Isabella Tsai, Y.-C.; Fantom, K.; Chung, C.-W.; Kümper, S.; Martino, L.; Thomas, D.A.; Eberl, H.C.; Muelbaier, M.; House, D.; et al. Fragment-Based Covalent Ligand Screening Enables Rapid Discovery of Inhibitors for the RBR E3 Ubiquitin Ligase HOIP. J. Am. Chem. Soc. 2019, 141, 2703–2712. [Google Scholar] [CrossRef] [Green Version]
- Patricelli, M.P.; Janes, M.R.; Li, L.-S.; Hansen, R.; Peters, U.; Kessler, L.V.; Chen, Y.; Kucharski, J.M.; Feng, J.; Ely, T.; et al. Selective Inhibition of Oncogenic KRAS Output with Small Molecules Targeting the Inactive State. Cancer Discov. 2016, 6, 316–329. [Google Scholar] [CrossRef] [Green Version]
- Kathman, S.G.; Span, I.; Smith, A.T.; Xu, Z.; Zhan, J.; Rosenzweig, A.C.; Statsyuk, A.V. A Small Molecule That Switches a Ubiquitin Ligase From a Processive to a Distributive Enzymatic Mechanism. J. Am. Chem. Soc. 2015, 137, 12442–12445. [Google Scholar] [CrossRef] [Green Version]
- Dubiella, C.; Pinch, B.J.; Koikawa, K.; Zaidman, D.; Poon, E.; Manz, T.D.; Nabet, B.; He, S.; Resnick, E.; Rogel, A.; et al. Sulfopin is a covalent inhibitor of Pin1 that blocks Myc-driven tumors in vivo. Nat. Chem. Biol. 2021, 17, 954–963. [Google Scholar] [CrossRef] [PubMed]
- Douangamath, A.; Fearon, D.; Gehrtz, P.; Krojer, T.; Lukacik, P.; Owen, C.D.; Resnick, E.; Strain-Damerell, C.; Aimon, A.; Ábrányi-Balogh, P.; et al. Crystallographic and electrophilic fragment screening of the SARS-CoV-2 main protease. Nat. Commun. 2020, 11, 5047. [Google Scholar] [CrossRef] [PubMed]
- Klein, B.A.; Reiz, B.; Robertson, I.M.; Irving, M.; Li, L.; Sun, Y.-B.; Sykes, B.D. Reversible Covalent Reaction of Levosimendan with Cardiac Troponin C in Vitro and in Situ. Biochemistry 2018, 57, 2256–2265. [Google Scholar] [CrossRef]
- Dhillon, S. Tirabrutinib: First Approval. Drugs 2020, 80, 835–840. [Google Scholar] [CrossRef] [PubMed]
- Hansen, R.; Peters, U.; Babbar, A.; Chen, Y.; Feng, J.; Janes, M.R.; Li, L.-S.; Ren, P.; Liu, Y.; Zarrinkar, P.P. The reactivity-driven biochemical mechanism of covalent KRASG12C inhibitors. Nat. Struct. Mol. Biol. 2018, 25, 454–462. [Google Scholar] [CrossRef] [PubMed]
- Li, K.S.; Quinn, J.G.; Saabye, M.J.; Guerrero, J.F.S.; Nonomiya, J.; Lian, Q.; Phung, W.; Izrayelit, Y.; Walters, B.T.; Gustafson, A.; et al. High-Throughput Kinetic Characterization of Irreversible Covalent Inhibitors of KRASG12C by Intact Protein MS and Targeted MRM. Anal. Chem. 2022, 94, 1230–1239. [Google Scholar] [CrossRef] [PubMed]
- Janes, M.R.; Zhang, J.; Li, L.-S.; Hansen, R.; Peters, U.; Guo, X.; Chen, Y.; Babbar, A.; Firdaus, S.J.; Darjania, L.; et al. Targeting KRAS Mutant Cancers with a Covalent G12C-Specific Inhibitor. Cell 2018, 172, 578–589.e517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canon, J.; Rex, K.; Saiki, A.Y.; Mohr, C.; Cooke, K.; Bagal, D.; Gaida, K.; Holt, T.; Knutson, C.G.; Koppada, N.; et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature 2019, 575, 217–223. [Google Scholar] [CrossRef]
- Hansen, R.; Firdaus, S.J.; Li, S.; Janes, M.R.; Zhang, J.; Liu, Y.; Zarrinkar, P.P. An Internally Controlled Quantitative Target Occupancy Assay for Covalent Inhibitors. Sci. Rep. 2018, 8, 14312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kantae, V.; Polanski, R.; Lewis, H.J.; Haider, A.; Barratt, D.; Srinivasan, B. Accelerating the Validation of Endogenous On-Target Engagement and In Cellulo Kinetic Assessment for Covalent Inhibitors of KRASG12C in Early Drug Discovery. ACS Chem. Biol. 2022, 17, 2366–2376. [Google Scholar] [CrossRef]
- Lito, P.; Solomon, M.; Li, L.-S.; Hansen, R.; Rosen, N. Allele-specific inhibitors inactivate mutant KRAS G12C by a trapping mechanism. Science 2016, 351, 604–608. [Google Scholar] [CrossRef] [Green Version]
- Fell, J.B.; Fischer, J.P.; Baer, B.R.; Blake, J.F.; Bouhana, K.; Briere, D.M.; Brown, K.D.; Burgess, L.E.; Burns, A.C.; Burkard, M.R.; et al. Identification of the Clinical Development Candidate MRTX849, a Covalent KRASG12C Inhibitor for the Treatment of Cancer. J. Med. Chem. 2020, 63, 6679–6693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simon, G.M.; Niphakis, M.J.; Cravatt, B.F. Determining target engagement in living systems. Nat. Chem. Biol. 2013, 9, 200–205. [Google Scholar] [CrossRef] [Green Version]
- McPherson, A.; Gavira, J.A. Introduction to protein crystallization. Acta Crystallogr. Sect. F 2014, 70, 2–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berman, H.; Henrick, K.; Nakamura, H. Announcing the worldwide Protein Data Bank. Nat. Struct. Mol. Biol. 2003, 10, 980. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Bender, A.T.; Gardberg, A.; Pereira, A.; Johnson, T.; Wu, Y.; Grenningloh, R.; Head, J.; Morandi, F.; Haselmayer, P.; Liu-Bujalski, L. Ability of Bruton’s Tyrosine Kinase Inhibitors to Sequester Y551 and Prevent Phosphorylation Determines Potency for Inhibition of Fc Receptor but not B-Cell Receptor Signaling. Mol. Pharmacol. 2017, 91, 208–219. [Google Scholar] [CrossRef] [Green Version]
- Engel, J.; Smith, S.; Lategahn, J.; Tumbrink, H.L.; Goebel, L.; Becker, C.; Hennes, E.; Keul, M.; Unger, A.; Müller, H.; et al. Structure-Guided Development of Covalent and Mutant-Selective Pyrazolopyrimidines to Target T790M Drug Resistance in Epidermal Growth Factor Receptor. J. Med. Chem. 2017, 60, 7725–7744. [Google Scholar] [CrossRef]
- Groll, M.; Huber, R.; Potts, B.C.M. Crystal Structures of Salinosporamide A (NPI-0052) and B (NPI-0047) in Complex with the 20S Proteasome Reveal Important Consequences of β-Lactone Ring Opening and a Mechanism for Irreversible Binding. J. Am. Chem. Soc. 2006, 128, 5136–5141. [Google Scholar] [CrossRef]
- Uhlenbrock, N.; Smith, S.; Weisner, J.; Landel, I.; Lindemann, M.; Le, T.A.; Hardick, J.; Gontla, R.; Scheinpflug, R.; Czodrowski, P.; et al. Structural and chemical insights into the covalent-allosteric inhibition of the protein kinase Akt. Chem. Sci. 2019, 10, 3573–3585. [Google Scholar] [CrossRef] [Green Version]
- Lockbaum, G.J.; Henes, M.; Lee, J.M.; Timm, J.; Nalivaika, E.A.; Thompson, P.R.; Kurt Yilmaz, N.; Schiffer, C.A. Pan-3C Protease Inhibitor Rupintrivir Binds SARS-CoV-2 Main Protease in a Unique Binding Mode. Biochemistry 2021, 60, 2925–2931. [Google Scholar] [CrossRef] [PubMed]
- London, N.; Miller, R.M.; Krishnan, S.; Uchida, K.; Irwin, J.J.; Eidam, O.; Gibold, L.; Cimermančič, P.; Bonnet, R.; Shoichet, B.K.; et al. Covalent docking of large libraries for the discovery of chemical probes. Nat. Chem. Biol. 2014, 10, 1066–1072. [Google Scholar] [CrossRef] [Green Version]
- Scarpino, A.; Ferenczy, G.G.; Keserű, G.M. Comparative Evaluation of Covalent Docking Tools. J. Chem. Inf. Model. 2018, 58, 1441–1458. [Google Scholar] [CrossRef]
- Gao, M.; Moumbock, A.F.A.; Qaseem, A.; Xu, Q.; Günther, S. CovPDB: A high-resolution coverage of the covalent protein–ligand interactome. Nucleic Acids Res. 2022, 50, D445–D450. [Google Scholar] [CrossRef]
- Shraga, A.; Olshvang, E.; Davidzohn, N.; Khoshkenar, P.; Germain, N.; Shurrush, K.; Carvalho, S.; Avram, L.; Albeck, S.; Unger, T.; et al. Covalent Docking Identifies a Potent and Selective MKK7 Inhibitor. Cell Chem. Biol. 2019, 26, 98–108.e105. [Google Scholar] [CrossRef] [PubMed]
- Wan, X.; Yang, T.; Cuesta, A.; Pang, X.; Balius, T.E.; Irwin, J.J.; Shoichet, B.K.; Taunton, J. Discovery of Lysine-Targeted eIF4E Inhibitors through Covalent Docking. J. Am. Chem. Soc. 2020, 142, 4960–4964. [Google Scholar] [CrossRef]
- Becker, D.; Kaczmarska, Z.; Arkona, C.; Schulz, R.; Tauber, C.; Wolber, G.; Hilgenfeld, R.; Coll, M.; Rademann, J. Irreversible inhibitors of the 3C protease of Coxsackie virus through templated assembly of protein-binding fragments. Nat. Commun. 2016, 7, 12761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serafim, R.A.M.; da Silva Santiago, A.; Schwalm, M.P.; Hu, Z.; dos Reis, C.V.; Takarada, J.E.; Mezzomo, P.; Massirer, K.B.; Kudolo, M.; Gerstenecker, S.; et al. Development of the First Covalent Monopolar Spindle Kinase 1 (MPS1/TTK) Inhibitor. J. Med. Chem. 2022, 65, 3173–3192. [Google Scholar] [CrossRef] [PubMed]
- Obaidat, A.; Weiss, J.; Wahlgren, B.; Manam, R.R.; Macherla, V.R.; McArthur, K.; Chao, T.-H.; Palladino, M.A.; Lloyd, G.K.; Potts, B.C.; et al. Proteasome Regulator Marizomib (NPI-0052) Exhibits Prolonged Inhibition, Attenuated Efflux, and Greater Cytotoxicity than Its Reversible Analogs. J. Pharmacol. Exp. Ther. 2011, 337, 479–486. [Google Scholar] [CrossRef] [PubMed]
- Law, S.; Andrault, P.-M.; Aguda, A.H.; Nguyen, N.T.; Kruglyak, N.; Brayer, G.D.; Brömme, D. Identification of mouse cathepsin K structural elements that regulate the potency of odanacatib. Biochem. J. 2017, 474, 851–864. [Google Scholar] [CrossRef] [Green Version]
- Forster, M.; Chaikuad, A.; Bauer, S.M.; Holstein, J.; Robers, M.B.; Corona, C.R.; Gehringer, M.; Pfaffenrot, E.; Ghoreschi, K.; Knapp, S.; et al. Selective JAK3 Inhibitors with a Covalent Reversible Binding Mode Targeting a New Induced Fit Binding Pocket. Cell Chem. Biol. 2016, 23, 1335–1340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wlodawer, A.; Minor, W.; Dauter, Z.; Jaskolski, M. Protein crystallography for aspiring crystallographers or how to avoid pitfalls and traps in macromolecular structure determination. FEBS J. 2013, 280, 5705–5736. [Google Scholar] [CrossRef] [Green Version]
- Rupp, B. Biomolecular Crystallography: Principles, Practice, and Application to Structural Biology, 1st ed.; Garland Science: New York, NY, USA, 2009. [Google Scholar]
- Taylor, G. Introduction to phasing. Acta Crystallogr. Sect. D 2010, 66, 325–338. [Google Scholar] [CrossRef]
- Hodel, A.; Kim, S.-H.; Brunger, A.T. Model bias in macromolecular crystal structures. Acta Crystallogr. Sect. A 1992, 48, 851–858. [Google Scholar] [CrossRef]
- Ferrall-Fairbanks, M.C.; Kieslich, C.A.; Platt, M.O. Reassessing enzyme kinetics: Considering protease-as-substrate interactions in proteolytic networks. Proc. Natl. Acad. Sci. USA 2020, 117, 3307. [Google Scholar] [CrossRef]
- Lee, G.M.; Balouch, E.; Goetz, D.H.; Lazic, A.; McKerrow, J.H.; Craik, C.S. Mapping Inhibitor Binding Modes on an Active Cysteine Protease via Nuclear Magnetic Resonance Spectroscopy. Biochemistry 2012, 51, 10087–10098. [Google Scholar] [CrossRef] [PubMed]
- Hassell, A.M.; An, G.; Bledsoe, R.K.; Bynum, J.M.; Carter, H.L., III; Deng, S.-J.J.; Gampe, R.T.; Grisard, T.E.; Madauss, K.P.; Nolte, R.T.; et al. Crystallization of protein-ligand complexes. Acta Crystallogr. Sect. D 2007, 63, 72–79. [Google Scholar] [CrossRef]
- Wienen-Schmidt, B.; Oebbeke, M.; Ngo, K.; Heine, A.; Klebe, G. Two Methods, One Goal: Structural Differences between Cocrystallization and Crystal Soaking to Discover Ligand Binding Poses. ChemMedChem 2021, 16, 292–300. [Google Scholar] [CrossRef] [PubMed]
- Wlodawer, A.; Minor, W.; Dauter, Z.; Jaskolski, M. Protein crystallography for non-crystallographers, or how to get the best (but not more) from published macromolecular structures. FEBS J. 2008, 275, 1–21. [Google Scholar] [CrossRef] [Green Version]
- Martz, E.; Sussman, J.L.; Decatur, W.; Hodis, E.; Jiang, Y.; Prilusky, J. Proteopedia: Resolution. Available online: https://proteopedia.org/wiki/index.php/Resolution (accessed on 21 August 2022).
- Woińska, M.; Grabowsky, S.; Dominiak, P.M.; Woźniak, K.; Jayatilaka, D. Hydrogen atoms can be located accurately and precisely by x-ray crystallography. Sci. Adv. 2016, 2, e1600192. [Google Scholar] [CrossRef] [Green Version]
- Lv, Z.; Yuan, L.; Atkison, J.H.; Williams, K.M.; Vega, R.; Sessions, E.H.; Divlianska, D.B.; Davies, C.; Chen, Y.; Olsen, S.K. Molecular mechanism of a covalent allosteric inhibitor of SUMO E1 activating enzyme. Nat. Commun. 2018, 9, 5145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harshbarger, W.; Miller, C.; Diedrich, C.; Sacchettini, J. Crystal Structure of the Human 20S Proteasome in Complex with Carfilzomib. Structure 2015, 23, 418–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ekkebus, R.; van Kasteren, S.I.; Kulathu, Y.; Scholten, A.; Berlin, I.; Geurink, P.P.; de Jong, A.; Goerdayal, S.; Neefjes, J.; Heck, A.J.R.; et al. On Terminal Alkynes That Can React with Active-Site Cysteine Nucleophiles in Proteases. J. Am. Chem. Soc. 2013, 135, 2867–2870. [Google Scholar] [CrossRef]
- Barbosa da Silva, E.; Dall, E.; Briza, P.; Brandstetter, H.; Ferreira, R.S. Cruzain structures: Apocruzain and cruzain bound to S-methyl thiomethanesulfonate and implications for drug design. Acta Crystallogr. Sect. F 2019, 75, 419–427. [Google Scholar] [CrossRef]
- Karala, A.R.; Ruddock, L.W. Does S-Methyl Methanethiosulfonate Trap the Thiol–Disulfide State of Proteins? Antioxid. Redox Signal. 2007, 9, 527–531. [Google Scholar] [CrossRef]
- Adams, J.; Kauffman, M. Development of the Proteasome Inhibitor Velcade™ (Bortezomib). Cancer Investig. 2004, 22, 304–311. [Google Scholar] [CrossRef] [PubMed]
- Kabir, M.L.; Wang, F.; Clayton, A.H.A. Intrinsically Fluorescent Anti-Cancer Drugs. Biology 2022, 11, 1135. [Google Scholar] [CrossRef] [PubMed]
- Wilson, J.N.; Liu, W.; Brown, A.S.; Landgraf, R. Binding-induced, turn-on fluorescence of the EGFR/ERBB kinase inhibitor, lapatinib. Org. Biomol. Chem. 2015, 13, 5006–5011. [Google Scholar] [CrossRef] [Green Version]
- Klüter , S.; Simard , J.R.; Rode, H.B.; Grütter, C.; Pawar, V.; Raaijmakers, H.C.A.; Barf, T.A.; Rabiller, M.; van Otterlo , W.A.L.; Rauh , D. Characterization of Irreversible Kinase Inhibitors by Directly Detecting Covalent Bond Formation: A Tool for Dissecting Kinase Drug Resistance. ChemBioChem 2010, 11, 2557–2566. [Google Scholar] [CrossRef] [PubMed]
- Hodge, C.D.; Edwards, R.A.; Markin, C.J.; McDonald, D.; Pulvino, M.; Huen, M.S.Y.; Zhao, J.; Spyracopoulos, L.; Hendzel, M.J.; Glover, J.N.M. Covalent Inhibition of Ubc13 Affects Ubiquitin Signaling and Reveals Active Site Elements Important for Targeting. ACS Chem. Biol. 2015, 10, 1718–1728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niu, L.-Y.; Chen, Y.-Z.; Zheng, H.-R.; Wu, L.-Z.; Tung, C.-H.; Yang, Q.-Z. Design strategies of fluorescent probes for selective detection among biothiols. Chem. Soc. Rev. 2015, 44, 6143–6160. [Google Scholar] [CrossRef]
- Kwan, A.H.; Mobli, M.; Gooley, P.R.; King, G.F.; Mackay, J.P. Macromolecular NMR spectroscopy for the non-spectroscopist. FEBS J. 2011, 278, 687–703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maity, S.; Gundampati, R.K.; Suresh Kumar, T.K. NMR Methods to Characterize Protein-Ligand Interactions. Nat. Prod. Commun. 2019, 14, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Keiffer, S.; Carneiro, M.G.; Hollander, J.; Kobayashi, M.; Pogoryelev, D.; Ab, E.; Theisgen, S.; Müller, G.; Siegal, G. NMR in target driven drug discovery: Why not? J. Biomol. NMR 2020, 74, 521–529. [Google Scholar] [CrossRef]
- Ishima, R. Protein-Inhibitor Interaction Studies Using NMR. Appl. NMR Spectrosc. 2015, 1, 143–181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugiki, T.; Furuita, K.; Fujiwara, T.; Kojima, C. Current NMR Techniques for Structure-Based Drug Discovery. Molecules 2018, 23, 148. [Google Scholar] [CrossRef] [Green Version]
- Krishnan, V.V.; Rupp, B. Macromolecular Structure Determination: Comparison of X-ray Crystallography and NMR Spectroscopy. In eLS; John Wiley & Sons: Chichester, UK, 2012. [Google Scholar]
- Schirò, A.; Carlon, A.; Parigi, G.; Murshudov, G.; Calderone, V.; Ravera, E.; Luchinat, C. On the complementarity of X-ray and NMR data. J. Struct. Biol. X 2020, 4, 100019. [Google Scholar] [CrossRef]
- Bjorndahl, T.C.; Andrew, L.C.; Semenchenko, V.; Wishart, D.S. NMR Solution Structures of the Apo and Peptide-Inhibited Human Rhinovirus 3C Protease (Serotype 14): Structural and Dynamic Comparison. Biochemistry 2007, 46, 12945–12958. [Google Scholar] [CrossRef]
- Zhulenkovs, D.; Rudevica, Z.; Jaudzems, K.; Turks, M.; Leonchiks, A. Discovery and structure–activity relationship studies of irreversible benzisothiazolinone-based inhibitors against Staphylococcus aureus sortase A transpeptidase. Bioorg. Med. Chem. 2014, 22, 5988–6003. [Google Scholar] [CrossRef] [PubMed]
- Jaudzems, K.; Kurbatska, V.; Jēkabsons, A.; Bobrovs, R.; Rudevica, Z.; Leonchiks, A. Targeting Bacterial Sortase A with Covalent Inhibitors: 27 New Starting Points for Structure-Based Hit-to-Lead Optimization. ACS Infect. Dis. 2020, 6, 186–194. [Google Scholar] [CrossRef]
- Sastry, M.; Fiala, R.; Lipman, R.; Tomasz, M.; Patel, D.J. Solution Structure of the Monoalkylated Mitomycin C–DNA Complex. J. Mol. Biol. 1995, 247, 338–359. [Google Scholar] [CrossRef]
- Lin, C.H.; Patel, D.J. Solution Structure of the Covalent Duocarmycin A-DNA Duplex Complex. J. Mol. Biol. 1995, 248, 162–179. [Google Scholar] [CrossRef] [PubMed]
- Meyer, B.; Peters, T. NMR Spectroscopy Techniques for Screening and Identifying Ligand Binding to Protein Receptors. Angew. Chem. Int. Ed. 2003, 42, 864–890. [Google Scholar] [CrossRef]
- Furukawa, A.; Konuma, T.; Yanaka, S.; Sugase, K. Quantitative analysis of protein–ligand interactions by NMR. Prog. Nucl. Magn. Reson. Spectrosc. 2016, 96, 47–57. [Google Scholar] [CrossRef] [PubMed]
- Olp, M.D.; Sprague, D.J.; Goetz, C.J.; Kathman, S.G.; Wynia-Smith, S.L.; Shishodia, S.; Summers, S.B.; Xu, Z.; Statsyuk, A.V.; Smith, B.C. Covalent-Fragment Screening of BRD4 Identifies a Ligandable Site Orthogonal to the Acetyl-Lysine Binding Sites. ACS Chem. Biol. 2020, 15, 1036–1049. [Google Scholar] [CrossRef] [PubMed]
- Becker, W.; Bhattiprolu, K.C.; Gubensäk, N.; Zangger, K. Investigating Protein–Ligand Interactions by Solution Nuclear Magnetic Resonance Spectroscopy. ChemPhysChem 2018, 19, 895–906. [Google Scholar] [CrossRef] [Green Version]
- Felli, I.C.; Pierattelli, R. 13C Direct Detected NMR for Challenging Systems. Chem. Rev. 2022, 122, 9468–9496. [Google Scholar] [CrossRef]
- Cook, E.C.; Usher, G.A.; Showalter, S.A. Chapter Five The Use of 13C Direct-Detect NMR to Characterize Flexible and Disordered Proteins. In Methods in Enzymology; Rhoades, E., Ed.; Academic Press: Cambridge, MA, USA, 2018; Volume 611, pp. 81–100. [Google Scholar]
- Skora, L.; Mestan, J.; Fabbro, D.; Jahnke, W.; Grzesiek, S. NMR reveals the allosteric opening and closing of Abelson tyrosine kinase by ATP-site and myristoyl pocket inhibitors. Proc. Natl. Acad. Sci. USA 2013, 110, E4437–E4445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Metzler, W.J.; Yanchunas, J.; Weigelt, C.; Kish, K.; Klei, H.E.; Xie, D.; Zhang, Y.; Corbett, M.; Tamura, J.K.; He, B.; et al. Involvement of DPP-IV catalytic residues in enzyme–saxagliptin complex formation. Protein Sci. 2008, 17, 240–250. [Google Scholar] [CrossRef] [Green Version]
- Harner, M.J.; Frank, A.O.; Fesik, S.W. Fragment-based drug discovery using NMR spectroscopy. J. Biomol. NMR 2013, 56, 65–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagana Gowda, G.A.; Pascua, V.; Neto, F.C.; Raftery, D. Hydrogen–Deuterium Addition and Exchange in N-Ethylmaleimide Reaction with Glutathione Detected by NMR Spectroscopy. ACS Omega 2022, 7, 26928–26935. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, S.; Miller, R.M.; Tian, B.; Mullins, R.D.; Jacobson, M.P.; Taunton, J. Design of Reversible, Cysteine-Targeted Michael Acceptors Guided by Kinetic and Computational Analysis. J. Am. Chem. Soc. 2014, 136, 12624–12630. [Google Scholar] [CrossRef] [Green Version]
- Boudreau, E.A.; Pelczer, I.; Borer, P.N.; Heffron, G.J.; LaPlante, S.R. Changes in drug 13C NMR chemical shifts as a tool for monitoring interactions with DNA. Biophys. Chem. 2004, 109, 333–344. [Google Scholar] [CrossRef] [PubMed]
- Yabe, Y.; Guillaume, D.; Rich, D.H. Irreversible inhibition of papain by epoxysuccinyl peptides. Carbon-13 NMR characterization of the site of alkylation. J. Am. Chem. Soc. 1988, 110, 4043–4044. [Google Scholar] [CrossRef]
- Transue, T.R.; Krahn, J.M.; Gabel, S.A.; DeRose, E.F.; London, R.E. X-ray and NMR Characterization of Covalent Complexes of Trypsin, Borate, and Alcohols. Biochemistry 2004, 43, 2829–2839. [Google Scholar] [CrossRef] [PubMed]
- Glynn, S.J.; Gaffney, K.J.; Sainz, M.A.; Louie, S.G.; Petasis, N.A. Molecular characterization of the boron adducts of the proteasome inhibitor bortezomib with epigallocatechin-3-gallate and related polyphenols. Org. Biomol. Chem. 2015, 13, 3887–3899. [Google Scholar] [CrossRef] [Green Version]
- Moon, J.B.; Coleman, R.S.; Hanzlik, R.P. Reversible Covalent Inhibition of Papain by a Peptide Nitrile. 13C NMR Evidence for a Thioimidate Ester Adduct. J. Am. Chem. Soc. 1986, 108, 1350–1351. [Google Scholar] [CrossRef]
- Thompson, S.K.; Halbert, S.M.; Bossard, M.J.; Tomaszek, T.A.; Levy, M.A.; Zhao, B.; Smith, W.W.; Abdel-Meguid, S.S.; Janson, C.A.; D’Alessio, K.J.; et al. Design of potent and selective human cathepsin K inhibitors that span the active site. Proc. Natl. Acad. Sci. USA 1997, 94, 14249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Falgueyret, J.-P.; Oballa, R.M.; Okamoto, O.; Wesolowski, G.; Aubin, Y.; Rydzewski, R.M.; Prasit, P.; Riendeau, D.; Rodan, S.B.; Percival, M.D. Novel, Nonpeptidic Cyanamides as Potent and Reversible Inhibitors of Human Cathepsins K and L. J. Med. Chem. 2001, 44, 94–104. [Google Scholar] [CrossRef]
- Sorsa, T.; Heikkinen, S.; Abbott, M.B.; Abusamhadneh, E.; Laakso, T.; Tilgmann, C.; Serimaa, R.; Annila, A.; Rosevear, P.R.; Drakenberg, T.; et al. Binding of Levosimendan, a Calcium Sensitizer, to Cardiac Troponin C. J. Biol. Chem. 2001, 276, 9337–9343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robertson, I.M.; Pineda-Sanabria, S.E.; Yan, Z.; Kampourakis, T.; Sun, Y.-B.; Sykes, B.D.; Irving, M. Reversible Covalent Binding to Cardiac Troponin C by the Ca2+-Sensitizer Levosimendan. Biochemistry 2016, 55, 6032–6045. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Patricelli, M.P.; Cravatt, B.F. Activity-based protein profiling: The serine hydrolases. Proc. Natl. Acad. Sci. USA 1999, 96, 14694–14699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cravatt, B.F.; Wright, A.T.; Kozarich, J.W. Activity-Based Protein Profiling: From Enzyme Chemistry to Proteomic Chemistry. Annu. Rev. Biochem. 2008, 77, 383–414. [Google Scholar] [CrossRef] [Green Version]
- Nomura, D.K.; Dix, M.M.; Cravatt, B.F. Activity-based protein profiling for biochemical pathway discovery in cancer. Nat. Rev. Cancer 2010, 10, 630–638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cravatt, B.F.; Hsu, K.-L.; Weerapana, E. (Eds.) Activity-Based Protein Profiling. In Current Topics in Microbiology and Immunology; Springer: Cham, Switzerland, 2019; Volume 420, pp. 1–417. [Google Scholar]
- Bachovchin, D.A.; Cravatt, B.F. The pharmacological landscape and therapeutic potential of serine hydrolases. Nat. Rev. Drug Discov. 2012, 11, 52–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kato, D.; Boatright, K.M.; Berger, A.B.; Nazif, T.; Blum, G.; Ryan, C.; Chehade, K.A.H.; Salvesen, G.S.; Bogyo, M. Activity-based probes that target diverse cysteine protease families. Nat. Chem. Biol. 2005, 1, 33–38. [Google Scholar] [CrossRef]
- Chakrabarty, S.; Kahler, J.P.; van de Plassche, M.A.T.; Vanhoutte, R.; Verhelst, S.H.L. Recent Advances in Activity-Based Protein Profiling of Proteases. Curr. Top. Microbiol. Immunol. 2019, 420, 253–281. [Google Scholar] [CrossRef]
- Hoch, D.G.; Abegg, D.; Adibekian, A. Cysteine-reactive probes and their use in chemical proteomics. Chem. Commun. 2018, 54, 4501–4512. [Google Scholar] [CrossRef]
- Hameed, D.S.; Sapmaz, A.; Ovaa, H. How Chemical Synthesis of Ubiquitin Conjugates Helps To Understand Ubiquitin Signal Transduction. Bioconjugate Chem. 2017, 28, 805–815. [Google Scholar] [CrossRef]
- Patricelli, M.P.; Szardenings, A.K.; Liyanage, M.; Nomanbhoy, T.K.; Wu, M.; Weissig, H.; Aban, A.; Chun, D.; Tanner, S.; Kozarich, J.W. Functional Interrogation of the Kinome Using Nucleotide Acyl Phosphates. Biochemistry 2007, 46, 350–358. [Google Scholar] [CrossRef] [PubMed]
- Deng, H.; Lei, Q.; Wu, Y.; He, Y.; Li, W. Activity-based protein profiling: Recent advances in medicinal chemistry. Eur. J. Med. Chem. 2020, 191, 112151. [Google Scholar] [CrossRef]
- Fang, H.; Peng, B.; Ong, S.Y.; Wu, Q.; Li, L.; Yao, S.Q. Recent advances in activity-based probes (ABPs) and affinity-based probes (AfBPs) for profiling of enzymes. Chem. Sci. 2021, 12, 8288–8310. [Google Scholar] [CrossRef] [PubMed]
- Backus, K.M.; Correia, B.E.; Lum, K.M.; Forli, S.; Horning, B.D.; González-Páez, G.E.; Chatterjee, S.; Lanning, B.R.; Teijaro, J.R.; Olson, A.J.; et al. Proteome-wide covalent ligand discovery in native biological systems. Nature 2016, 534, 570–574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lanning, B.R.; Whitby, L.R.; Dix, M.M.; Douhan, J.; Gilbert, A.M.; Hett, E.C.; Johnson, T.O.; Joslyn, C.; Kath, J.C.; Niessen, S.; et al. A road map to evaluate the proteome-wide selectivity of covalent kinase inhibitors. Nat. Chem. Biol. 2014, 10, 760–767. [Google Scholar] [CrossRef] [Green Version]
- van Esbroeck, A.C.M.; Janssen, A.P.A.; Cognetta, A.B.; Ogasawara, D.; Shpak, G.; van der Kroeg, M.; Kantae, V.; Baggelaar, M.P.; de Vrij, F.M.S.; Deng, H.; et al. Activity-based protein profiling reveals off-target proteins of the FAAH inhibitor BIA 10-2474. Science 2017, 356, 1084–1087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bird, R.E.; Lemmel, S.A.; Yu, X.; Zhou, Q.A. Bioorthogonal Chemistry and Its Applications. Bioconjugate Chem. 2021, 32, 2457–2479. [Google Scholar] [CrossRef]
- Rostovtsev, V.V.; Green, L.G.; Fokin, V.V.; Sharpless, K.B. A Stepwise Huisgen Cycloaddition Process: Copper(I)-Catalyzed Regioselective “Ligation” of Azides and Terminal Alkynes. Angew. Chem. Int. Ed. 2002, 41, 2596–2599. [Google Scholar] [CrossRef]
- Parker, C.G.; Pratt, M.R. Click Chemistry in Proteomic Investigations. Cell 2020, 180, 605–632. [Google Scholar] [CrossRef] [PubMed]
- Smeenk, M.L.W.J.; Agramunt, J.; Bonger, K.M. Recent developments in bioorthogonal chemistry and the orthogonality within. Curr. Opin. Chem. Biol. 2021, 60, 79–88. [Google Scholar] [CrossRef] [PubMed]
- Agard, N.J.; Prescher, J.A.; Bertozzi, C.R. A Strain-Promoted [3 + 2] Azide−Alkyne Cycloaddition for Covalent Modification of Biomolecules in Living Systems. J. Am. Chem. Soc. 2004, 126, 15046–15047. [Google Scholar] [CrossRef]
- Oliveira, B.L.; Guo, Z.; Bernardes, G.J.L. Inverse electron demand Diels–Alder reactions in chemical biology. Chem. Soc. Rev. 2017, 46, 4895–4950. [Google Scholar] [CrossRef] [Green Version]
- van Rooden, E.J.; Florea, B.I.; Deng, H.; Baggelaar, M.P.; van Esbroeck, A.C.M.; Zhou, J.; Overkleeft, H.S.; van der Stelt, M. Mapping in vivo target interaction profiles of covalent inhibitors using chemical proteomics with label-free quantification. Nat. Protoc. 2018, 13, 752–767. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Jesson, M.I.; Seneviratne, U.I.; Lin, T.H.; Sharif, M.N.; Xue, L.; Nguyen, C.; Everley, R.A.; Trujillo, J.I.; Johnson, D.S.; et al. PF-06651600, a Dual JAK3/TEC Family Kinase Inhibitor. ACS Chem. Biol. 2019, 14, 1235–1242. [Google Scholar] [CrossRef]
- Yang, P.-Y.; Liu, K.; Ngai, M.H.; Lear, M.J.; Wenk, M.R.; Yao, S.Q. Activity-Based Proteome Profiling of Potential Cellular Targets of Orlistat An FDA-Approved Drug with Anti-Tumor Activities. J. Am. Chem. Soc. 2010, 132, 656–666. [Google Scholar] [CrossRef]
- Martin, J.G.; Ward, J.A.; Feyertag, F.; Zhang, L.; Couvertier, S.; Guckian, K.; Huber, K.V.M.; Johnson, D.S. Chemoproteomic Profiling of Covalent XPO1 Inhibitors to Assess Target Engagement and Selectivity. ChemBioChem 2021, 22, 2116–2123. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Hou, X.; Kummari, E.; Borazjani, A.; Edelmann, M.J.; Ross, M.K. Endocannabinoid hydrolases in avian HD11 macrophages identified by chemoproteomics: Inactivation by small-molecule inhibitors and pathogen-induced downregulation of their activity. Mol. Cell. Biochem. 2018, 444, 125–141. [Google Scholar] [CrossRef]
- Gjonaj, L.; Sapmaz, A.; Flierman, D.; Janssen, G.M.C.; van Veelen, P.A.; Ovaa, H. Development of a DUB-selective fluorogenic substrate. Chem. Sci. 2019, 10, 10290–10296. [Google Scholar] [CrossRef]
- van Rooden, E.J.; van Esbroeck, A.C.M.; Baggelaar, M.P.; Deng, H.; Florea, B.I.; Marques, A.R.A.; Ottenhoff, R.; Boot, R.G.; Overkleeft, H.S.; Aerts, J.M.F.G.; et al. Chemical Proteomic Analysis of Serine Hydrolase Activity in Niemann-Pick Type C Mouse Brain. Front. Neurosci. 2018, 12, 440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Blankman, J.L.; Cravatt, B.F. A Functional Proteomic Strategy to Discover Inhibitors for Uncharacterized Hydrolases. J. Am. Chem. Soc. 2007, 129, 9594–9595. [Google Scholar] [CrossRef] [PubMed]
- van der Wel, T.; Hilhorst, R.; den Dulk, H.; van den Hooven, T.; Prins, N.M.; Wijnakker, J.A.P.M.; Florea, B.I.; Lenselink, E.B.; van Westen, G.J.P.; Ruijtenbeek, R.; et al. Chemical genetics strategy to profile kinase target engagement reveals role of FES in neutrophil phagocytosis. Nat. Commun. 2020, 11, 3216. [Google Scholar] [CrossRef] [PubMed]
- Kooij, R.; Liu, S.; Sapmaz, A.; Xin, B.-T.; Janssen, G.M.C.; van Veelen, P.A.; Ovaa, H.; Dijke, P.t.; Geurink, P.P. Small-Molecule Activity-Based Probe for Monitoring Ubiquitin C-Terminal Hydrolase L1 (UCHL1) Activity in Live Cells and Zebrafish Embryos. J. Am. Chem. Soc. 2020, 142, 16825–16841. [Google Scholar] [CrossRef]
- Leal, J.; Martínez-Díez, M.; García-Hernández, V.; Moneo, V.; Domingo, A.; Bueren-Calabuig, J.; Negri, A.; Gago, F.; Guillén-Navarro, M.; Avilés, P.; et al. PM01183, a new DNA minor groove covalent binder with potent in vitro and in vivo anti-tumour activity. Br. J. Pharmacol. 2010, 161, 1099–1110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, Y.; Kim, R.Q.; Kooij, R.; Ovaa, H.; Sapmaz, A.; Geurink, P.P. Chemical Toolkit for PARK7: Potent, Selective, and High-Throughput. J. Med. Chem. 2022, 65, 13288–13304. [Google Scholar] [CrossRef]
- Dana, D.; Pathak, S.K. A Review of Small Molecule Inhibitors and Functional Probes of Human Cathepsin L. Molecules 2020, 25, 698. [Google Scholar] [CrossRef] [Green Version]
- Bogyo, M.; Verhelst, S.; Bellingard-Dubouchaud, V.; Toba, S.; Greenbaum, D. Selective targeting of lysosomal cysteine proteases with radiolabeled electrophilic substrate analogs. Chem. Biol. 2000, 7, 27–38. [Google Scholar] [CrossRef]
- Pellegatti, M. Preclinical in vivo ADME studies in drug development: A critical review. Expert Opin. Drug Metab. Toxicol. 2012, 8, 161–172. [Google Scholar] [CrossRef]
- Harding, C.R.; Scott, I.R. Fluorography—Limitations on its use for the quantitative detection of 3H- and 14C-labeled proteins in polyacrylamide gels. Anal. Biochem. 1983, 129, 371–376. [Google Scholar] [CrossRef] [PubMed]
- Miyahisa, I.; Sameshima, T.; Hixon, M.S. Rapid Determination of the Specificity Constant of Irreversible Inhibitors (kinact/KI) by Means of an Endpoint Competition Assay. Angew. Chem. Int. Ed. 2015, 54, 14099–14102. [Google Scholar] [CrossRef] [PubMed]
- Tsou, H.-R.; Overbeek-Klumpers, E.G.; Hallett, W.A.; Reich, M.F.; Floyd, M.B.; Johnson, B.D.; Michalak, R.S.; Nilakantan, R.; Discafani, C.; Golas, J.; et al. Optimization of 6,7-Disubstituted-4-(arylamino)quinoline-3-carbonitriles as Orally Active, Irreversible Inhibitors of Human Epidermal Growth Factor Receptor-2 Kinase Activity. J. Med. Chem. 2005, 48, 1107–1131. [Google Scholar] [CrossRef] [PubMed]
- Scicinski, J.; Oronsky, B.; Taylor, M.; Luo, G.; Musick, T.; Marini, J.; Adams, C.M.; Fitch, W.L. Preclinical Evaluation of the Metabolism and Disposition of RRx-001, a Novel Investigative Anticancer Agent. Drug Metab. Dispos. 2012, 40, 1810–1816. [Google Scholar] [CrossRef] [Green Version]
- McClure, R.A.; Williams, J.D. Impact of Mass Spectrometry-Based Technologies and Strategies on Chemoproteomics as a Tool for Drug Discovery. ACS Med. Chem. Lett. 2018, 9, 785–791. [Google Scholar] [CrossRef]
- Counihan, J.L.; Ford, B.; Nomura, D.K. Mapping proteome-wide interactions of reactive chemicals using chemoproteomic platforms. Curr. Opin. Chem. Biol. 2016, 30, 68–76. [Google Scholar] [CrossRef] [Green Version]
- Maurais, A.J.; Weerapana, E. Reactive-cysteine profiling for drug discovery. Curr. Opin. Chem. Biol. 2019, 50, 29–36. [Google Scholar] [CrossRef]
- Neilson, K.A.; Ali, N.A.; Muralidharan, S.; Mirzaei, M.; Mariani, M.; Assadourian, G.; Lee, A.; van Sluyter, S.C.; Haynes, P.A. Less label, more free: Approaches in label-free quantitative mass spectrometry. Proteomics 2011, 11, 535–553. [Google Scholar] [CrossRef]
- Rozanova, S.; Barkovits, K.; Nikolov, M.; Schmidt, C.; Urlaub, H.; Marcus, K. Quantitative Mass Spectrometry-Based Proteomics: An Overview. In Quantitative Methods in Proteomics; Marcus, K., Eisenacher, M., Sitek, B., Eds.; Humana Press: New York, NY, USA, 2021; pp. 85–100. [Google Scholar]
- Wang, C.; Weerapana, E.; Blewett, M.M.; Cravatt, B.F. A chemoproteomic platform to quantitatively map targets of lipid-derived electrophiles. Nat. Methods 2014, 11, 79–85. [Google Scholar] [CrossRef] [Green Version]
- Benns, H.J.; Wincott, C.J.; Tate, E.W.; Child, M.A. Activity- and reactivity-based proteomics: Recent technological advances and applications in drug discovery. Curr. Opin. Chem. Biol. 2021, 60, 20–29. [Google Scholar] [CrossRef]
- Weerapana, E.; Wang, C.; Simon, G.M.; Richter, F.; Khare, S.; Dillon, M.B.D.; Bachovchin, D.A.; Mowen, K.; Baker, D.; Cravatt, B.F. Quantitative reactivity profiling predicts functional cysteines in proteomes. Nature 2010, 468, 790–795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuljanin, M.; Mitchell, D.C.; Schweppe, D.K.; Gikandi, A.S.; Nusinow, D.P.; Bulloch, N.J.; Vinogradova, E.V.; Wilson, D.L.; Kool, E.T.; Mancias, J.D.; et al. Reimagining high-throughput profiling of reactive cysteines for cell-based screening of large electrophile libraries. Nat. Biotechnol. 2021, 39, 630–641. [Google Scholar] [CrossRef] [PubMed]
- Boatner, L.; Palafox, M.; Schweppe, D.; Backus, K. CysDB: A Human Cysteine Database based on Experimental Quantitative Chemoproteomics. ChemRxiv 2022. [Google Scholar] [CrossRef]
- Hsu, J.-L.; Chen, S.-H. Stable isotope dimethyl labelling for quantitative proteomics and beyond. Philos. Trans. R. Soc. A 2016, 374, 20150364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adibekian, A.; Martin, B.R.; Wang, C.; Hsu, K.-L.; Bachovchin, D.A.; Niessen, S.; Hoover, H.; Cravatt, B.F. Click-generated triazole ureas as ultrapotent in vivo–active serine hydrolase inhibitors. Nat. Chem. Biol. 2011, 7, 469–478. [Google Scholar] [CrossRef]
- Mann, M. Functional and quantitative proteomics using SILAC. Nat. Rev. Mol. Cell Biol. 2006, 7, 952–958. [Google Scholar] [CrossRef]
- Ong, S.-E.; Blagoev, B.; Kratchmarova, I.; Kristensen, D.B.; Steen, H.; Pandey, A.; Mann, M. Stable Isotope Labeling by Amino Acids in Cell Culture, SILAC, as a Simple and Accurate Approach to Expression Proteomics. Mol. Cell. Proteom. 2002, 1, 376–386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Browne, C.M.; Jiang, B.; Ficarro, S.B.; Doctor, Z.M.; Johnson, J.L.; Card, J.D.; Sivakumaren, S.C.; Alexander, W.M.; Yaron, T.M.; Murphy, C.J.; et al. A Chemoproteomic Strategy for Direct and Proteome-Wide Covalent Inhibitor Target-Site Identification. J. Am. Chem. Soc. 2019, 141, 191–203. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Sun, Y.; Zhang, T.; Shu, L.; Roepstorff, P.; Yang, F. Quantitative Proteomics Using Isobaric Labeling: A Practical Guide. Genom. Proteom. Bioinf. 2021, 19, 689–706. [Google Scholar] [CrossRef] [PubMed]
- Weerapana, E.; Speers, A.E.; Cravatt, B.F. Tandem orthogonal proteolysis-activity-based protein profiling (TOP-ABPP)—A general method for mapping sites of probe modification in proteomes. Nat. Protoc. 2007, 2, 1414–1425. [Google Scholar] [CrossRef]
- Zanon, P.R.A.; Lewald, L.; Hacker, S.M. Isotopically Labeled Desthiobiotin Azide (isoDTB) Tags Enable Global Profiling of the Bacterial Cysteinome. Angew. Chem. Int. Ed. 2020, 59, 2829–2836. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Gao, J.; Che, J.; Jia, G.; Wang, C. A Dimethyl-Labeling-Based Strategy for Site-Specifically Quantitative Chemical Proteomics. Anal. Chem. 2018, 90, 9576–9582. [Google Scholar] [CrossRef]
- Hsu, J.-L.; Huang, S.-Y.; Chow, N.-H.; Chen, S.-H. Stable-Isotope Dimethyl Labeling for Quantitative Proteomics. Anal. Chem. 2003, 75, 6843–6852. [Google Scholar] [CrossRef]
- Boersema, P.J.; Raijmakers, R.; Lemeer, S.; Mohammed, S.; Heck, A.J.R. Multiplex peptide stable isotope dimethyl labeling for quantitative proteomics. Nat. Protoc. 2009, 4, 484–494. [Google Scholar] [CrossRef]
- Li, N.; Kuo, C.-L.; Paniagua, G.; van den Elst, H.; Verdoes, M.; Willems, L.I.; van der Linden, W.A.; Ruben, M.; van Genderen, E.; Gubbens, J.; et al. Relative quantification of proteasome activity by activity-based protein profiling and LC-MS/MS. Nat. Protoc. 2013, 8, 1155–1168. [Google Scholar] [CrossRef]
- Baggelaar, M.P.; Chameau, P.J.P.; Kantae, V.; Hummel, J.; Hsu, K.-L.; Janssen, F.; van der Wel, T.; Soethoudt, M.; Deng, H.; den Dulk, H.; et al. Highly Selective, Reversible Inhibitor Identified by Comparative Chemoproteomics Modulates Diacylglycerol Lipase Activity in Neurons. J. Am. Chem. Soc. 2015, 137, 8851–8857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vinogradova, E.V.; Zhang, X.; Remillard, D.; Lazar, D.C.; Suciu, R.M.; Wang, Y.; Bianco, G.; Yamashita, Y.; Crowley, V.M.; Schafroth, M.A.; et al. An Activity-Guided Map of Electrophile-Cysteine Interactions in Primary Human T Cells. Cell 2020, 182, 1009–1026.e1029. [Google Scholar] [CrossRef]
- Werner, T.; Becher, I.; Sweetman, G.; Doce, C.; Savitski, M.M.; Bantscheff, M. High-Resolution Enabled TMT 8-plexing. Anal. Chem. 2012, 84, 7188–7194. [Google Scholar] [CrossRef] [PubMed]
- Zanon, P.R.A.; Yu, F.; Musacchio, P.; Lewald, L.; Zollo, M.; Krauskopf, K.; Mrdović, D.; Raunft, P.; Maher, T.E.; Cigler, M.; et al. Profiling the Proteome-Wide Selectivity of Diverse Electrophiles. ChemRxiv 2021. [Google Scholar] [CrossRef]
- Adibekian, A.; Martin, B.R.; Chang, J.W.; Hsu, K.-L.; Tsuboi, K.; Bachovchin, D.A.; Speers, A.E.; Brown, S.J.; Spicer, T.; Fernandez-Vega, V.; et al. Confirming Target Engagement for Reversible Inhibitors in Vivo by Kinetically Tuned Activity-Based Probes. J. Am. Chem. Soc. 2012, 134, 10345–10348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Senkane, K.; Vinogradova, E.V.; Suciu, R.M.; Crowley, V.M.; Zaro, B.W.; Bradshaw, J.M.; Brameld, K.A.; Cravatt, B.F. The Proteome-Wide Potential for Reversible Covalency at Cysteine. Angew. Chem. Int. Ed. 2019, 58, 11385–11389. [Google Scholar] [CrossRef]
- Blum, G.; Mullins, S.R.; Keren, K.; Fonovič, M.; Jedeszko, C.; Rice, M.J.; Sloane, B.F.; Bogyo, M. Dynamic imaging of protease activity with fluorescently quenched activity-based probes. Nat. Chem. Biol. 2005, 1, 203–209. [Google Scholar] [CrossRef]
- Edgington-Mitchell, L.E.; Bogyo, M.; Verdoes, M. Live Cell Imaging and Profiling of Cysteine Cathepsin Activity Using a Quenched Activity-Based Probe. In Activity-Based Proteomics: Methods and Protocols; Overkleeft, H.S., Florea, B.I., Eds.; Springer: New York, NY, USA, 2017; pp. 145–159. [Google Scholar]
- Blum, G.; Weimer, R.M.; Edgington, L.E.; Adams, W.; Bogyo, M. Comparative Assessment of Substrates and Activity Based Probes as Tools for Non-Invasive Optical Imaging of Cysteine Protease Activity. PLoS ONE 2009, 4, e6374. [Google Scholar] [CrossRef] [Green Version]
- Blum, G.; von Degenfeld, G.; Merchant, M.J.; Blau, H.M.; Bogyo, M. Noninvasive optical imaging of cysteine protease activity using fluorescently quenched activity-based probes. Nat. Chem. Biol. 2007, 3, 668–677. [Google Scholar] [CrossRef] [PubMed]
- van Rooden, E.J.; Kohsiek, M.; Kreekel, R.; van Esbroeck, A.C.M.; van den Nieuwendijk, A.M.C.H.; Janssen, A.P.A.; van den Berg, R.J.B.H.N.; Overkleeft, H.S.; van der Stelt, M. Design and Synthesis of Quenched Activity-based Probes for Diacylglycerol Lipase and α,β-Hydrolase Domain Containing Protein 6. Chem. Asian J. 2018, 13, 3491–3500. [Google Scholar] [CrossRef]
- Serim, S.; Baer, P.; Verhelst, S.H.L. Mixed alkyl aryl phosphonate esters as quenched fluorescent activity-based probes for serine proteases. Org. Biomol. Chem. 2015, 13, 2293–2299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verdoes, M.; Oresic Bender, K.; Segal, E.; van der Linden, W.A.; Syed, S.; Withana, N.P.; Sanman, L.E.; Bogyo, M. Improved Quenched Fluorescent Probe for Imaging of Cysteine Cathepsin Activity. J. Am. Chem. Soc. 2013, 135, 14726–14730. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Liu, H.; Pan, Z. A general approach for the development of fluorogenic probes suitable for no-wash imaging of kinases in live cells. Chem. Commun. 2014, 50, 15319–15322. [Google Scholar] [CrossRef] [PubMed]
- Janssen, A.P.A.; van Hengst, J.M.A.; Béquignon, O.J.M.; Deng, H.; van Westen, G.J.P.; van der Stelt, M. Structure Kinetics Relationships and Molecular Dynamics Show Crucial Role for Heterocycle Leaving Group in Irreversible Diacylglycerol Lipase Inhibitors. J. Med. Chem. 2019, 62, 7910–7922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petri, L.; Ábrányi-Balogh, P.; Varga, P.R.; Imre, T.; Keserű, G.M. Comparative reactivity analysis of small-molecule thiol surrogates. Bioorg. Med. Chem. 2020, 28, 115357. [Google Scholar] [CrossRef] [PubMed]
- Reddi, R.N.; Resnick, E.; Rogel, A.; Rao, B.V.; Gabizon, R.; Goldenberg, K.; Gurwicz, N.; Zaidman, D.; Plotnikov, A.; Barr, H.; et al. Tunable Methacrylamides for Covalent Ligand Directed Release Chemistry. J. Am. Chem. Soc. 2021, 143, 4979–4992. [Google Scholar] [CrossRef] [PubMed]
- Kawahata, W.; Asami, T.; Fujii, I.; Sawa, M. ‘Turn On/Off’ fluorescence probe for the screening of unactivated Bruton’s tyrosine kinase. Bioorg. Med. Chem. Lett. 2015, 25, 2141–2145. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Gao, X.; Shi, W.; Ma, H. Design Strategies for Water-Soluble Small Molecular Chromogenic and Fluorogenic Probes. Chem. Rev. 2014, 114, 590–659. [Google Scholar] [CrossRef]
- Shie, J.-J.; Liu, Y.-C.; Lee, Y.-M.; Lim, C.; Fang, J.-M.; Wong, C.-H. An Azido-BODIPY Probe for Glycosylation: Initiation of Strong Fluorescence upon Triazole Formation. J. Am. Chem. Soc. 2014, 136, 9953–9961. [Google Scholar] [CrossRef]
- Blau, R.; Epshtein, Y.; Pisarevsky, E.; Tiram, G.; Israeli Dangoor, S.; Yeini, E.; Krivitsky, A.; Eldar-Boock, A.; Ben-Shushan, D.; Gibori, H.; et al. Image-guided surgery using near-infrared Turn-ON fluorescent nanoprobes for precise detection of tumor margins. Theranostics 2018, 8, 3437–3460. [Google Scholar] [CrossRef]
- Ben-Nun, Y.; Merquiol, E.; Brandis, A.; Turk, B.; Scherz, A.; Blum, G. Photodynamic Quenched Cathepsin Activity Based Probes for Cancer Detection and Macrophage Targeted Therapy. Theranostics 2015, 5, 847–862. [Google Scholar] [CrossRef] [Green Version]
- Weiss-Sadan, T.; Ben-Nun, Y.; Maimoun, D.; Merquiol, E.; Abd-Elrahman, I.; Gotsman, I.; Blum, G. A Theranostic Cathepsin Activity-Based Probe for Noninvasive Intervention in Cardiovascular Diseases. Theranostics 2019, 9, 5731–5738. [Google Scholar] [CrossRef]
- Evans, E.K.; Tester, R.; Aslanian, S.; Karp, R.; Sheets, M.; Labenski, M.T.; Witowski, S.R.; Lounsbury, H.; Chaturvedi, P.; Mazdiyasni, H.; et al. Inhibition of Btk with CC-292 Provides Early Pharmacodynamic Assessment of Activity in Mice and Humans. J. Pharmacol. Exp. Ther. 2013, 346, 219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Callaway, E. Revolutionary cryo-EM is taking over structural biology. Nature 2020, 578, 201. [Google Scholar] [CrossRef] [Green Version]
- Chiu, Y.-H.; Ko, K.-T.; Yang, T.-J.; Wu, K.-P.; Ho, M.-R.; Draczkowski, P.; Hsu, S.-T.D. Direct Visualization of a 26 kDa Protein by Cryo-Electron Microscopy Aided by a Small Scaffold Protein. Biochemistry 2021, 60, 1075–1079. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Huynh, D.T.; Yeates, T.O. A 3.8 Å resolution cryo-EM structure of a small protein bound to an imaging scaffold. Nat. Commun. 2019, 10, 1864. [Google Scholar] [CrossRef] [Green Version]
- Borgnia, M.J.; Banerjee, S.; Merk, A.; Matthies, D.; Bartesaghi, A.; Rao, P.; Pierson, J.; Earl, L.A.; Falconieri, V.; Subramaniam, S.; et al. Using Cryo-EM to Map Small Ligands on Dynamic Metabolic Enzymes: Studies with Glutamate Dehydrogenase. Mol. Pharmacol. 2016, 89, 645. [Google Scholar] [CrossRef] [Green Version]
- Toelzer, C.; Gupta, K.; Yadav, S.K.N.; Borucu, U.; Davidson, A.D.; Kavanagh Williamson, M.; Shoemark, D.K.; Garzoni, F.; Staufer, O.; Milligan, R.; et al. Free fatty acid binding pocket in the locked structure of SARS-CoV-2 spike protein. Science 2020, 370, 725–730. [Google Scholar] [CrossRef] [PubMed]
- Greber, B.J.; Perez-Bertoldi, J.M.; Lim, K.; Iavarone, A.T.; Toso, D.B.; Nogales, E. The cryoelectron microscopy structure of the human CDK-activating kinase. Proc. Natl. Acad. Sci. USA 2020, 117, 22849–22857. [Google Scholar] [CrossRef]
- Robertson, M.J.; van Zundert, G.C.P.; Borrelli, K.; Skiniotis, G. GemSpot: A Pipeline for Robust Modeling of Ligands into Cryo-EM Maps. Structure 2020, 28, 707–716.e703. [Google Scholar] [CrossRef]
- Chengalroyen, M.D.; Mason, M.K.; Borsellini, A.; Tassoni, R.; Abrahams, G.L.; Lynch, S.; Ahn, Y.-M.; Ambler, J.; Young, K.; Crowley, B.M.; et al. DNA-Dependent Binding of Nargenicin to DnaE1 Inhibits Replication in Mycobacterium tuberculosis. ACS Infect. Dis. 2022, 8, 612–625. [Google Scholar] [CrossRef]
- Lindberg, P.; Nordberg, P.; Alminger, T.; Braendstroem, A.; Wallmark, B. The mechanism of action of the antisecretory agent omeprazole. J. Med. Chem. 1986, 29, 1327–1329. [Google Scholar] [CrossRef]
- Mukherjee, H.; Grimster, N.P. Beyond cysteine: Recent developments in the area of targeted covalent inhibition. Curr. Opin. Chem. Biol. 2018, 44, 30–38. [Google Scholar] [CrossRef]
- Jones, L.H. Chapter Four Design of next-generation covalent inhibitors: Targeting residues beyond cysteine. In Annual Reports in Medicinal Chemistry; Ward, R.A., Grimster, N.P., Eds.; Academic Press: Cambridge, MA, USA, 2021; Volume 56, pp. 95–134. [Google Scholar]
- Gambini, L.; Baggio, C.; Udompholkul, P.; Jossart, J.; Salem, A.F.; Perry, J.J.P.; Pellecchia, M. Covalent Inhibitors of Protein–Protein Interactions Targeting Lysine, Tyrosine, or Histidine Residues. J. Med. Chem. 2019, 62, 5616–5627. [Google Scholar] [CrossRef] [Green Version]
- Pettinger, J.; Jones, K.; Cheeseman, M.D. Lysine-Targeting Covalent Inhibitors. Angew. Chem. Int. Ed. 2017, 56, 15200–15209. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Guiley, K.Z.; Shokat, K.M. Chemical acylation of an acquired serine suppresses oncogenic signaling of K-Ras(G12S). Nat. Chem. Biol. 2022, 18, 1177–1183. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Morstein, J.; Ecker, A.K.; Guiley, K.Z.; Shokat, K.M. Chemoselective Covalent Modification of K-Ras(G12R) with a Small Molecule Electrophile. J. Am. Chem. Soc. 2022, 144, 15916–15921. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Sun, J.; Zhu, C.; Tang, G.; Wang, W.; Xu, M.; Xiang, M.; Zhang, C.-J.; Zhang, Z.-M.; Gao, L.; et al. Cell-Active, Reversible, and Irreversible Covalent Inhibitors That Selectively Target the Catalytic Lysine of BCR-ABL Kinase. Angew. Chem. Int. Ed. 2022, 61, e202203878. [Google Scholar] [CrossRef]
- Lill, J.R.; Mathews, W.R.; Rose, C.M.; Schirle, M. Proteomics in the pharmaceutical and biotechnology industry: A look to the next decade. Expert Rev. Proteom. 2021, 18, 503–526. [Google Scholar] [CrossRef]
- Aranda, J.; Orozco, M. RNA-Dependent RNA Polymerase From SARS-CoV-2. Mechanism Of Reaction And Inhibition By Remdesivir. bioRxiv 2020. [Google Scholar] [CrossRef]
- Zhao, Y.; Fang, C.; Zhang, Q.; Zhang, R.; Zhao, X.; Duan, Y.; Wang, H.; Zhu, Y.; Feng, L.; Zhao, J.; et al. Crystal structure of SARS-CoV-2 main protease in complex with protease inhibitor PF-07321332. Protein Cell 2022, 13, 689–693. [Google Scholar] [CrossRef]
- Kalyukina, M.; Yosaatmadja, Y.; Middleditch, M.J.; Patterson, A.V.; Smaill, J.B.; Squire, C.J. TAS-120 Cancer Target Binding: Defining Reactivity and Revealing the First Fibroblast Growth Factor Receptor 1 (FGFR1) Irreversible Structure. ChemMedChem 2019, 14, 494–500. [Google Scholar] [CrossRef] [PubMed]










| Prerequisites | Compatibility | Structural | Characterization | ||||||
| Detection Method | Ligand Resynthesis | Protein Optimization | Reversible Covalent | Whole proteome a | Modified Amino Acid b | Bond Isoform | Covalent Occupancy | Reversibility | Notes |
| 2. Mass Spectrometry | Relatively fast and easy. Bottom-up MS(/MS) compatible with large proteins (in mixtures) | ||||||||
| 2.1. Top-down MS | − | + | + | ~ c | − | − | + | + | |
| 2.2. Bottom-up MS | − | − | ~ | + | ~ d | − | ~ | − | |
| 2.3. MS/MS | − | − | ~ | + | + | − | + | − | |
| 3. Protein Crystallography | − | + | ++ | - | + | + | - | − | Most informative but laborious |
| 4. Intrinsic Fluorescence/Absorbance | − e | − | + | - | − | − | ~ | + | Limited ligand compatibility |
| 5. Nuclear Magnetic Resonance | + | ~ | ++ | + | ~ | ++ | + | + | Compatible with labile adduct detection in solution |
| 6. Activity-based Protein Profiling | Detection of modified (off-) target proteins in whole proteomes | ||||||||
| 6.1. Gel Electrophoresis | + | − | ~ | ++ | − | − | + | + | |
| 6.2. Chemoproteomic Platforms | + | − | ~ | ++ | + | − | ~ f | + | |
| 6.3. Homogeneous/plate-based | + | − | + | ++ | − | − | + | − | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mons, E.; Kim, R.Q.; Mulder, M.P.C. Technologies for Direct Detection of Covalent Protein–Drug Adducts. Pharmaceuticals 2023, 16, 547. https://doi.org/10.3390/ph16040547
Mons E, Kim RQ, Mulder MPC. Technologies for Direct Detection of Covalent Protein–Drug Adducts. Pharmaceuticals. 2023; 16(4):547. https://doi.org/10.3390/ph16040547
Chicago/Turabian StyleMons, Elma, Robbert Q. Kim, and Monique P. C. Mulder. 2023. "Technologies for Direct Detection of Covalent Protein–Drug Adducts" Pharmaceuticals 16, no. 4: 547. https://doi.org/10.3390/ph16040547
APA StyleMons, E., Kim, R. Q., & Mulder, M. P. C. (2023). Technologies for Direct Detection of Covalent Protein–Drug Adducts. Pharmaceuticals, 16(4), 547. https://doi.org/10.3390/ph16040547