_Kim.png)
In Silico Identification of Promising New Pyrazole Derivative-Based Small Molecules for Modulating CRMP2, C-RAF, CYP17, VEGFR, C-KIT, and HDAC—Application towards Cancer Therapeutics

 , ,
, ,  ,
,  ,
,  ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Molecular Docking
2.1.1. Ligand (Pyrazole Derivatives) Preparation
2.1.2. Protein Structure Selection and Preparation
2.1.3. Configuration File Generation and Molecular Docking Execution
2.2. Molecular Dynamic Simulation
2.3. In Silico Drug-Likeness and ADME_T Profiles Estimation
3. Results and Discussion
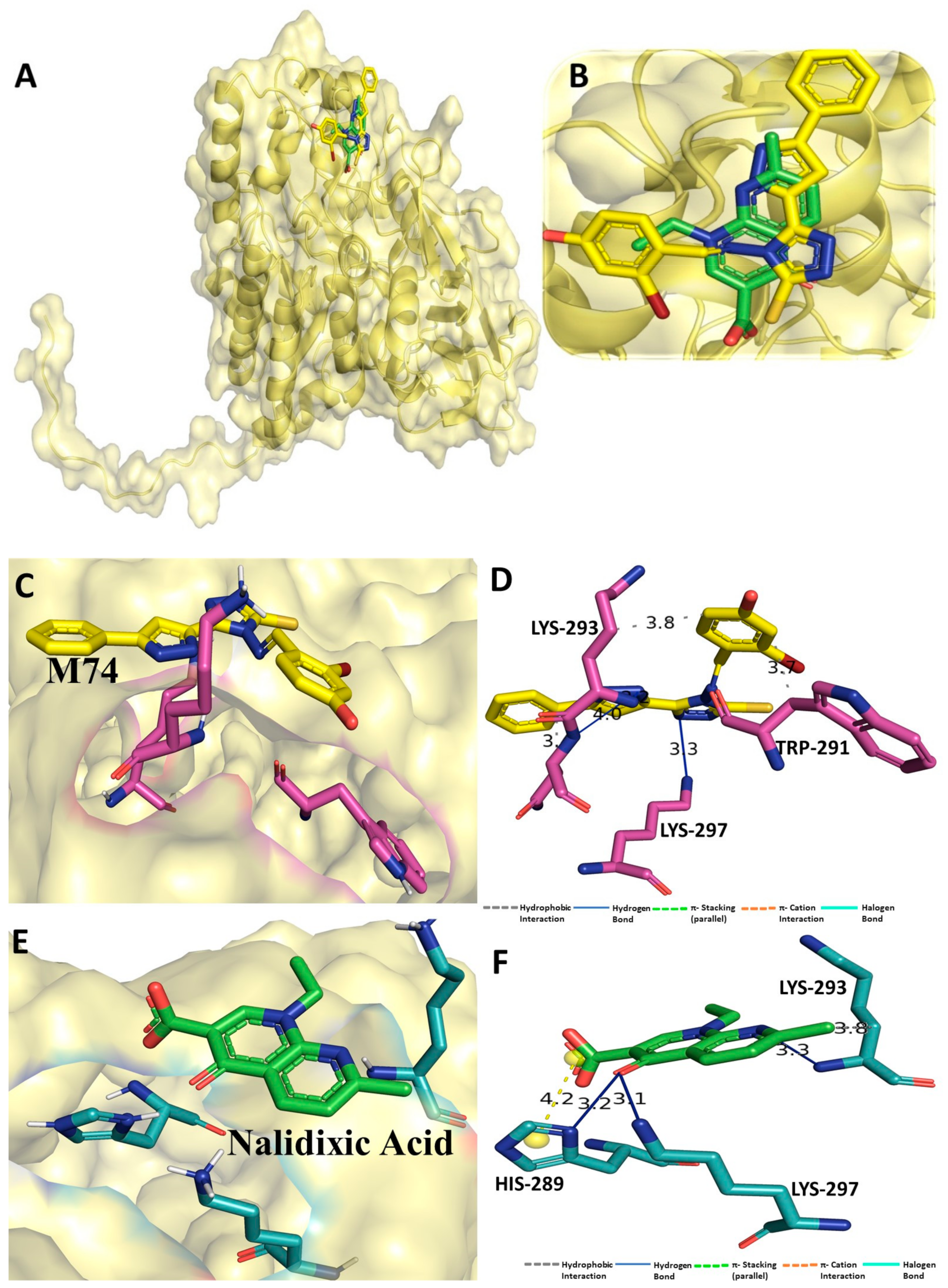
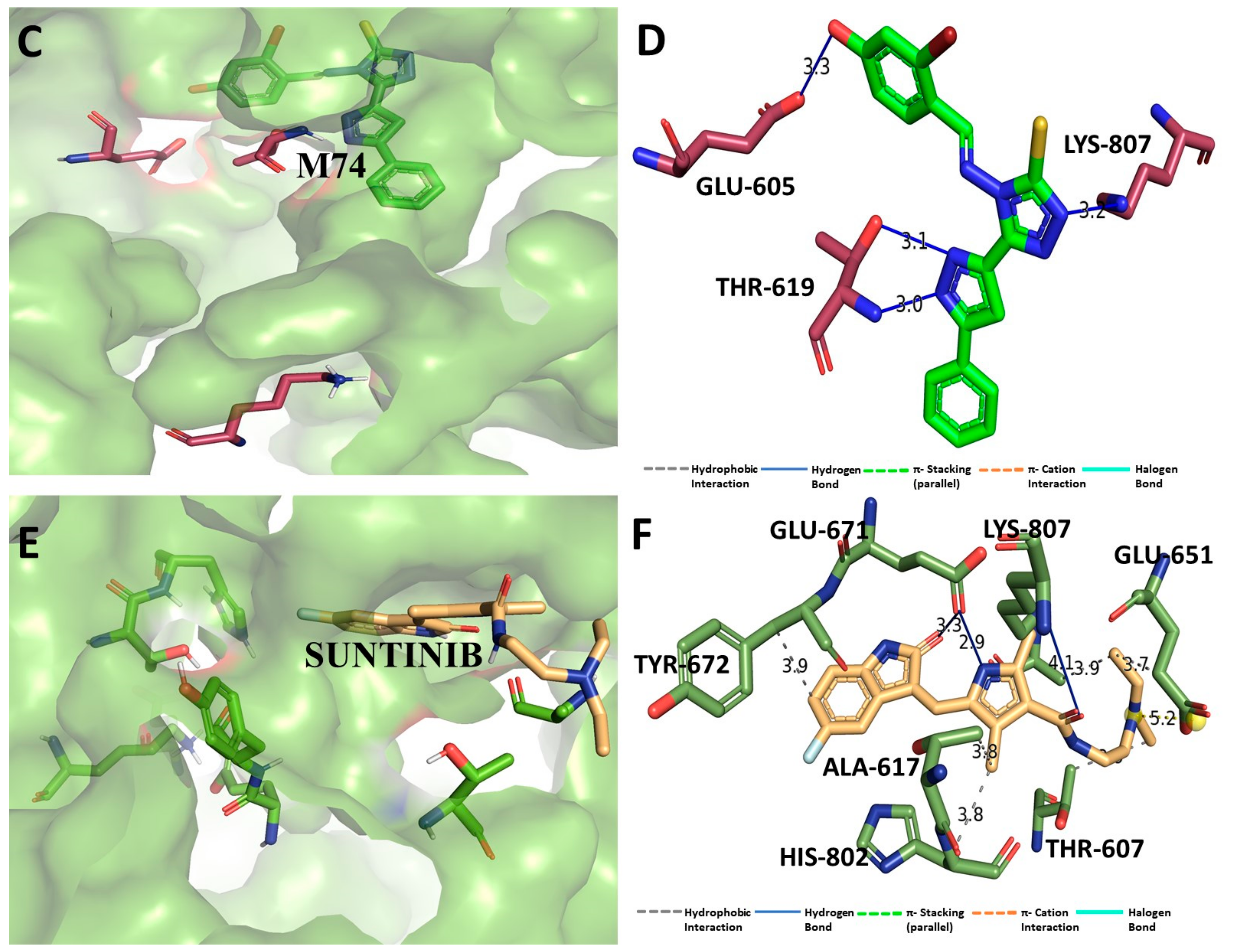
3.1. Binding Interaction Profile Analysis of CRMP2 and Pyrazole Derivative Complex
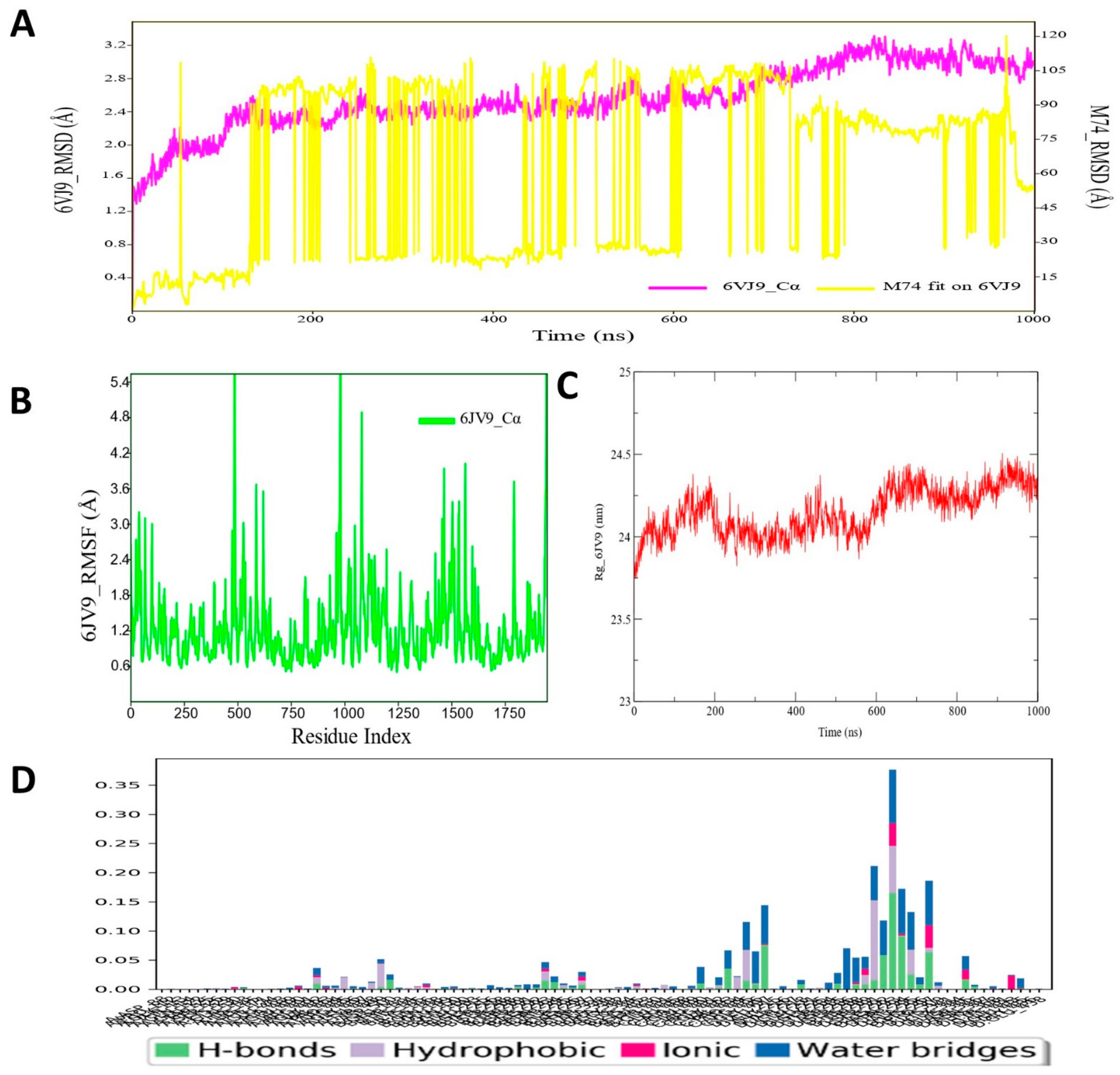
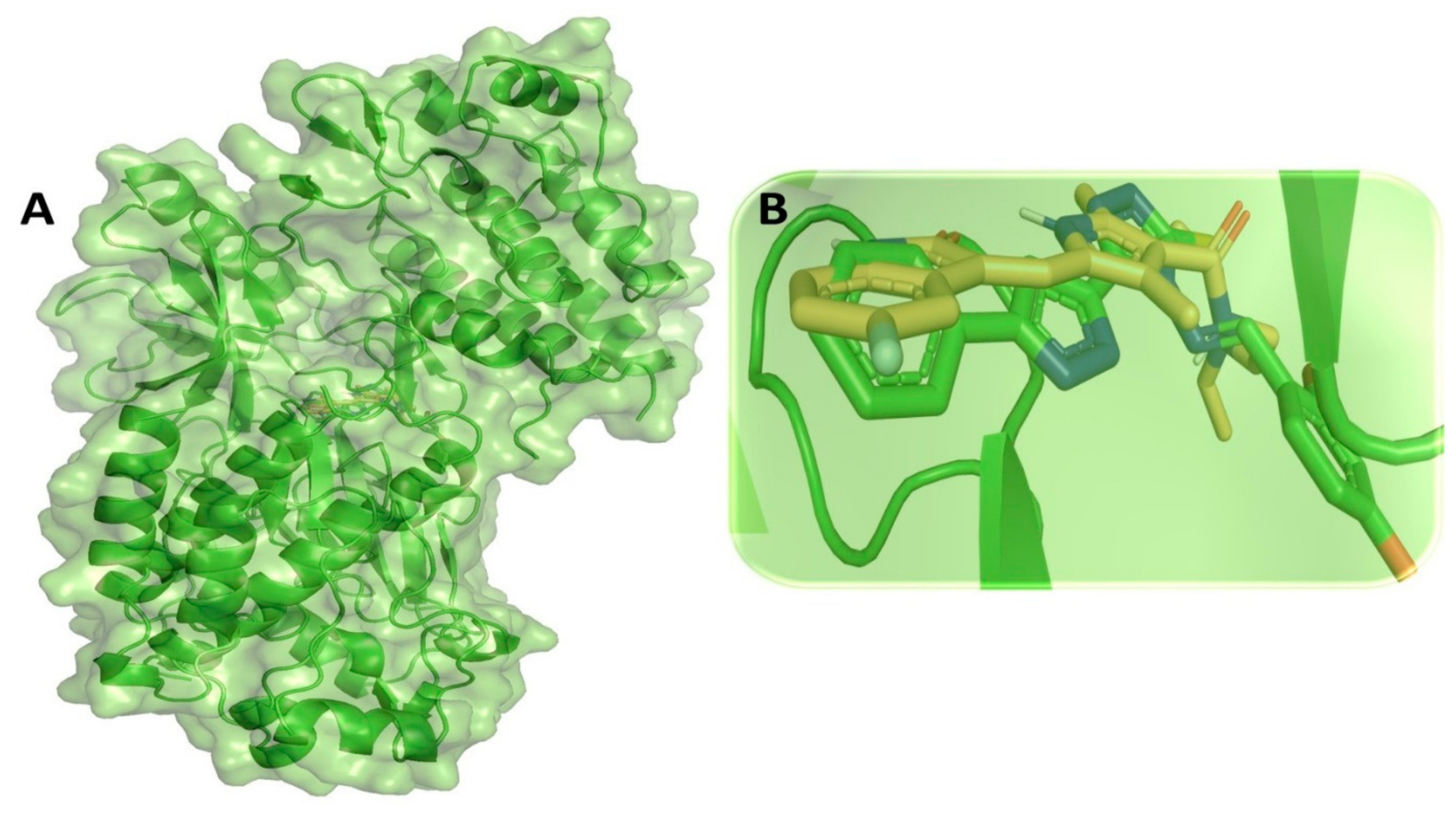
3.2. Molecular Dynamic Simulation Analysis of CRMP2–Pyrazole Derivative M74 Complex
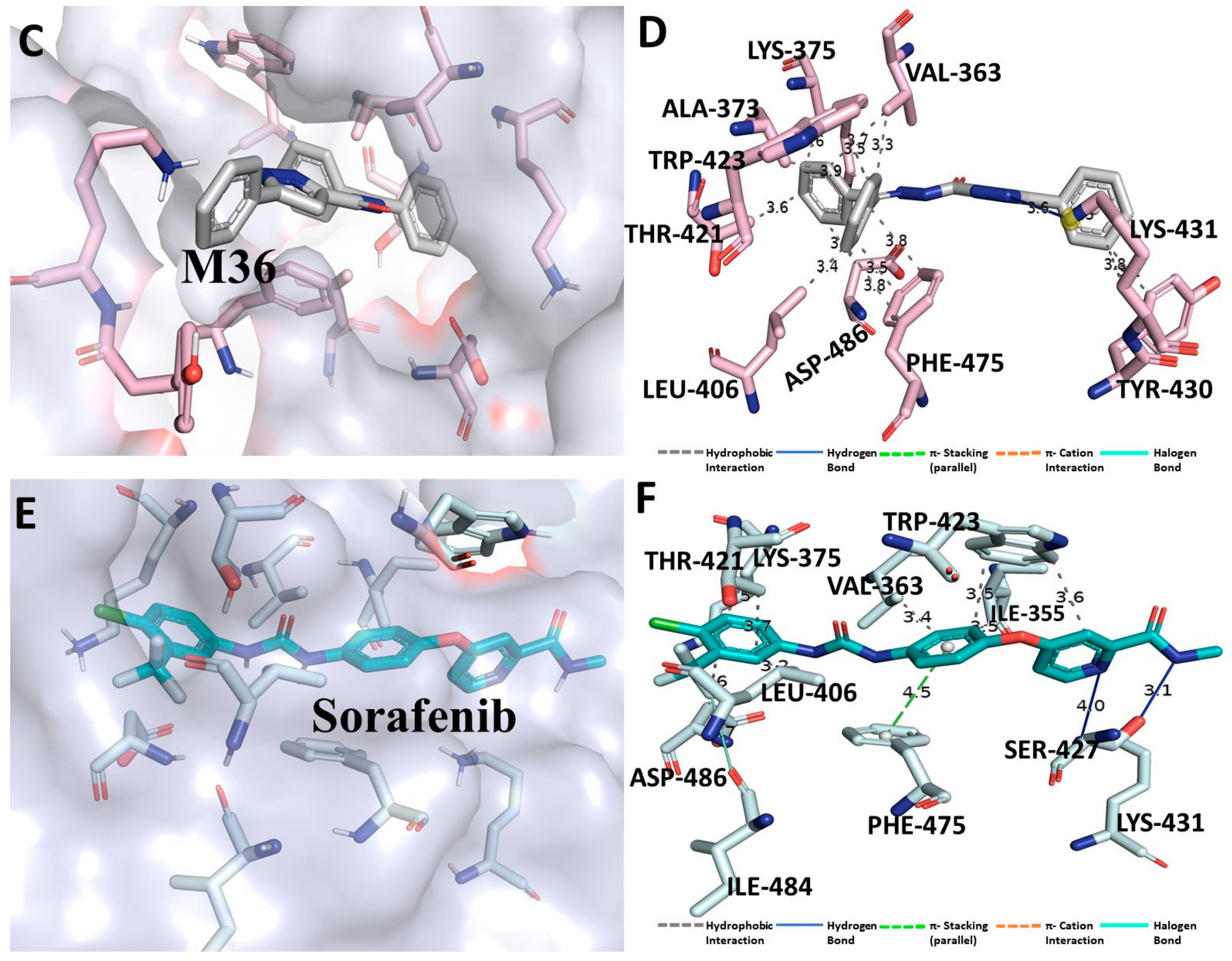
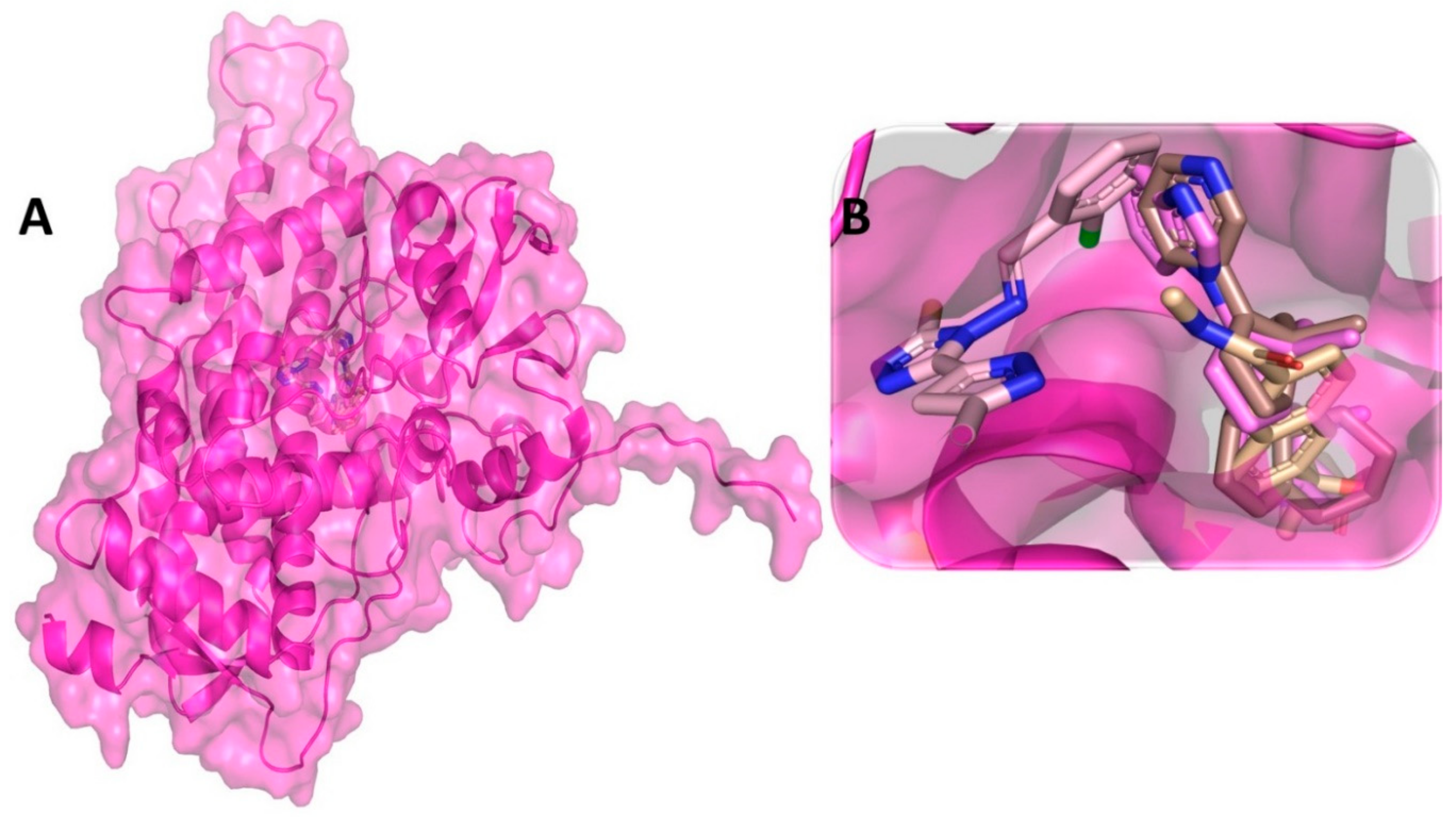
3.3. Binding Interaction Profile of C-RAF and Pyrazole Derivative Complex
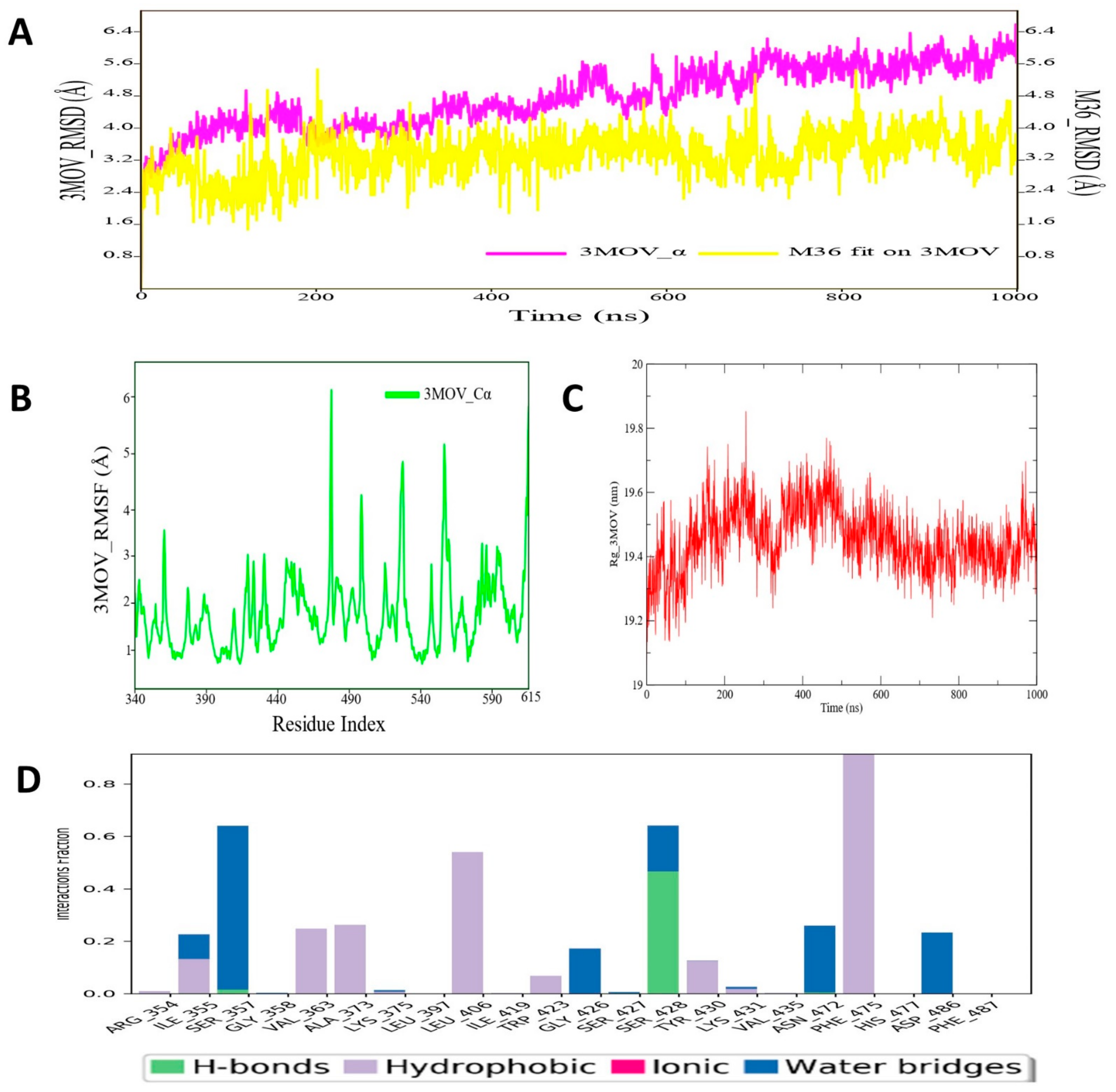
3.4. Molecular Dynamic Simulation Analysis of C-RAF–Pyrazole Derivative M36 Complex
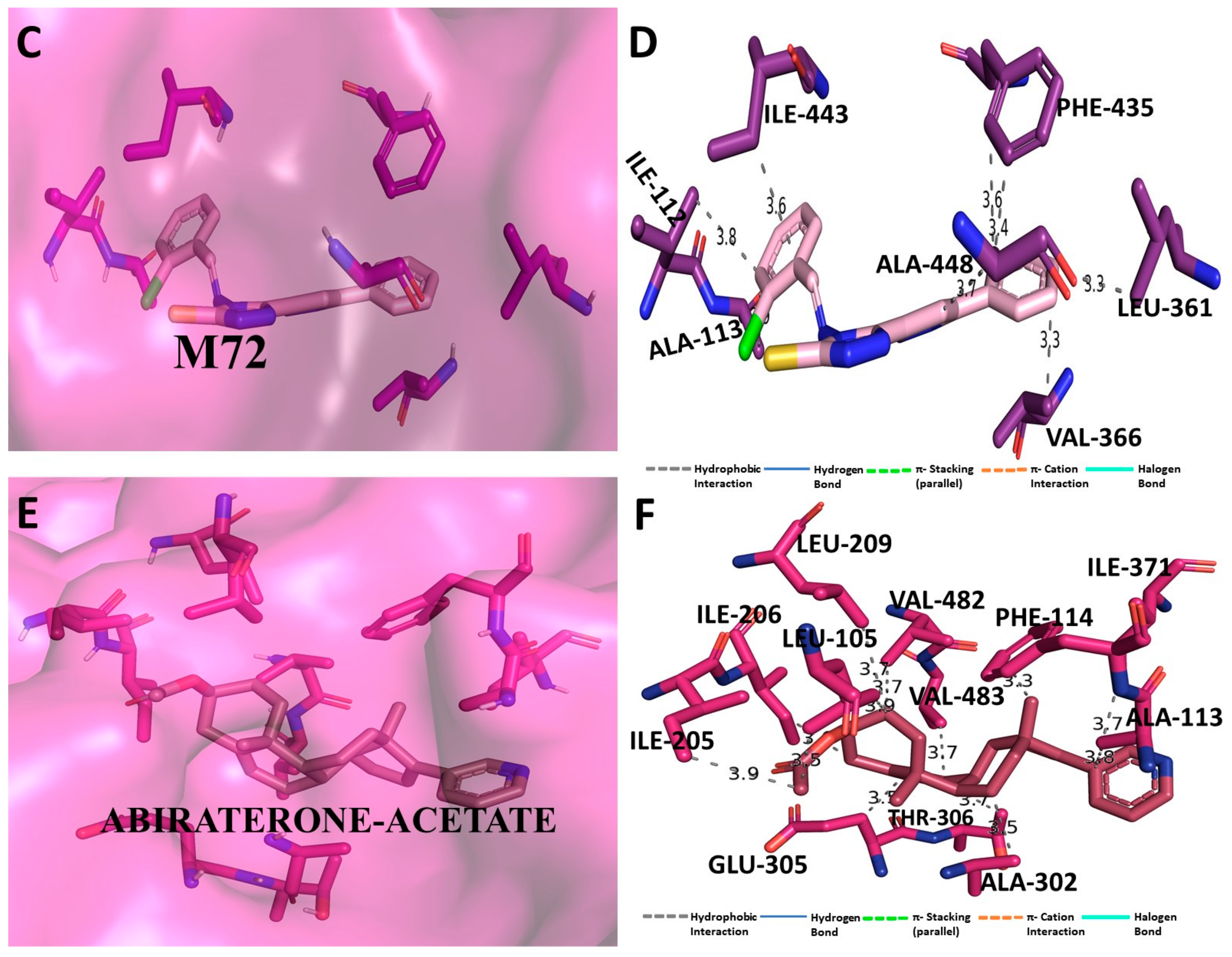
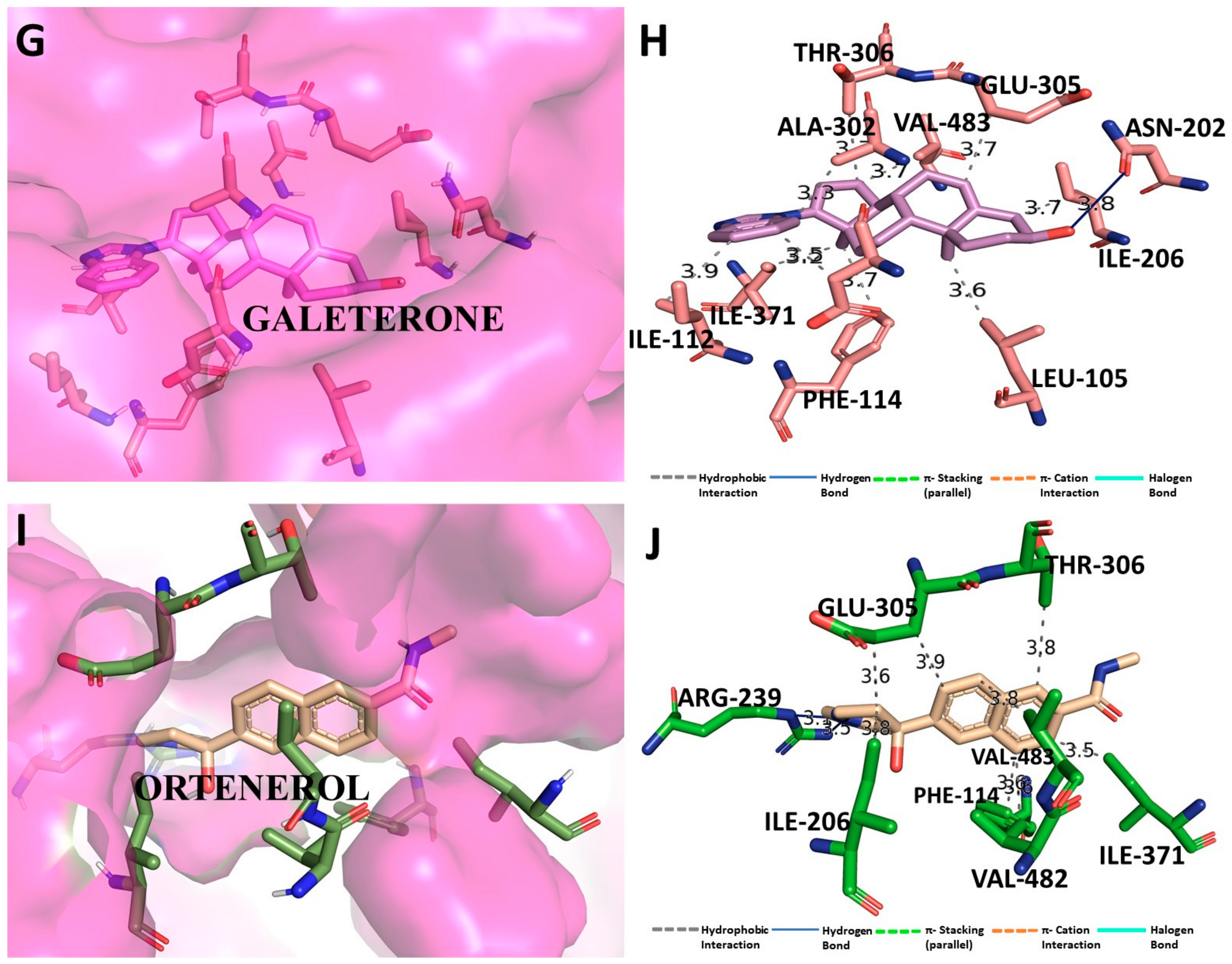
3.5. Binding Interaction Profile Analysis of CYP17 and Pyrazole Derivative Complex
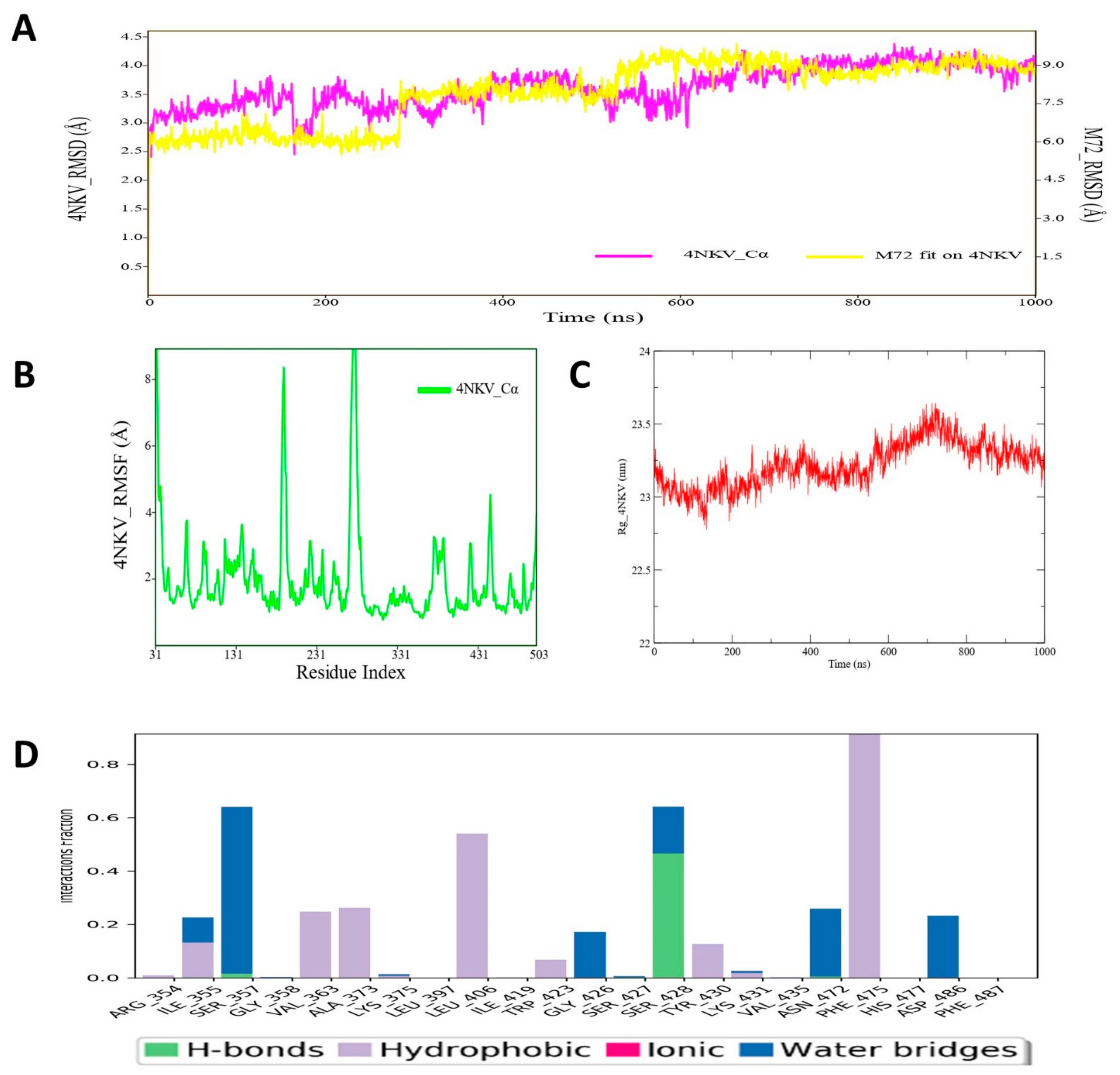
3.6. Molecular Dynamic Simulation Analysis of CYP17–Pyrazole Derivative M72 Complex
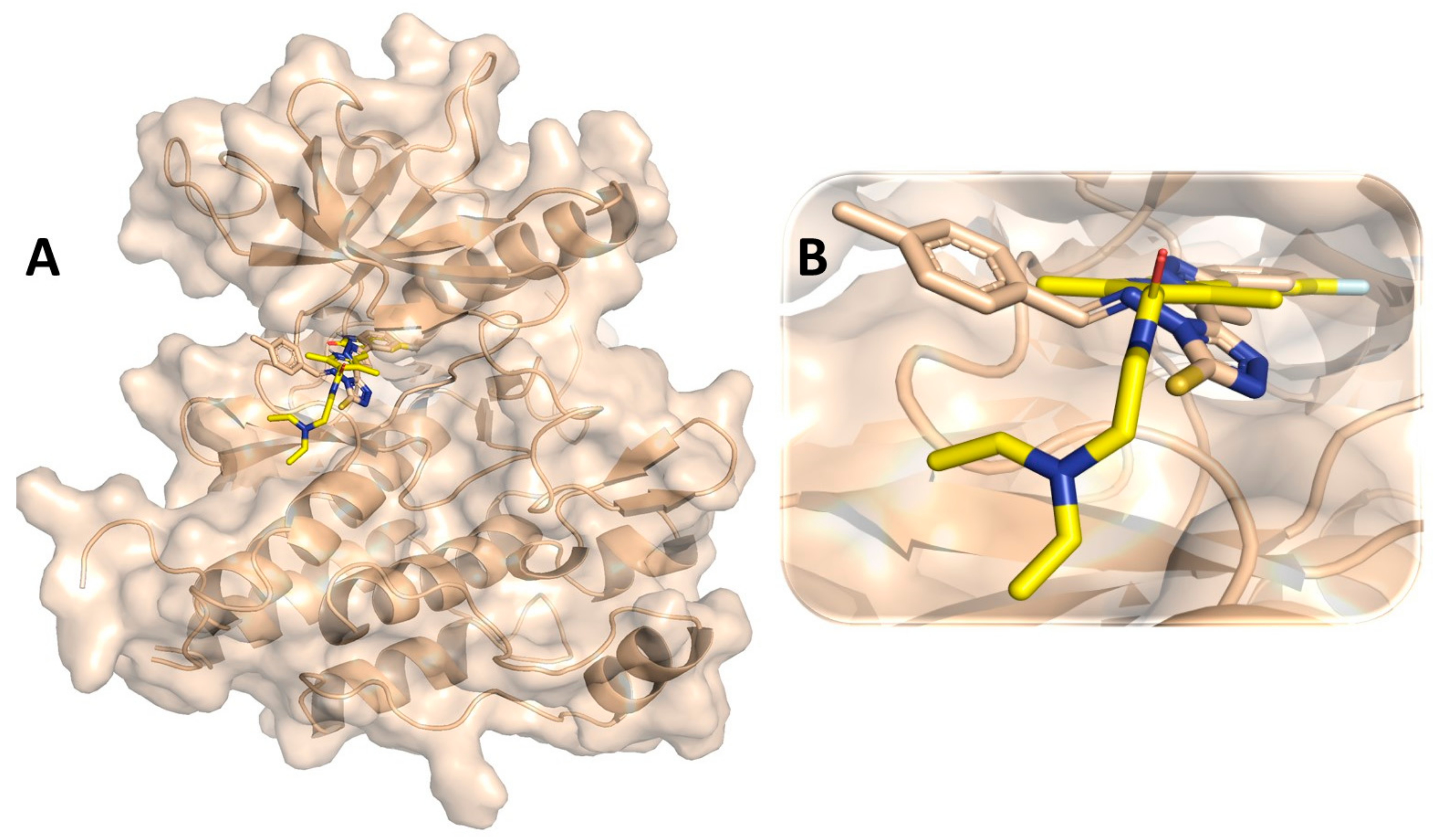
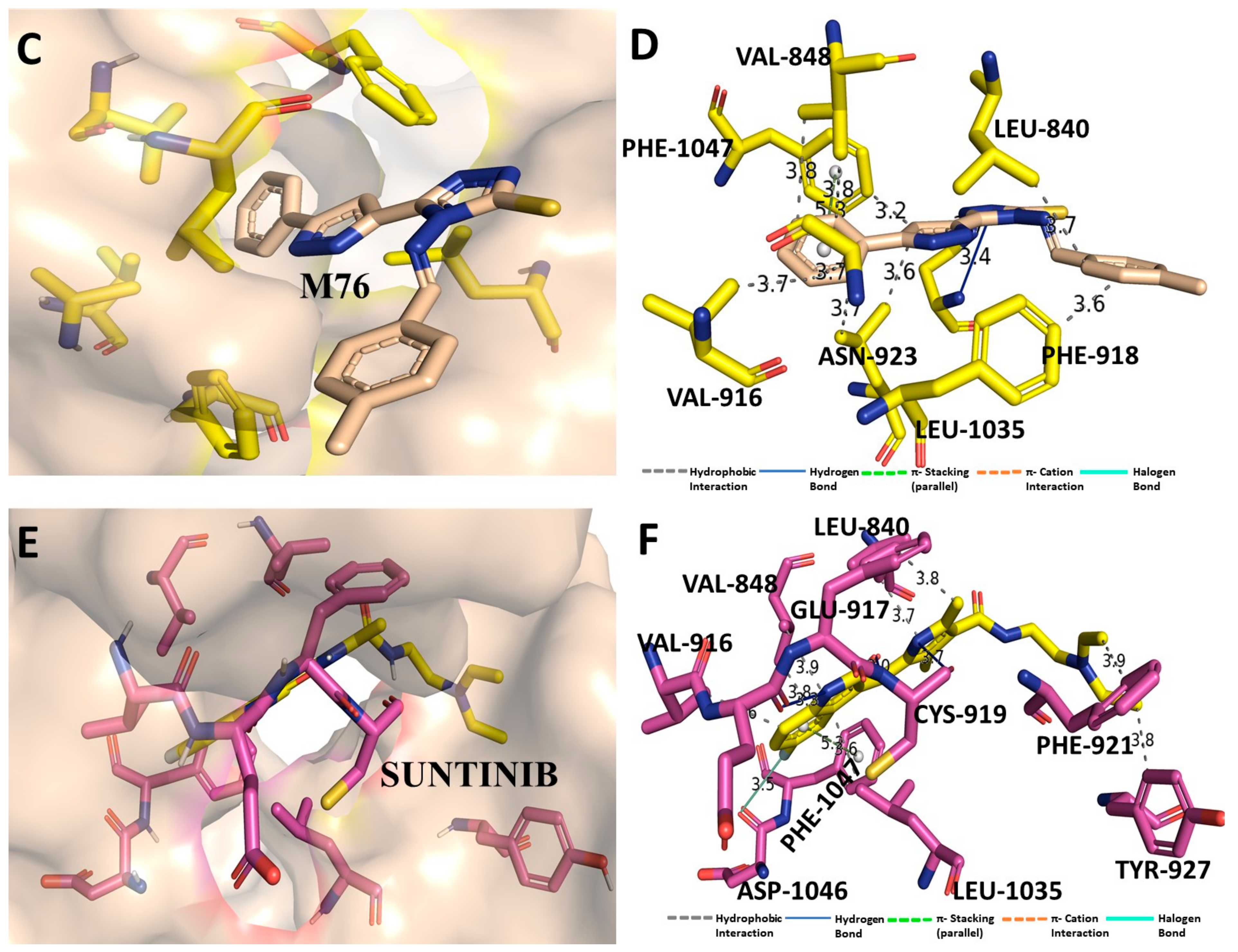
3.7. Binding Interaction Profile Analysis of VEGFR and Pyrazole Derivative Complex
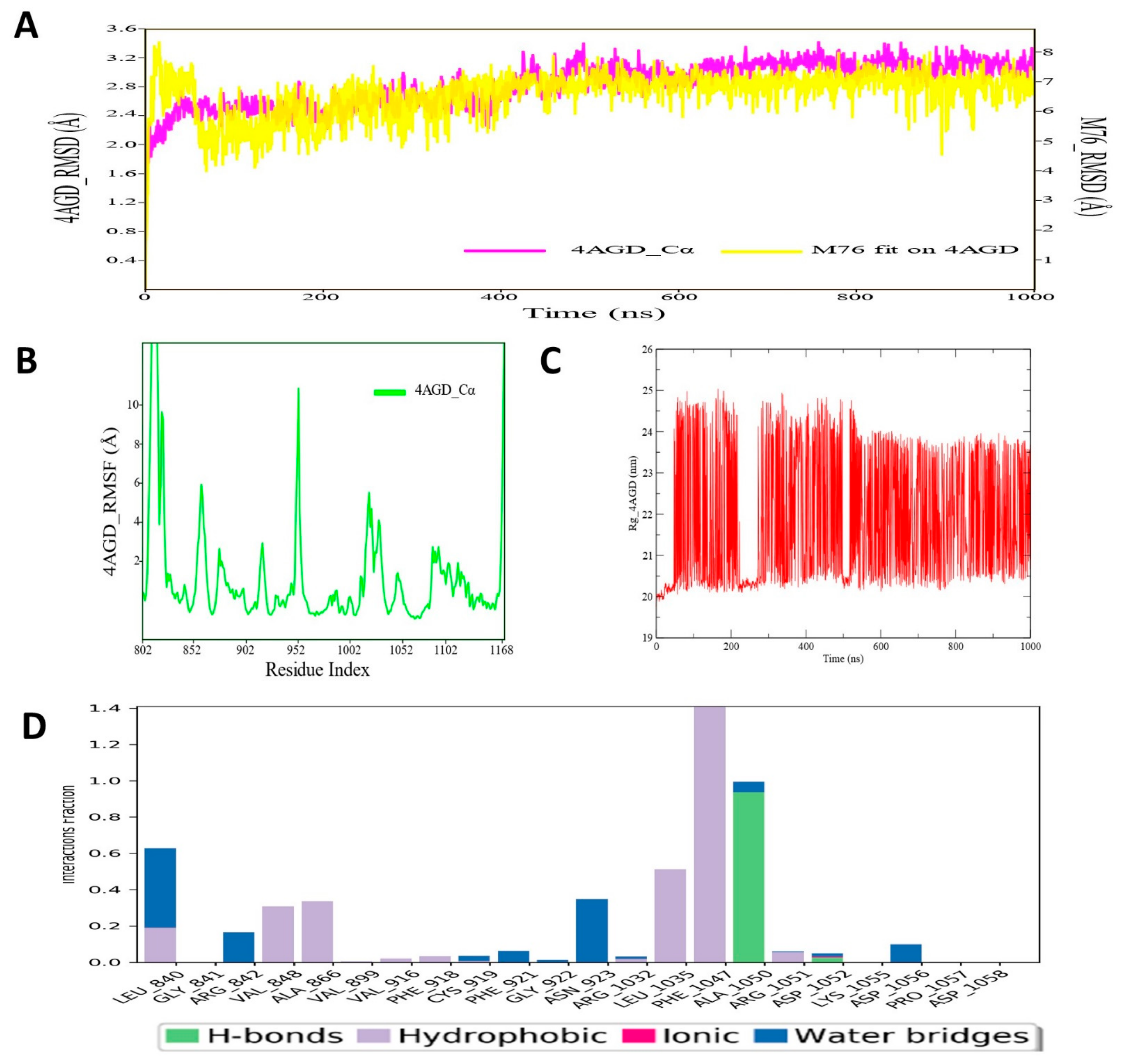
3.8. Molecular Dynamic Simulation Analysis of VEGFR–Pyrazole Derivative M76 Complex
3.9. Binding Interaction Profile Analysis of c-Kit–Pyrazole Derivative Complex
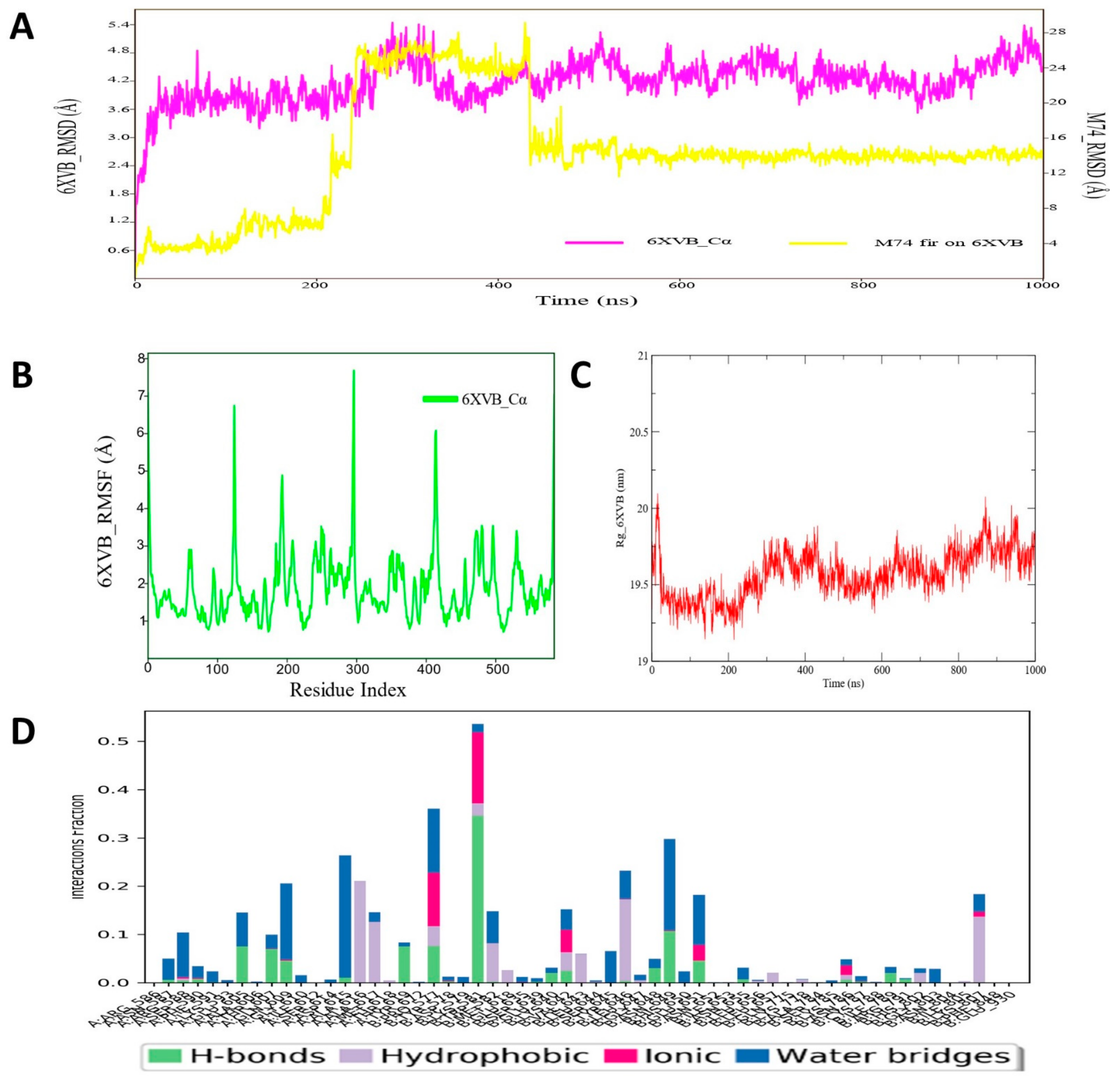
3.10. Molecular Dynamic Simulation Analyses of c-Kit–Pyrazole Derivative M74 Complex
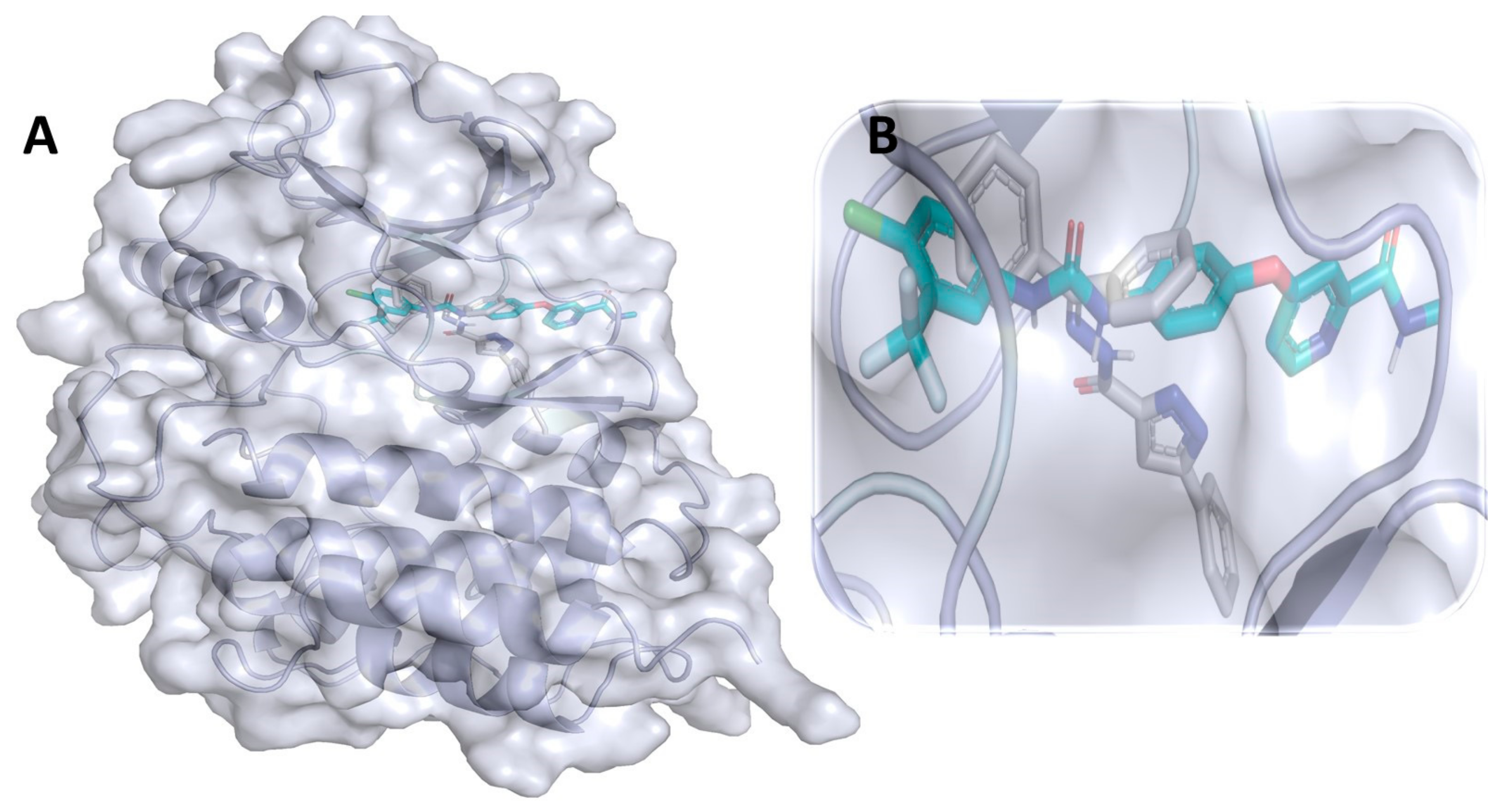
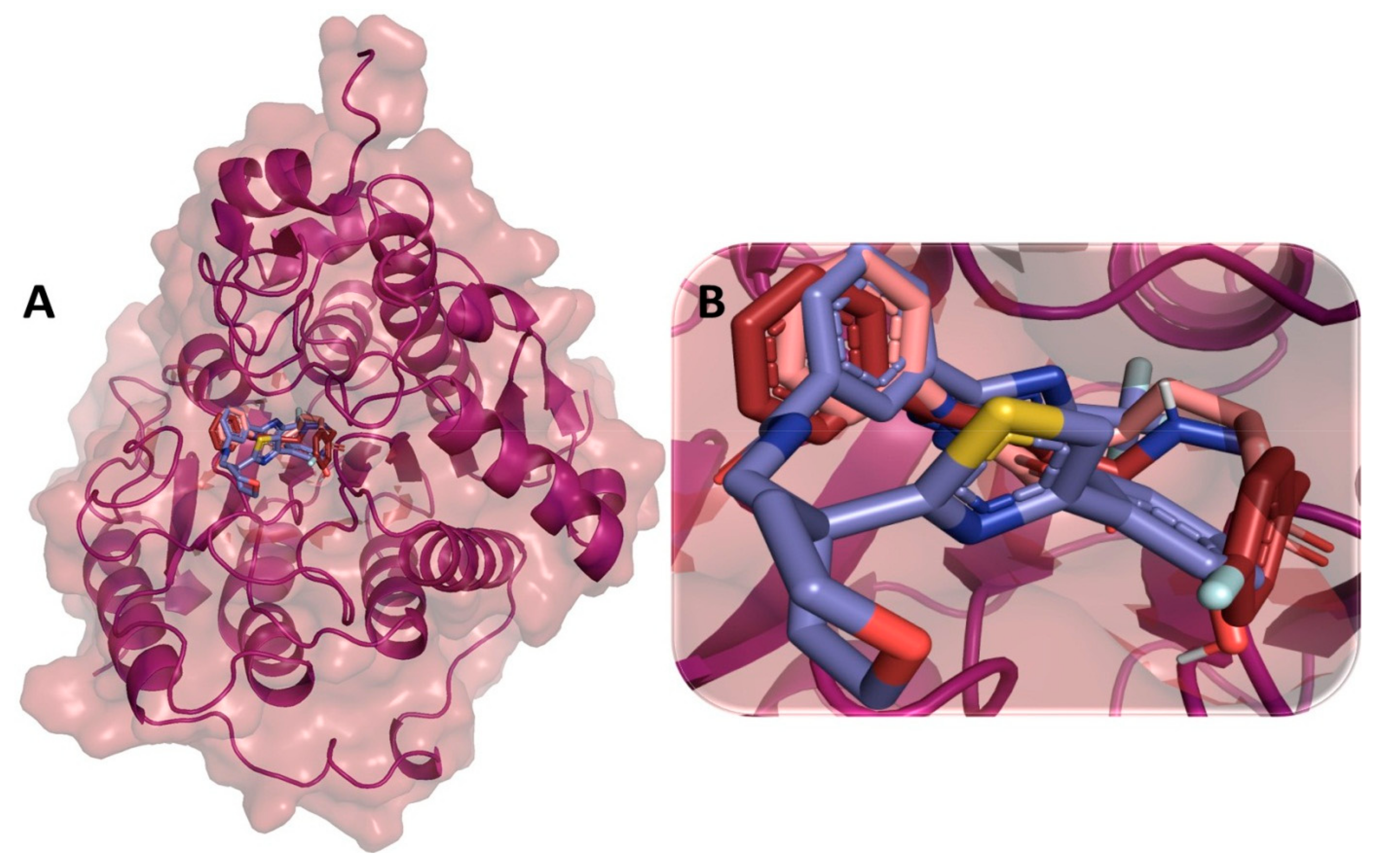
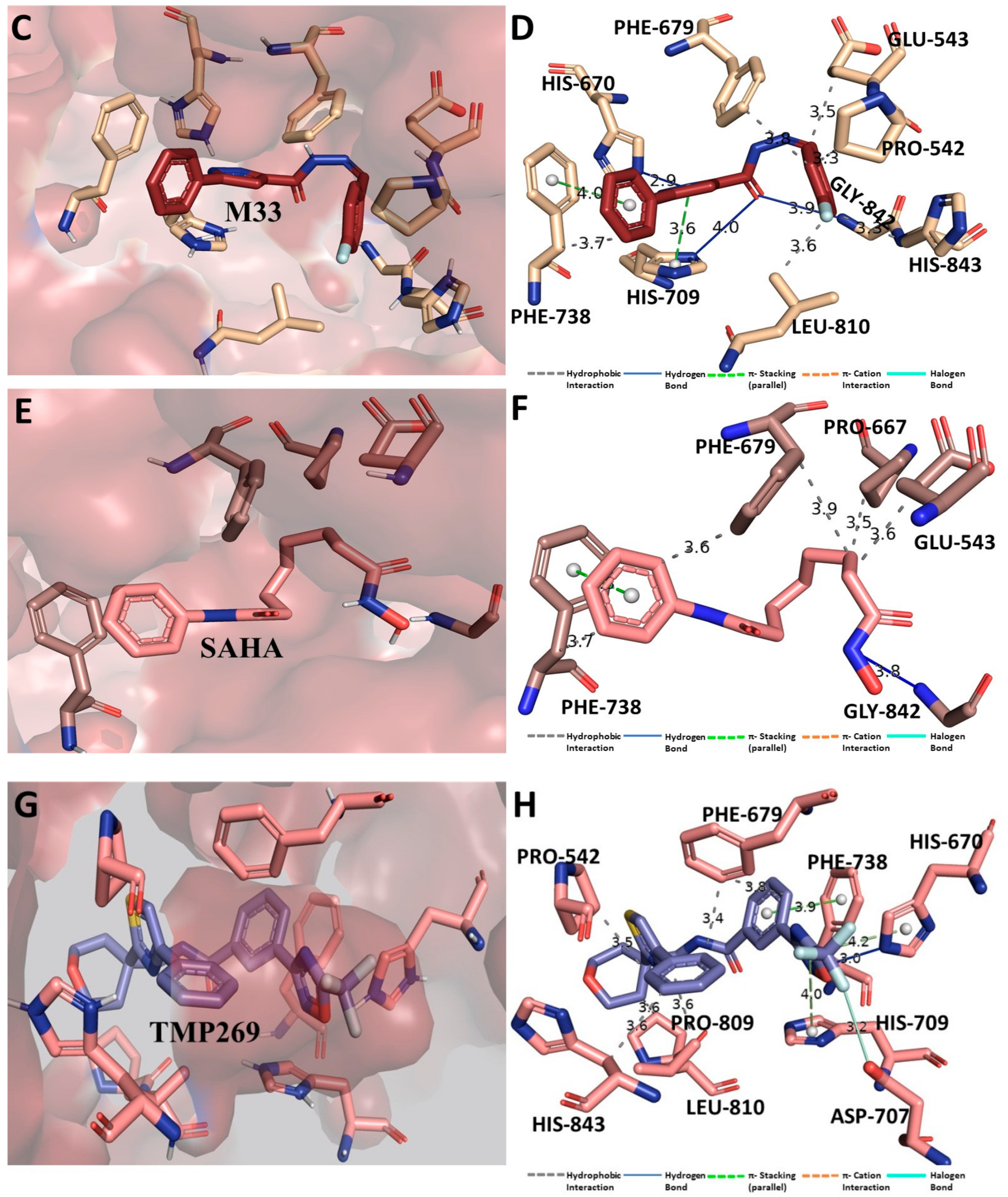
3.11. Binding Interaction Profile Analysis of HDAC–Pyrazole Derivative Complex
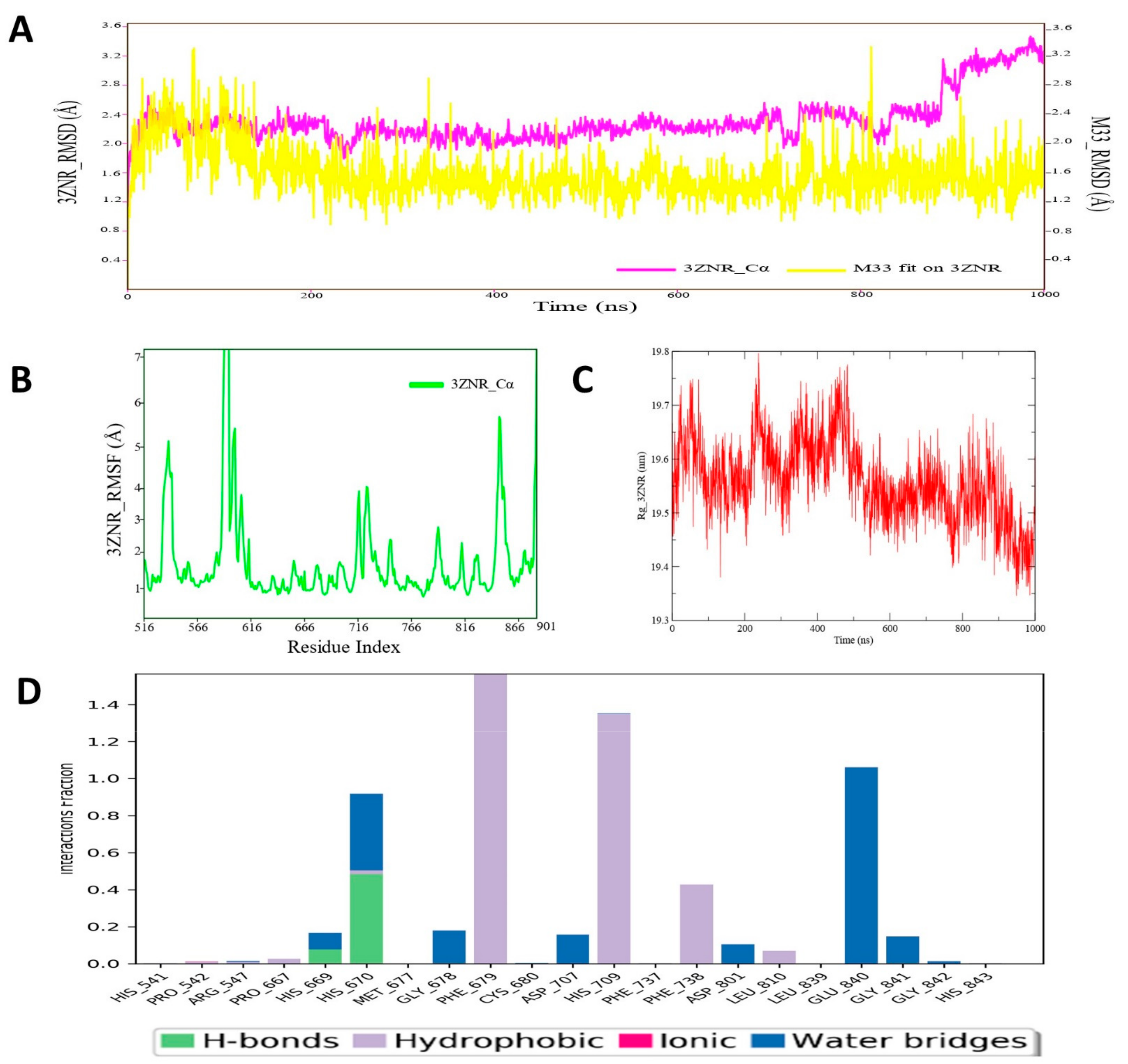
3.12. Molecular Dynamic Simulation Analysis of HDAC–Pyrazole Derivative M33 Complex
3.13. ADME_T Prediction
3.13.1. Absorption Profile
3.13.2. Distribution Profile
3.13.3. Metabolism Profile
3.13.4. Excretion Profile
3.13.5. Toxicity Profile
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Yacoub, E.; Abdul-Wahab, O.M.S.; Al-Shyarba, M.H.; Mardassi, B.B.A. The Relationship between Mycoplasmas and Cancer: Is It Fact or Fiction? Narrative Review and Update on the Situation. J. Oncol. 2021, 2021, 9986550. [Google Scholar] [CrossRef] [PubMed]
- Yahya, E.B.; Alqadhi, A.M. Recent trends in cancer therapy: A review on the current state of gene delivery. Life Sci. 2021, 269, 119087. [Google Scholar] [CrossRef] [PubMed]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Kocyigit, U.M.; Budak, Y.; Gürdere, M.B.; Tekin, Ş.; Köprülü, T.K.; Ertürk, F.; Ceylan, M. Synthesis, characterization, anticancer, antimicrobial and carbonic anhydrase inhibition profiles of novel (3aR,4S,7R,7aS)-2-(4-((E)-3-(3-aryl)acryloyl)phenyl)-3a,4,7,7a-tetrahydro-1H-4,7 methanoisoindole-1,3(2H)-dione derivatives. Bioorg Chem. 2017, 70, 118–125. [Google Scholar] [CrossRef]
- Dastan, T.; Kocyigit, U.M.; Durna Dastan, S.; Canturk Kilickaya, P.; Taslimi, P.; Cevik, O.; Cetin, A. Investigation of acetylcholinesterase and mammalian DNA topoisomerases, carbonic anhydrase inhibition profiles, and cytotoxic activity of novel bis(α-aminoalkyl)phosphinic acid derivatives against human breast cancer. J. Biochem. Mol. Toxicol. 2017, 31, e2171. [Google Scholar] [CrossRef]
- Wesdorp, N.J.; Hellingman, T.; Jansma, E.P.; van Waesberghe, J.-H.T.M.; Boellaard, R.; Punt, C.J.A.; Huiskens, J.; Kazemier, G. Advanced analytics and artificial intelligence in gastrointestinal cancer: A systematic review of radiomics predicting response to treatment. Eur. J. Nucl. Med. Mol. Imaging 2020, 48, 1785–1794. [Google Scholar] [CrossRef]
- Karrouchi, K.; Fettach, S.; Radi, S.; Yousfi, E.B.; Taoufik, J.; Mabkhot, Y.N.; Alterary, S.; Faouzi, M.E.A.; Ansar, M. Synthesis, Characterization, Free-radical Scavenging Capacity and Antioxidant Activity of Novel Series of Hydrazone, 1,3,4-oxadiazole and 1,2,4- triazole Derived from 3,5-dimethyl-1H-pyrazole. Lett. Drug Des. Discov. 2019, 16, 712–720. [Google Scholar] [CrossRef]
- Karrouchi, K.; Chemlal, L.; Taoufik, J.; Cherrah, Y.; Radi, S.; Faouzi, M.E.A.; Ansar, M. Synthèse, activités anti-oxydantes et analgésiques de bases de Schiff dérivées du 4-amino-1,2,4-triazole porteur d’un noyau pyrazole. Ann. Pharm. Fr. 2016, 74, 431–438. [Google Scholar] [CrossRef]
- Pillai, R.R.; Karrouchi, K.; Fettach, S.; Armaković, S.; Armaković, S.J.; Brik, Y.; Taoufik, J.; Radi, S.; El Abbes Faouzi, M.; Ansar, M. Synthesis, spectroscopic characterization, reactive properties by DFT calculations, molecular dynamic simulations and biological evaluation of Schiff bases tethered 1,2,4-triazole and pyrazole rings. J. Mol. Struct. 2019, 1177, 47–54. [Google Scholar] [CrossRef]
- Karrouchi, K.; Yousfi, E.B.; Sebbar, N.K.; Ramli, Y.; Taoufik, J.; Ouzidan, Y.; Ansar, M.; Mabkhot, Y.N.; Ghabbour, H.A.; Radi, S. New Pyrazole-Hydrazone Derivatives: X-ray Analysis, Molecular Structure Investigation via Density Functional Theory (DFT) and Their High In-Situ Catecholase Activity. Int. J. Mol. Sci. 2017, 18, 2215. [Google Scholar] [CrossRef]
- Karrouchi, K.; Brandán, S.A.; Sert, Y.; El-Marzouqi, H.; Radi, S.; Ferbinteanu, M.; Faouzi, M.E.A.; Garcia, Y.; Ansar, M. Synthesis, X-ray structure, vibrational spectroscopy, DFT, biological evaluation and molecular docking studies of (E)-N’-(4-(dimethylamino)benzylidene)-5-methyl-1H-pyrazole-3-carbohydrazide. J. Mol. Struct. 2020, 1219, 128541. [Google Scholar] [CrossRef]
- Karrouchi, K.; Brandán, S.A.; Sert, Y.; El Karbane, M.; Radi, S.; Ferbinteanu, M.; Garcia, Y.; Ansar, M. Synthesis, structural, molecular docking and spectroscopic studies of (E)-N’-(4-methoxybenzylidene)-5-methyl-1H-pyrazole-3-carbohydrazide. J. Mol. Struct. 2021, 1225, 129072. [Google Scholar] [CrossRef]
- Karrouchi, K.; Brandán, S.A.; Hassan, M.; Bougrin, K.; Radi, S.; Ferbinteanu, M.; Garcia, Y.; Ansar, M. Synthesis, X-ray, spectroscopy, molecular docking and DFT calculations of (E)-N’-(2,4-dichlorobenzylidene)-5-phenyl-1H-pyrazole-3-carbohydrazide. J. Mol. Struct. 2021, 1228, 129714. [Google Scholar] [CrossRef]
- Karrouchi, K.; Ansar, M.; Radi, S.; Saadi, M.; El Ammari, L. Crystal structure ofN′-diphenylmethylidene-5-methyl-1H-pyrazole-3-carbohydrazide. Acta Crystallogr. Sect. E Crystallogr. Commun. 2015, 71, o890–o891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karrouchi, K.; Radi, S.; Ansar, M.; Taoufik, J.; Ghabbour, H.A.; Mabkhot, Y.N. Crystal structure of N′-(4-(dimethylamino)benzylidene)-5-phenyl-1H-pyrazole-3-carbohydrazide, C19H19N5O. Zeitschrift Fur Krist -New Cryst. Struct. 2016, 231, 883–886. [Google Scholar] [CrossRef]
- Karrouchi, K.; Radi, S.; Ansar, M.; Taoufik, J.; Ghabbour, H.A.; Mabkhot, Y.N. Crystal structure of N′-(4-methoxybenzylidene)-5-phenyl-1H-pyrazole-3-carbohydrazide, C18H16N4O2. Zeitschrift Fur Krist -New Cryst. Struct. 2016, 231, 835–837. [Google Scholar] [CrossRef] [Green Version]
- Karrouchi, K.; Radi, S.; Ansar, M.; Taoufik, J.; Ghabbour, H.A.; Mabkhot, Y.N. Crystal structure of N′-(4-nitrobenzylidene)-5-phenyl-1H-pyrazole-3-carbohydrazide, C17H13N5O3. Zeitschrift Fur Krist -New Cryst. Struct. 2016, 231, 839–841. [Google Scholar] [CrossRef]
- Karrouchi, K.; Fettach, S.; Anouar, E.H.; Tüzün, B.; Radi, S.; Alharthi, A.I.; Ghabbour, H.A.; Mabkhot, Y.N.; Faouzi, M.E.A.; Ansar, M.; et al. Synthesis, crystal structure, DFT, α-glucosidase and α-amylase inhibition and molecular docking studies of (E)-N’-(4-chlorobenzylidene)-5-phenyl-1H-pyrazole-3-carbohydrazide. J. Mol. Struct. 2021, 1245, 131067. [Google Scholar] [CrossRef]
- Lin, B.; Li, Y.; Wang, T.; Qiu, Y.; Chen, Z.; Zhao, K.; Lu, N. CRMP2 is a therapeutic target that suppresses the aggressiveness of breast cancer cells by stabilizing RECK. Oncogene 2020, 39, 6024–6040. [Google Scholar] [CrossRef]
- Hwang, Y.H.; Choi, J.Y.; Kim, S.; Chung, E.S.; Kim, T.; Koh, S.S.; Park, Y.M. Over-expression of c-raf-1 proto-oncogene in liver cirrhosis and hepatocellular carcinoma. Hepatol. Res. 2004, 29, 113–121. [Google Scholar] [CrossRef]
- Vasaitis, T.S.; Bruno, R.D.; Njar, V.C. CYP17 inhibitors for prostate cancer therapy. J. Steroid Biochem. Mol. Biol. 2011, 125, 23–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, Z.-H.; Han, E.M.; Lee, E.S.; Kim, C.W.; Kim, H.K.; Kim, I.; Kim, Y.-S. A distinct expression pattern and point mutation of c-KIT in papillary renal cell carcinomas. Mod. Pathol. 2004, 17, 611–616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iacovelli, R.; Sternberg, C.N.; Porta, C.; Verzoni, E.; de Braud, F.G.M.; Escudier, B.; Procopio, G. Inhibition of the VEGF/VEGFR Pathway Improves Survival in Advanced Kidney Cancer: A Systematic Review and Meta-Analysis. Curr. Drug Targets 2015, 16, 164–170. [Google Scholar] [CrossRef] [PubMed]
- Xiong, K.; Zhang, H.; Du, Y.; Tian, J.; Ding, S. Identification of HDAC9 as a viable therapeutic target for the treatment of gastric cancer. Exp. Mol. Med. 2019, 51, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Lei, J.; Zhong, J.; Hao, J.; Liu, Z.; Zhang, P.; Wu, L.; Yan, L.; Zhu, J.; Zeng, Y.; Lixue, W.; et al. Hepatocellular carcinoma cases with high levels of c-Raf-1 expression may benefit from postoperative adjuvant sorafenib after hepatic resection even with high risk of recurrence. Oncotarget 2016, 7, 42598–42607. [Google Scholar] [CrossRef] [Green Version]
- Keating, G.M.; Santoro, A. Sorafenib: A review of its use in advanced hepatocellular carcinoma. Drugs 2009, 69, 223–240. [Google Scholar] [CrossRef]
- Wilhelm, S.M.; Adnane, L.; Newell, P.; Villanueva, A.; Llovet, J.M.; Lynch, M. Preclinical overview of sorafenib, a multikinase inhibitor that targets both Raf and VEGF and PDGF receptor tyrosine kinase signaling. Mol. Cancer Ther. 2008, 7, 3129–3140. [Google Scholar] [CrossRef] [Green Version]
- Yin, L.; Hu, Q. CYP17 inhibitors—Abiraterone, C17,20-lyase inhibitors and multi-targeting agents. Nat. Rev. Urol. 2014, 11, 32–42. [Google Scholar] [CrossRef]
- Ang, J.E.; Olmos, D.; De Bono, J.S. CYP17 blockade by abiraterone: Further evidence for frequent continued hormone-dependence in castration-resistant prostate cancer. Br. J. Cancer 2009, 100, 671–675. [Google Scholar] [CrossRef] [Green Version]
- Malikova, J.; Brixius-Anderko, S.; Udhane, S.S.; Parween, S.; Dick, B.; Bernhardt, R.; Pandey, A.V. CYP17A1 inhibitor abiraterone, an anti-prostate cancer drug, also inhibits the 21-hydroxylase activity of CYP21A2. J. Steroid Biochem. Mol. Biol. 2017, 174, 192–200. [Google Scholar] [CrossRef]
- De Bono, J.S.; Logothetis, C.J.; Molina, A.; Fizazi, K.; North, S.; Chu, L.; Chi, K.N.; Jones, R.J.; Goodman, O.B., Jr.; Saad, F.; et al. Abiraterone and Increased Survival in Metastatic Prostate Cancer. N. Engl. J. Med. 2011, 364, 1995–2005. [Google Scholar] [CrossRef] [PubMed]
- Attard, G.; Reid, A.H.; Yap, T.A.; Raynaud, F.; Dowsett, M.; Settatree, S.; Barrett, M.; Parker, C.; Martins, V.; Folkerd, E.; et al. Phase I Clinical Trial of a Selective Inhibitor of CYP17, Abiraterone Acetate, Confirms That Castration-Resistant Prostate Cancer Commonly Remains Hormone Driven. J. Clin. Oncol. 2008, 26, 4563–4571. [Google Scholar] [CrossRef] [PubMed]
- Van Hook, K.; Huang, T.; Alumkal, J.J. Orteronel for the treatment of prostate cancer. Futur. Oncol. 2014, 10, 803–811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saad, F.; Fizazi, K.; Jinga, V.; Efstathiou, E.; Fong, P.C.; Hart, L.L.; Jones, R.; McDermott, R.; Wirth, M.; Suzuki, K.; et al. Orteronel plus prednisone in patients with chemotherapy-naive metastatic castration-resistant prostate cancer (ELM-PC 4): A double-blind, multicentre, phase 3, randomised, placebo-controlled trial. Lancet Oncol. 2015, 16, 338–348. [Google Scholar] [CrossRef]
- Antonarakis, E.; Bastos, D. Galeterone for the treatment of advanced prostate cancer: The evidence to date. Drug Des. Dev. Ther. 2016, 10, 2289–2297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gan, H.K.; Seruga, B.; Knox, J.J. Sunitinib in solid tumors. Expert Opin. Investig. Drugs 2009, 18, 821–834. [Google Scholar] [CrossRef] [PubMed]
- Wayback Machine n.d. Available online: http://web.archive.org/ (accessed on 31 October 2021).
- Porta, C.; Paglino, C.; Imarisio, I.; Ganini, C.; Sacchi, L.; Quaglini, S.; Giunta, V.; DE Amici, M. Changes in Circulating Pro-Angiogenic Cytokines, other than VEGF, before Progression to Sunitinib Therapy in Advanced Renal Cell Carcinoma Patients. Oncology 2012, 84, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Lobera, M.; Madauss, K.P.; Pohlhaus, D.T.; Wright, Q.; Trocha, M.; Schmidt, D.R.; Baloglu, E.; Trump, R.P.; Head, M.S.; Hofmann, G.A.; et al. Selective class IIa histone deacetylase inhibition via a nonchelating zinc-binding group. Nat. Chem. Biol. 2013, 9, 319–325. [Google Scholar] [CrossRef]
- Muha, V.; Williamson, R.; Hills, R.; McNeilly, A.; McWilliams, T.; Alonso, J.; Schimpl, M.; Leney, A.C.; Heck, A.; Sutherland, C.; et al. Loss of CRMP2 O-GlcNAcylation leads to reduced novel object recognition performance in mice. Open Biol. 2019, 9, 190192. [Google Scholar] [CrossRef] [Green Version]
- Shimada, K.; Ishikawa, T.; Nakamura, F.; Shimizu, D.; Chishima, T.; Ichikawa, Y.; Sasaki, T.; Endo, I.; Nagashima, Y.; Goshima, Y. Collapsin response mediator protein 2 is involved in regulating breast cancer progression. Breast Cancer 2013, 21, 715–723. [Google Scholar] [CrossRef]
- Force, T.; Bonventre, J.V.; Heidecker, G.; Rapp, U.; Avruch, J.; Kyriakis, J.M. Enzymatic characteristics of the c-Raf-1 proteinkinase. Proc. Natl. Acad. Sci. USA 1994, 91, 1270–1274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maurer, G.; Tarkowski, B.; Baccarini, M. Raf kinases in cancer–roles and therapeutic opportunities. Oncogene 2011, 30, 3477–3488. [Google Scholar] [CrossRef] [Green Version]
- Migliaccio, N.; Sanges, C.; Ruggiero, I.; Martucci, N.M.; Rippa, E.; Arcari, P.; Lamberti, A. Raf kinases in signal transduction and interaction with translation machinery. Biomol. Concepts 2013, 4, 391–399. [Google Scholar] [CrossRef]
- Beeram, M.; Patnaik, A.; Rowinsky, E.K. Raf: A Strategic Target for Therapeutic Development Against Cancer. J. Clin. Oncol. 2005, 23, 6771–6790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edwards, J.; Krishna, N.S.; Witton, C.J.; Bartlett, J.M.S. Gene amplifications associated with the development of hormone-resistant prostate cancer. Clin. Cancer Res. 2003, 9, 5271–5281. [Google Scholar] [PubMed]
- Guengerich, F.P. Cytochrome P450 and Chemical Toxicology. Chem. Res. Toxicol. 2008, 21, 70–83. [Google Scholar] [CrossRef] [PubMed]
- Alex, A.B.; Pal, S.K.; Agarwal, N. CYP17 inhibitors in prostate cancer: Latest evidence and clinical potential. Ther. Adv. Med. Oncol. 2016, 8, 267–275. [Google Scholar] [CrossRef] [Green Version]
- Bryce, A.H.; Ryan, C.J. Development and Clinical Utility of Abiraterone Acetate as an Androgen Synthesis Inhibitor. Clin. Pharmacol. Ther. 2012, 91, 101–108. [Google Scholar] [CrossRef]
- Sobti, R.C.; Gupta, L.; Thakur, H.; Seth, A.; Singh, S.K.; Kaur, P. CYP17 gene polymorphism and its association in north Indian prostate cancer patients. Anticancer Res. 2009, 29, 1659–1663. [Google Scholar]
- Attard, G.; Reid, A.H.; Olmos, D.; de Bono, J.S. Antitumor Activity with CYP17 Blockade Indicates That Castration-Resistant Prostate Cancer Frequently Remains Hormone Driven. Cancer Res. 2009, 69, 4937–4940. [Google Scholar] [CrossRef] [Green Version]
- Petrunak, E.M.; DeVore, N.M.; Porubsky, P.R.; Scott, E.E. Structures of Human Steroidogenic Cytochrome P450 17A1 with Substrates. J. Biol. Chem. 2014, 289, 32952–32964. [Google Scholar] [CrossRef] [PubMed]
- Marech, I.; Gadaleta, C.D.; Ranieri, G. Possible Prognostic and Therapeutic Significance of c-KIT Expression, Mast Cell Count and Microvessel Density in Renal Cell Carcinoma. Int. J. Mol. Sci. 2014, 15, 13060–13076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zimpfer, A.; Janke, S.; Hühns, M.; Schneider, B.; Kundt, G.; Zettl, H.; Kilic, E.; Maruschke, M.; Hakenberg, O.W.; Erbersdobler, A. C-KIT overexpression is not associated with KIT gene mutations in chromophobe renal cell carcinoma or renal oncocytoma. Pathol.—Res. Pr. 2014, 210, 521–525. [Google Scholar] [CrossRef] [PubMed]
- Weber, R.; Medinger, M.; Kleinschmidt, M.; Filipovic, D.; Schaefer, H.E.; Unger, C. Immunohistochemical analysis of c-KIT (CD117) expression in solid tumors. J. Clin. Oncol. 2004, 22, 9642. [Google Scholar] [CrossRef]
- Bierer, S.; Herrmann, E.; Köpke, T.; Neumann, J.; Eltze, E.; Hertle, L.; Wülfing, C. Lymphangiogenesis in kidney cancer: Expression of VEGF-C, VEGF-D and VEGFR-3 in clear cell and papillary renal cell carcinoma. Oncol. Rep. 1994, 20, 721–725. [Google Scholar] [CrossRef] [Green Version]
- Scartozzi, M.; Bianconi, M.; Faloppi, L.; Loretelli, C.; Bittoni, A.; Del Prete, M.; Giampieri, R.; Maccaroni, E.; Nicoletti, S.; Burattini, L.; et al. VEGF and VEGFR polymorphisms affect clinical outcome in advanced renal cell carcinoma patients receiving first-line sunitinib. Br. J. Cancer 2013, 108, 1126–1132. [Google Scholar] [CrossRef] [Green Version]
- Kluger, H.M.; Siddiqui, S.F.; Angeletti, C.; Sznol, M.; Kelly, W.K.; Molinaro, A.M.; Camp, R.L. Classification of renal cell carcinoma based on expression of VEGF and VEGF receptors in both tumor cells and endothelial cells. Lab. Investig. 2008, 88, 962–972. [Google Scholar] [CrossRef] [Green Version]
- Amnekar, R.V.; Khan, S.A.; Rashid, M.; Khade, B.; Thorat, R.; Gera, P.; Shrikhande, S.V.; Smoot, D.T.; Ashktorab, H.; Gupta, S. Histone deacetylase inhibitor pre-treatment enhances the efficacy of DNA-interacting chemotherapeutic drugs in gastric cancer. World J. Gastroenterol. 2020, 26, 598–613. [Google Scholar] [CrossRef]
- Ropero, S.; Esteller, M. The role of histone deacetylases (HDACs) in human cancer. Mol. Oncol. 2007, 1, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Seto, E. HDACs and HDAC Inhibitors in Cancer Development and Therapy. Cold Spring Harb. Perspect. Med. 2016, 6, a026831. [Google Scholar] [CrossRef] [Green Version]
- Bassett, S.A.; Barnett, M.P.G. The Role of Dietary Histone Deacetylases (HDACs) Inhibitors in Health and Disease. Nutrients 2014, 6, 4273–4301. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.-J.; Greégoire, S. Class II Histone Deacetylases: From Sequence to Function, Regulation, and Clinical Implication. Mol. Cell. Biol. 2005, 25, 2873–2884. [Google Scholar] [CrossRef] [Green Version]
- Gallinari, P.; Di Marco, S.; Jones, P.; Pallaoro, M.; Steinkühler, C. HDACs, histone deacetylation and gene transcription: From molecular biology to cancer therapeutics. Cell Res. 2007, 17, 195–211. [Google Scholar] [CrossRef]
- Li, G.; Tian, Y.; Zhu, W.-G. The Roles of Histone Deacetylases and Their Inhibitors in Cancer Therapy. Front. Cell Dev. Biol. 2020, 8, 1004. [Google Scholar] [CrossRef]
- Caslini, C.; Hong, S.; Ban, Y.J.; Chen, X.S.; Ince, T.A. HDAC7 regulates histone 3 lysine 27 acetylation and transcriptional activity at super-enhancer-associated genes in breast cancer stem cells. Oncogene 2019, 38, 6599–6614. [Google Scholar] [CrossRef] [Green Version]
- Lei, Y.; Liu, L.; Zhang, S.; Guo, S.; Li, X.; Wang, J.; Su, B.; Fang, Y.; Chen, X.; Ke, H.; et al. Hdac7 promotes lung tumorigenesis by inhibiting Stat3 activation. Mol. Cancer 2017, 16, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Q.-G.; Xiao, T.; Zhu, W.; Yu, Z.-Z.; Huang, X.-P.; Yi, H.; Lu, S.-S.; Tang, Y.-Y.; Huang, W.; Xiao, Z.-Q. HDAC7 promotes the oncogenicity of nasopharyngeal carcinoma cells by miR-4465-EphA2 signaling axis. Cell Death Dis. 2020, 11, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Moreno, D.A.; Scrideli, C.A.; Cortez, M.A.A.; De Paula Queiroz, R.; Valera, E.T.; Da Silva Silveira, V.; Yunes, J.A.; Brandalise, S.R.; Tone, L.G. research paper: Differential expression of HDAC3, HDAC7 and HDAC9 is associated with prognosis and survival in childhood acute lymphoblastic leukaemia. Br. J. Haematol. 2010, 150, 665–673. [Google Scholar] [CrossRef] [PubMed]
- Ouaïssi, M.; Silvy, F.; Loncle, C.; Da Silva, D.F.; Abreu, C.M.; Martinez, E.; Berthézene, P.; Cadra, S.; Le Treut, Y.P.; Hardwigsen, J.; et al. Further Characterization of HDAC and SIRT Gene Expression Patterns in Pancreatic Cancer and Their Relation to Disease Outcome. PLoS ONE 2014, 9, e108520. [Google Scholar] [CrossRef]
- Sang, Y.; Sun, L.; Wu, Y.; Yuan, W.; Liu, Y.; Li, S.-W. Histone deacetylase 7 inhibits plakoglobin expression to promote lung cancer cell growth and metastasis. Int. J. Oncol. 2019, 54, 1112–1122. [Google Scholar] [CrossRef] [Green Version]
- Uzelac, B.; Krivokuca, A.; Susnjar, S.; Milovanovic, Z.; Supic, G. Histone Deacetylase 7 Gene Overexpression Is Associated with Poor Prognosis of Triple-Negative Breast Cancer Patients. Genet. Test. Mol. Biomarkers 2021, 25, 227–235. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Cao, F.; Yu, X.; Zhou, P.; Di, Q.; Lei, J.; Tai, Y.; Wu, H.; Li, X.; Wang, X.; et al. The expression of HDAC7 in cancerous gastric tissues is positively associated with distant metastasis and poor patient prognosis. Clin. Transl. Oncol. 2017, 19, 1045–1054. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Chen, Q.; Xie, Z.; Ai, J.; Tong, L.; Ding, J.; Geng, M. The role of histone deacetylase 7 (HDAC7) in cancer cell proliferation: Regulation on c-Myc. J. Mol. Med. 2011, 89, 279–289. [Google Scholar] [CrossRef] [PubMed]
- Cutano, V.; Di Giorgio, E.; Minisini, M.; Picco, R.; Dalla, E.; Brancolini, C. HDAC7-mediated control of tumour microenvironment maintains proliferative and stemness competence of human mammary epithelial cells. Mol. Oncol. 2019, 13, 1651–1668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaudhary, M.; Kumar, N.; Baldi, A.; Chandra, R.; Babu, M.A.; Madan, J. Chloro and bromo-pyrazole curcumin Knoevenagel condensates augmented anticancer activity against human cervical cancer cells: Design, synthesis, in silico docking and in vitro cytotoxicity analysis. J. Biomol. Struct. Dyn. 2020, 38, 200–218. [Google Scholar] [CrossRef]
- Zhou, S.; Xu, L.; Cao, M.; Wang, Z.; Xiao, D.; Xu, S.; Deng, J.; Hu, X.; He, C.; Tao, T.; et al. Anticancer properties of novel pyrazole-containing biguanide derivatives with activating the adenosine monophosphate-activated protein kinase signaling pathway. Arch. Pharm. 2019, 352, e1900075. [Google Scholar] [CrossRef]
- Ran, F.; Liu, Y.; Zhang, D.; Liu, M.; Zhao, G. Discovery of novel pyrazole derivatives as potential anticancer agents in MCL. Bioorganic Med. Chem. Lett. 2019, 29, 1060–1064. [Google Scholar] [CrossRef]
- Xiao, D.; He, F.; Peng, D.; Zou, M.; Peng, J.; Liu, P.; Liu, Y.; Liu, Z. Synthesis and Anticancer Activity of 9-O-Pyrazole Alkyl Substituted Berberine Derivatives. Anti-Cancer Agents Med. Chem. 2018, 18, 1639–1648. [Google Scholar] [CrossRef]
- Ganguly, S.; Jacob, S.K. Therapeutic Outlook of Pyrazole Analogs: A Mini Review. Mini-Reviews Med. Chem. 2017, 17, 959–983. [Google Scholar] [CrossRef]
- Khan, M.F.; Alam, M.M.; Verma, G.; Akhtar, W.; Akhter, M.; Shaquiquzzaman, M. The therapeutic voyage of pyrazole and its analogs: A review. Eur. J. Med. Chem. 2016, 120, 170–201. [Google Scholar] [CrossRef]
- Shi, J.B.; Tang, W.J.; Qi, X.B.; Li, R.; Liu, X.H. Novel pyrazole-5-carboxamide and pyrazole–pyrimidine derivatives: Synthesis and anticancer activity. Eur. J. Med. Chem. 2015, 90, 889–896. [Google Scholar] [CrossRef] [PubMed]
- Kumar, H.; Saini, D.; Jain, S.; Jain, N. Pyrazole scaffold: A remarkable tool in the development of anticancer agents. Eur. J. Med. Chem. 2013, 70, 248–258. [Google Scholar] [CrossRef] [PubMed]
- Bennani, F.E.; Doudach, L.; Cherrah, Y.; Ramli, Y.; Karrouchi, K.; Ansar, M.; Faouzi, M.E.A. Overview of recent developments of pyrazole derivatives as an anticancer agent in different cell line. Bioorganic Chem. 2020, 97, 103470. [Google Scholar] [CrossRef] [PubMed]
- Cai, W.; Wu, J.; Sun, Y.; Liu, A.; Wang, R.; Ma, Y.; Wang, S.; Dong, W. Synthesis, evaluation, molecular dynamics simulation and targets identification of novel pyrazole-containing imide derivatives. J. Biomol. Struct. Dyn. 2021, 39, 2176–2188. [Google Scholar] [CrossRef]
- Azimi, F.; Ghasemi, J.B.; Azizian, H.; Najafi, M.; Faramarzi, M.A.; Saghaei, L.; Sadeghi-Aliabadi, H.; Larijani, B.; Hassanzadeh, F.; Mahdavi, M. Design and synthesis of novel pyrazole-phenyl semicarbazone derivatives as potential α-glucosidase inhibitor: Kinetics and molecular dynamics simulation study. Int. J. Biol. Macromol. 2021, 166, 1082–1095. [Google Scholar] [CrossRef]
- Thillainayagam, M.; Ramaiah, S.; Anbarasu, A. Molecular docking and dynamics studies on novel benzene sulfonamide substituted pyrazole-pyrazoline analogues as potent inhibitors of Plasmodium falciparum Histo aspartic protease. J. Biomol. Struct. Dyn. 2020, 38, 3235–3245. [Google Scholar] [CrossRef] [PubMed]
- Philoppes, J.N.; Khedr, M.A.; Hassan, M.; Kamel, G.; Lamie, P.F. New pyrazolopyrimidine derivatives with anticancer activity: Design, synthesis, PIM-1 inhibition, molecular docking study and molecular dynamics. Bioorganic Chem. 2020, 100, 103944. [Google Scholar] [CrossRef]
- Peytam, F.; Adib, M.; Shourgeshty, R.; Mohammadi-Khanaposhtani, M.; Jahani, M.; Imanparast, S.; Faramarzi, M.A.; Mahdavi, M.; Moghadamnia, A.A.; Rastegar, H.; et al. Design and synthesis of new imidazo[1,2-b]pyrazole derivatives, in vitro α-glucosidase inhibition, kinetic and docking studies. Mol. Divers. 2020, 24, 69–80. [Google Scholar] [CrossRef]
- Czaja, K.; Kujawski, J.; Kamel, K.; Bernard, M.K. Selected arylsulphonyl pyrazole derivatives as potential Chk1 kinase ligands—Computational investigations. J. Mol. Model. 2020, 26, 1–11. [Google Scholar] [CrossRef]
- Hassan, G.S.; Rahman, D.E.A.; Abdelmajeed, E.A.; Refaey, R.H.; Salem, M.A.; Nissan, Y.M. New pyrazole derivatives: Synthesis, anti-inflammatory activity, cycloxygenase inhibition assay and evaluation of mPGES. Eur. J. Med. Chem. 2019, 171, 332–342. [Google Scholar] [CrossRef]
- Elsayed, N.M.; Serya, R.A.; Tolba, M.F.; Ahmed, M.; Barakat, K.; El Ella, D.A.A.; Abouzid, K.A. Design, synthesis, biological evaluation and dynamics simulation of indazole derivatives with antiangiogenic and antiproliferative anticancer activity. Bioorganic Chem. 2019, 82, 340–359. [Google Scholar] [CrossRef] [PubMed]
- Nossier, E.S.; El-Karim, S.S.A.; Khalifa, N.M.; El-Sayed, A.S.; Hassan, E.S.I.; El-Hallouty, S.M. Kinase Inhibitory Activities and Molecular Docking of a Novel Series of Anticancer Pyrazole Derivatives. Molecules 2018, 23, 3074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caballero, J.; Alzate-Morales, J.H.; Vergara-Jaque, A. Investigation of the Differences in Activity between Hydroxycycloalkyl N1 Substituted Pyrazole Derivatives As Inhibitors of B-Raf Kinase by Using Docking, Molecular Dynamics, QM/MM, and Fragment-Based De Novo Design: Study of Binding Mode of Diastereomer Compounds. J. Chem. Inf. Model. 2011, 51, 2920–2931. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Brittain, J.M.; Jarecki, B.W.; Park, K.D.; Wilson, S.M.; Wang, B.; Hale, R.; Meroueh, S.O.; Cummins, T.R.; Khanna, R. In Silico Docking and Electrophysiological Characterization of Lacosamide Binding Sites on Collapsin Response Mediator Protein-2 Identifies a Pocket Important in Modulating Sodium Channel Slow Inactivation. J. Biol. Chem. 2010, 285, 25296–25307. [Google Scholar] [CrossRef] [Green Version]
- Brown, T. ChemDraw. Sci. Teach. 2014, 81, 67. [Google Scholar]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4 Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [Green Version]
- Toyoshima, M.; Jiang, X.; Ogawa, T.; Ohnishi, T.; Yoshihara, S.; Balan, S.; Yoshikawa, T.; Hirokawa, N. Enhanced carbonyl stress induces irreversible multimerization of CRMP2 in schizophrenia pathogenesis. Life Sci. Alliance 2019, 2, e201900478. [Google Scholar] [CrossRef]
- Hatzivassiliou, G.; Song, K.; Yen, I.; Brandhuber, B.J.; Anderson, D.J.; Alvarado, R.; Ludlam, M.J.C.; Stokoe, D.; Gloor, S.L.; Vigers, G.; et al. RAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growth. Nature 2010, 464, 431–435. [Google Scholar] [CrossRef] [Green Version]
- McTigue, M.; Murray, B.W.; Chen, J.H.; Deng, Y.-L.; Solowiej, J.; Kania, R.S. Molecular conformations, interactions, and properties associated with drug efficiency and clinical performance among VEGFR TK inhibitors. Proc. Natl. Acad. Sci. USA 2012, 109, 18281–18289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McAulay, K.; Hoyt, E.A.; Thomas, M.; Schimpl, M.; Bodnarchuk, M.S.; Lewis, H.J.; Kettle, J.G. Alkynyl Benzoxazines and Dihy-droquinazolines as Cysteine Targeting Covalent Warheads and Their Application in Identification of Selective Irreversible Kinase Inhibitors. J. Am. Chem. Soc. 2020, 142, 10358–10372. [Google Scholar] [CrossRef]
- Alder, B.J.; Wainwright, T.E. Studies in molecular dynamics. I. General method. J. Chem. Phys. 1959, 31, 459–466. [Google Scholar] [CrossRef] [Green Version]
- Desmond Molecular Dynamics System. D. E. Shaw Research, New York, NY, 2021. Maestro-Desmond Interoperability Tools, Schrödinger, New York, NY. 2021. Available online: https://www.schrodinger.com/products/desmond (accessed on 1 November 2021).
- Shivakumar, D.; Williams, J.; Wu, Y.; Damm, W.; Shelley, J.; Sherman, W. Prediction of Absolute Solvation Free Energies using Molecular Dynamics Free Energy Perturbation and the OPLS Force Field. J. Chem. Theory Comput. 2010, 6, 1509–1519. [Google Scholar] [CrossRef] [PubMed]
- Martyna, G.J.; Klein, M.L.; Tuckerman, M.E. Nosé–Hoover chains: The canonical ensemble via continuous dynamics. J. Chem. Phys. 1992, 97, 2635–2643. [Google Scholar] [CrossRef]
- Tuckerman, M.; Berne, B.J.; Martyna, G.J. Reversible multiple time scale molecular dynamics. J. Chem. Phys. 1992, 97, 1990–2001. [Google Scholar] [CrossRef] [Green Version]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef] [Green Version]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [Green Version]
- Pires, D.E.V.; Blundell, T.L.; Ascher, D.B. pkCSM: Predicting Small-Molecule Pharmacokinetic and Toxicity Properties Using Graph-Based Signatures. J. Med. Chem. 2015, 58, 4066–4072. [Google Scholar] [CrossRef]
- Lipinski, C.A. Lead- and drug-like compounds: The rule-of-five revolution. Drug Discov. Today Technol. 2004, 1, 337–341. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, H.-Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular Properties That Influence the Oral Bioavailability of Drug Candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef] [PubMed]
- Jabbarzadeh Kaboli, P.; Ismail, P.; Ling, K.-H. Molecular modeling, dynamics simulations, and binding efficiency of berberine derivatives: A new group of RAF inhibitors for cancer treatment. PLoS ONE 2018, 13, e0193941. [Google Scholar] [CrossRef] [PubMed]
- Al-Masoudi, N.A.; Ali, D.S.; Saeed, B.; Hartmann, R.W.; Engel, M.; Rashid, S.; Saeed, A. New CYP17 hydroxylase inhibitors: Synthesis, biological evaluation, QSAR, and molecular docking study of new pregnenolone analogs. Arch. Der Pharm. 2014, 347, 896–907. [Google Scholar] [CrossRef]
- Kang, D.; Pang, X.; Lian, W.; Xu, L.; Wang, J.; Jia, H.; Zhang, B.; Liu, A.-L.; Du, G.-H. Discovery of VEGFR2 inhibitors by integrating naïve Bayesian classification, molecular docking and drug screening approaches. RSC Adv. 2018, 8, 5286–5297. [Google Scholar] [CrossRef] [Green Version]
- Meng, F. Molecular Dynamics Simulation of VEGFR2 with Sorafenib and Other Urea-Substituted Aryloxy Compounds. J. Theor. Chem. 2013, 2013, 739574. [Google Scholar] [CrossRef] [Green Version]
- Mak, J.Y.W.; Wu, K.-C.; Gupta, P.K.; Barbero, S.; McLaughlin, M.G.; Lucke, A.J.; Tng, J.; Lim, J.; Loh, Z.; Sweet, M.J.; et al. HDAC7 Inhibition by Phenacetyl and Phenylbenzoyl Hydroxamates. J. Med. Chem. 2021, 64, 2186–2204. [Google Scholar] [CrossRef]




















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Proteins | PDB IDs | References | Resolution (Å) | Residue’s Count |
|---|---|---|---|---|
| CRMP2 | 6JV9 | [100] | 2.26 | 540 |
| C-RAF | 3OMV | [101] | 2.4 | 95 |
| CYP17 | 4NKV | [52] | 2.65 | 494 |
| VEGFR | 4AGD | [102] | 2.81 | 353 |
| C-KIT | 6XVB | [103] | 2.15 | 328 |
| HDAC | 3ZNR | [39] | 2.4 | 423 |
| Compounds | Lipinski Rule of 5 | Veber’s Rule | ||||||
|---|---|---|---|---|---|---|---|---|
| MW | LogP | HBA | HBD | Lipinski’s Rule Violations | NRB | Surface Area | Vebers’ Violations | |
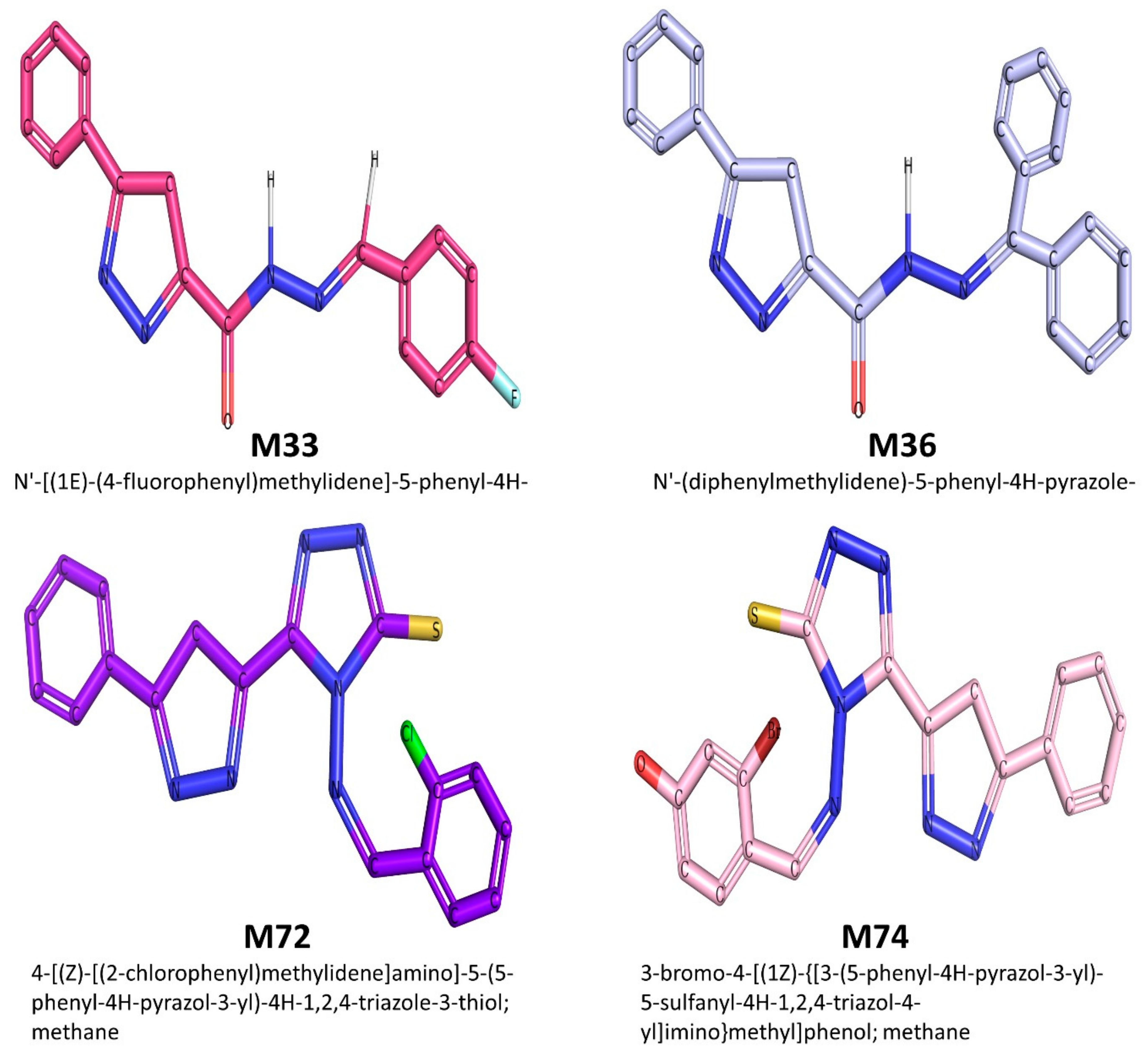
| M33 | 308.316 | 2.5248 | 4 | 4 | Suitable | 1 | 131.811 | Suitable |
| M36 | 366.424 | 3.8042 | 5 | 4 | Suitable | 1 | 162.702 | Suitable |
| M72 | 380.864 | 3.6995 | 4 | 7 | Suitable | 1 | 159.911 | Suitable |
| M74 | 441.314 | 3.5142 | 4 | 8 | Suitable | 2 | 168.269 | Suitable |
| M76 | 360.446 | 3.35452 | 4 | 7 | Suitable | 1 | 155.973 | Suitable |
| ABSORPTION | |||||||
|---|---|---|---|---|---|---|---|
| Compounds | Water Solubility (Log mol/L) | CaCO2 Permeability (cm/s) | Intestinal Absorption (Human) in % | Skin Permeability | P-Glycoprotein Substrate | P-Glycoprotein II Inhibitor | P-Glycoprotein I Inhibitor |
| M33 | −3.78 | 0.91 | 92.749 | −2.774 | − | + | − |
| M36 | −4.472 | 1.144 | 94.034 | −2.656 | − | + | + |
| M72 | −4.969 | 0.953 | 91.145 | −2.878 | − | + | + |
| M74 | −4.813 | 0.964 | 88.182 | −2.986 | − | + | + |
| M76 | −4.527 | 0.976 | 92.592 | −2.889 | − | + | + |
| DITRIBUTION | ||||
|---|---|---|---|---|
| Molecules | VDss (Human) L kg−1 | Fraction Unbound (Human) | BBB Permeability | CNS Permeability |
| M33 | −0.021 | 0.053 | 0.011 | −2.229 |
| M36 | 0.088 | 0.008 | −0.046 | −2.502 |
| M72 | −0.365 | 0.035 | −0.752 | −1.946 |
| M74 | −0.623 | 0.058 | −0.996 | −2.113 |
| M76 | −0.342 | 0.050 | −0.587 | −1.986 |
| METABOLISM | |||||
|---|---|---|---|---|---|
| Molecules | CYP1A2 Inhibitor | CYP2C19 Inhibitor | CYP2C9 Inhibitor | CYP2D6 Inhibitor | CYP3A4 Inhibitor |
| M33 | Inhibitor | Inhibitor | Non-Inhibitor | Non-Inhibitor | Non-Inhibitor |
| M36 | Inhibitor | Inhibitor | Inhibitor | Non-Inhibitor | Non-Inhibitor |
| M72 | Inhibitor | Inhibitor | Inhibitor | Non-Inhibitor | Non-Inhibitor |
| M74 | Inhibitor | Inhibitor | Inhibitor | Non-Inhibitor | Non-Inhibitor |
| M76 | Inhibitor | Inhibitor | Inhibitor | Non-Inhibitor | Non-Inhibitor |
| EXCRETION | ||
|---|---|---|
| Molecules | Total Drug Clearance | Renal OCT2 Substrate Inhibitor |
| M33 | 0.543 | Yes |
| M36 | 0.809 | No |
| M72 | 0.322 | No |
| M74 | 0.031 | No |
| M76 | 0.276 | No |
| TOXICITY | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Molecules | AMES Toxicity | Max. Tolerated Dose (Human) | hERG I Inhibitor | hERG II Inhibitor | Oral Rat Acute Toxicity (LD50) | Oral Rat Chronic Toxicity (LOAEL) | Hepatotoxicity | Skin Sensitization | T. Pyriformis Toxicity | Minnow Toxicity |
| M33 | No | 0.091 | No | No | 2.472 | 1.379 | No | No | 1.994 | 1.289 |
| M36 | No | 0.225 | No | Yes | 2.276 | 2.261 | No | No | 0.894 | 0.054 |
| M72 | No | −0.127 | No | Yes | 2.751 | 1.531 | Yes | No | 1.207 | 0.478 |
| M74 | No | −0.142 | No | Yes | 2.776 | 1.556 | Yes | No | 0.898 | 0.748 |
| M76 | No | −0.119 | No | Yes | 2.615 | 1.649 | Yes | No | 1.253 | 0.768 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bennani, F.E.; Karrouchi, K.; Doudach, L.; Scrima, M.; Rahman, N.; Rastrelli, L.; Tallei, T.E.; Rudd, C.E.; Faouzi, M.E.A.; Ansar, M. In Silico Identification of Promising New Pyrazole Derivative-Based Small Molecules for Modulating CRMP2, C-RAF, CYP17, VEGFR, C-KIT, and HDAC—Application towards Cancer Therapeutics. Curr. Issues Mol. Biol. 2022, 44, 5312-5351. https://doi.org/10.3390/cimb44110361
Bennani FE, Karrouchi K, Doudach L, Scrima M, Rahman N, Rastrelli L, Tallei TE, Rudd CE, Faouzi MEA, Ansar M. In Silico Identification of Promising New Pyrazole Derivative-Based Small Molecules for Modulating CRMP2, C-RAF, CYP17, VEGFR, C-KIT, and HDAC—Application towards Cancer Therapeutics. Current Issues in Molecular Biology. 2022; 44(11):5312-5351. https://doi.org/10.3390/cimb44110361
Chicago/Turabian StyleBennani, Fatima Ezzahra, Khalid Karrouchi, Latifa Doudach, Mario Scrima, Noor Rahman, Luca Rastrelli, Trina Ekawati Tallei, Christopher E. Rudd, My El Abbes Faouzi, and M’hammed Ansar. 2022. "In Silico Identification of Promising New Pyrazole Derivative-Based Small Molecules for Modulating CRMP2, C-RAF, CYP17, VEGFR, C-KIT, and HDAC—Application towards Cancer Therapeutics" Current Issues in Molecular Biology 44, no. 11: 5312-5351. https://doi.org/10.3390/cimb44110361
APA StyleBennani, F. E., Karrouchi, K., Doudach, L., Scrima, M., Rahman, N., Rastrelli, L., Tallei, T. E., Rudd, C. E., Faouzi, M. E. A., & Ansar, M. (2022). In Silico Identification of Promising New Pyrazole Derivative-Based Small Molecules for Modulating CRMP2, C-RAF, CYP17, VEGFR, C-KIT, and HDAC—Application towards Cancer Therapeutics. Current Issues in Molecular Biology, 44(11), 5312-5351. https://doi.org/10.3390/cimb44110361