
Prior Treatment with AICAR Causes the Selective Phosphorylation of mTOR Substrates in C2C12 Cells



Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
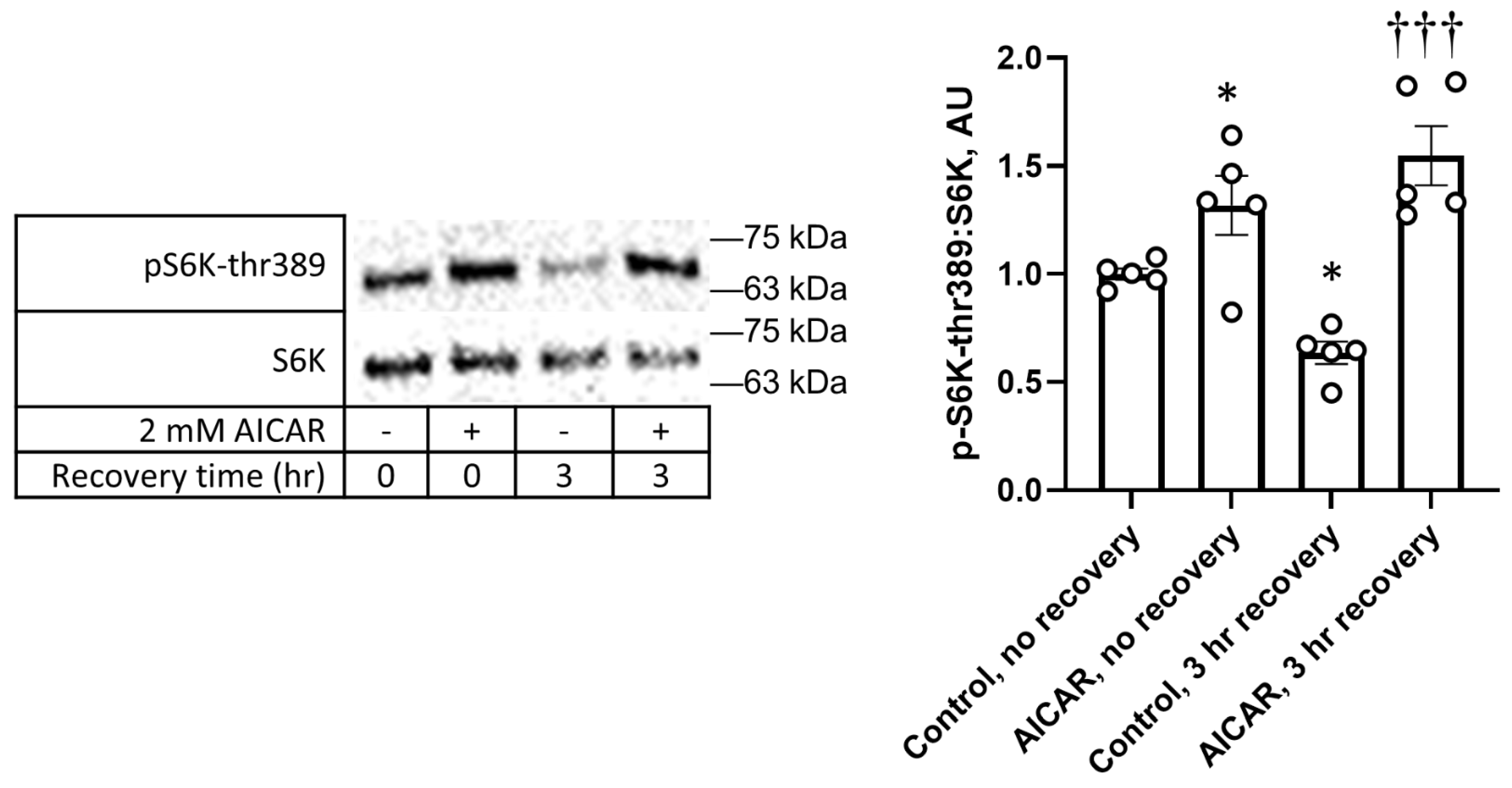{kind=link}
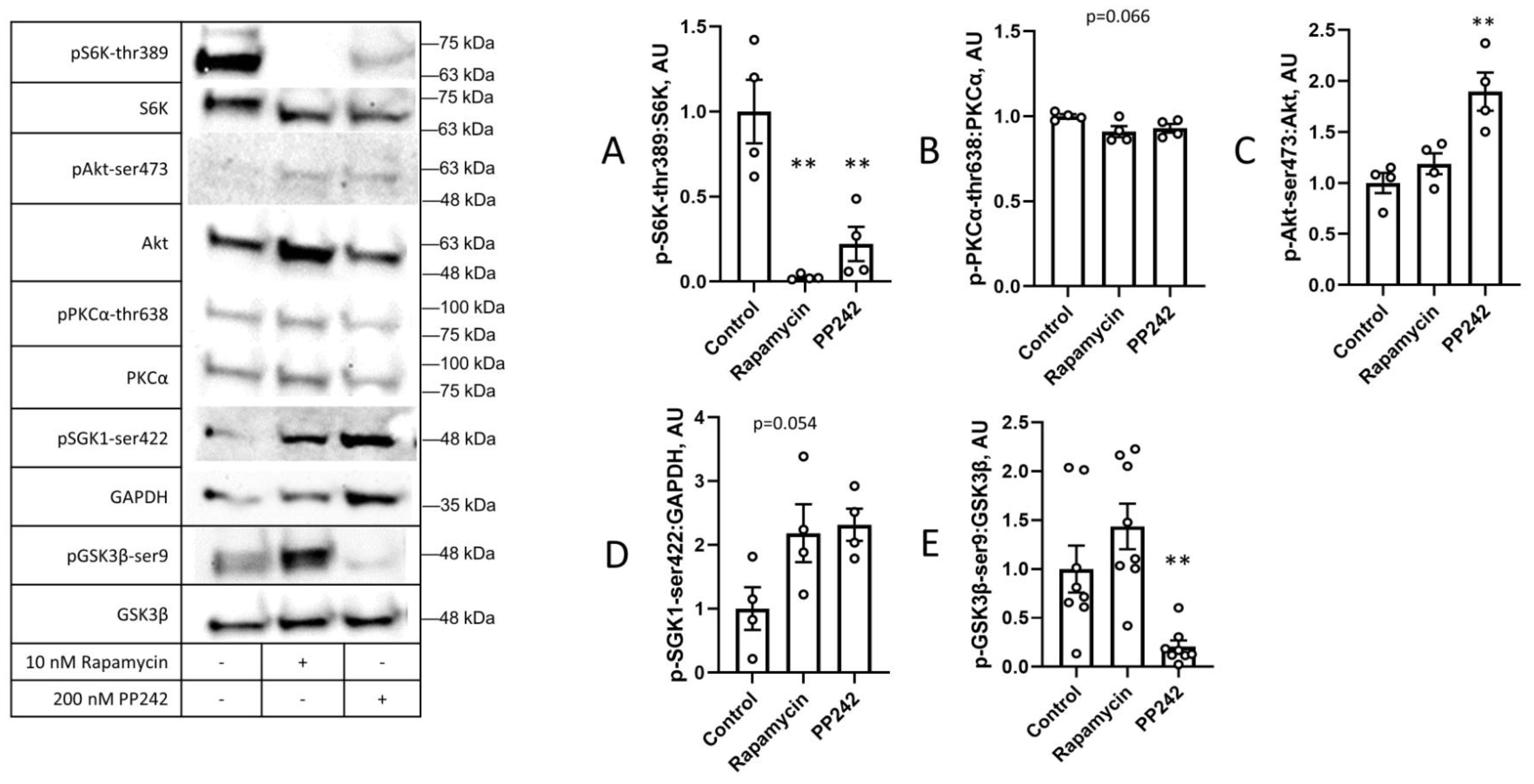{kind=link}
1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Cells Studied
2.3. Cell Plating and Harvest
2.4. Enzymatic Activity Assay
2.5. Western Blot
2.6. Statistics
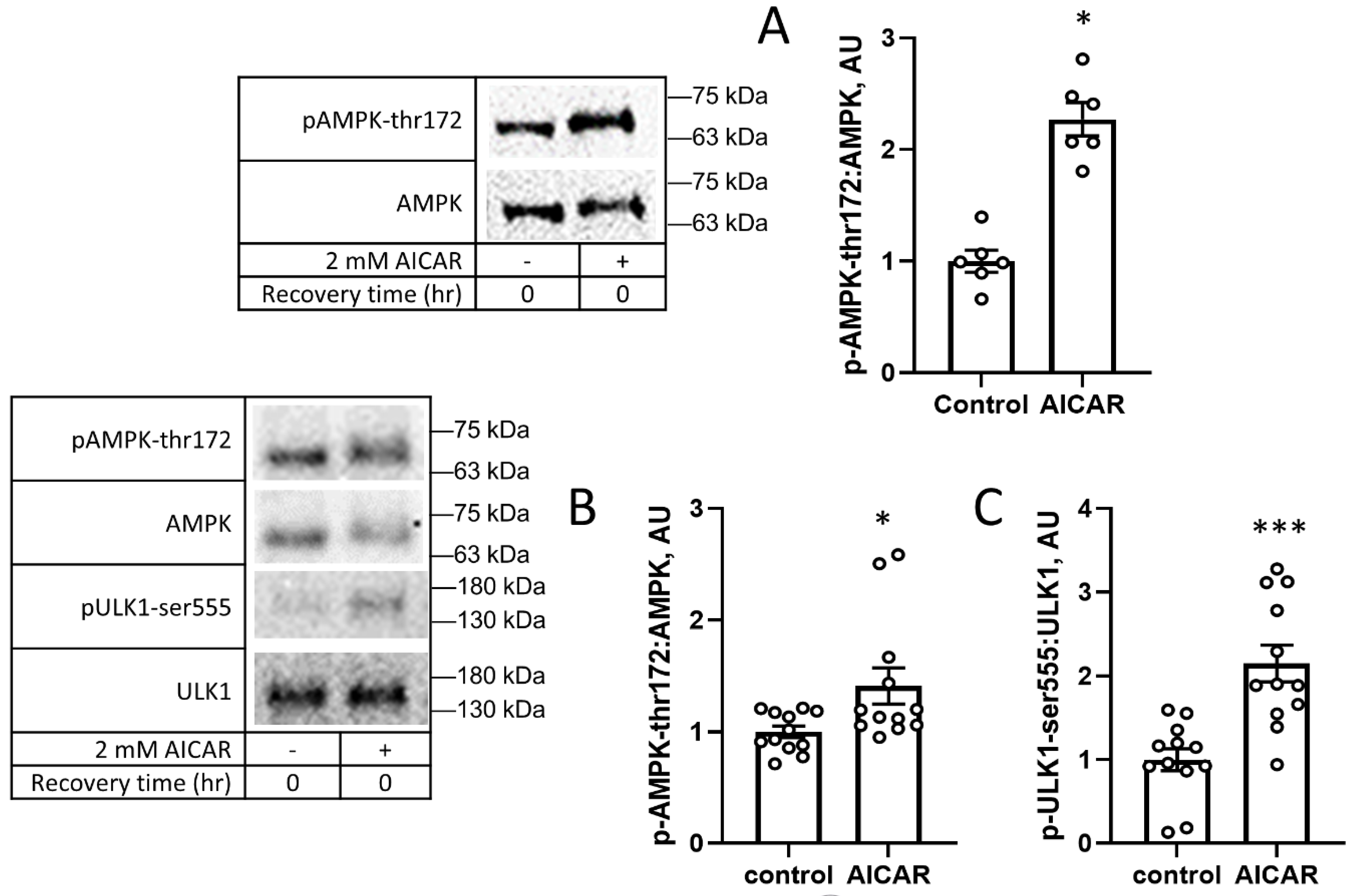
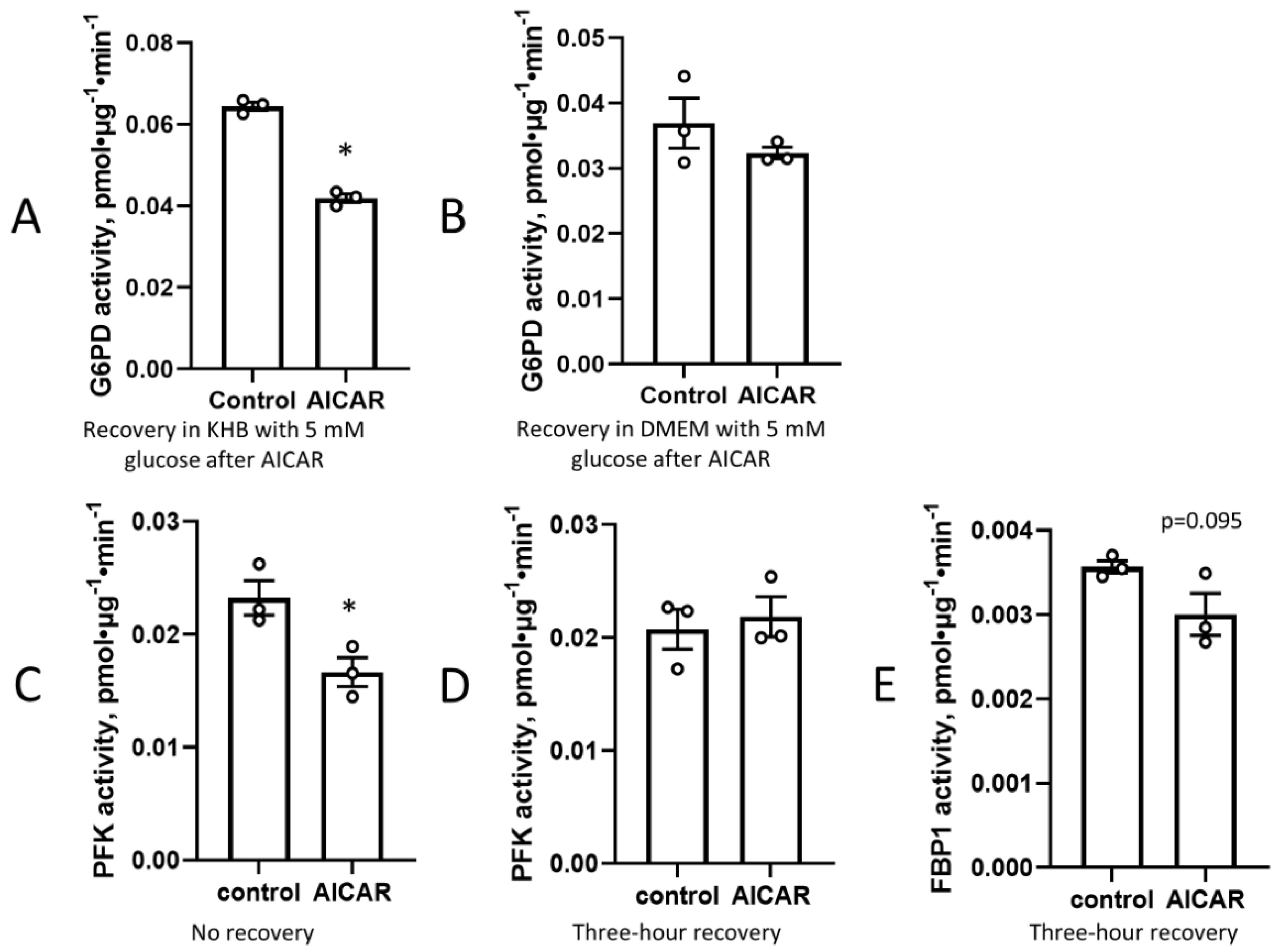
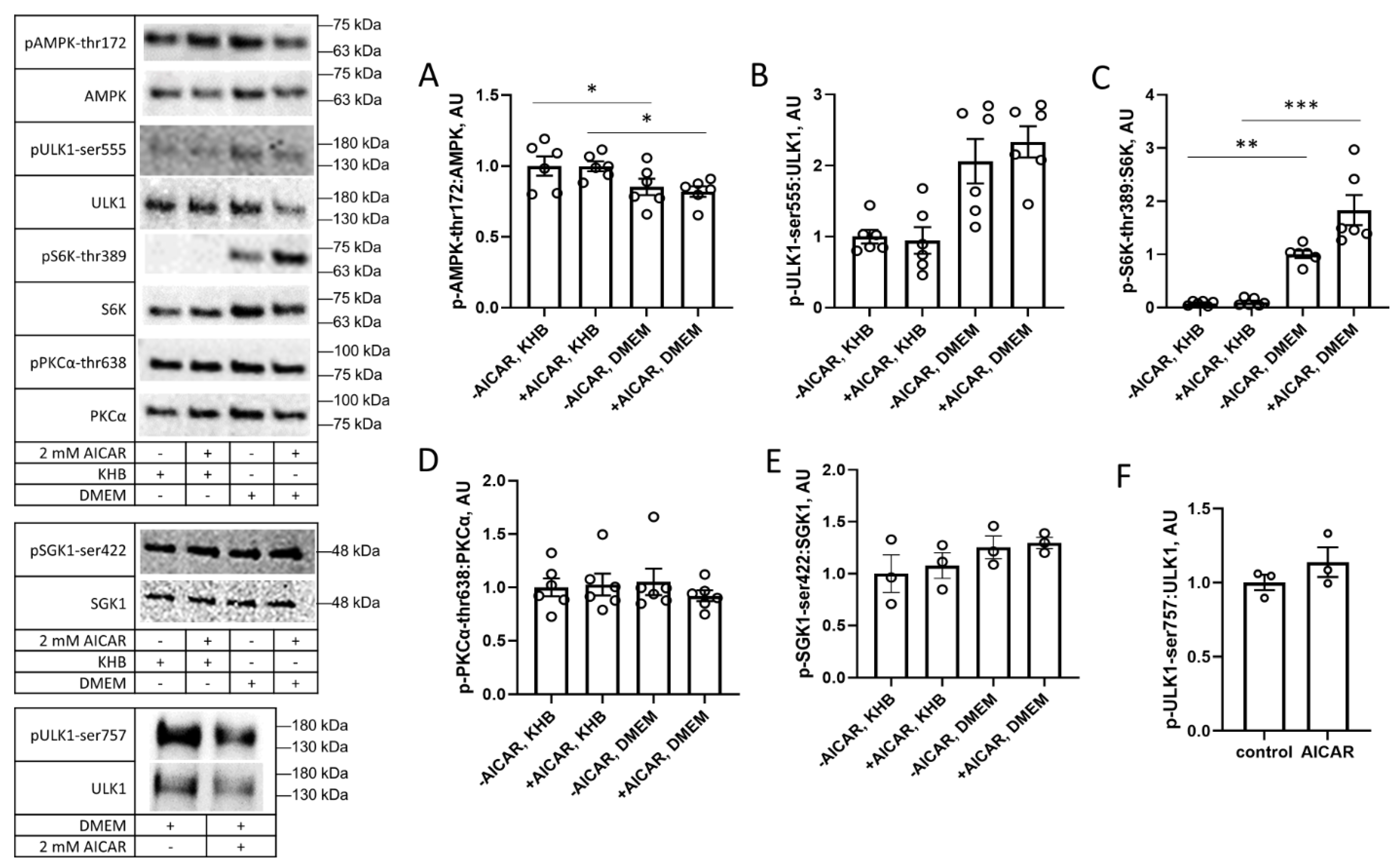
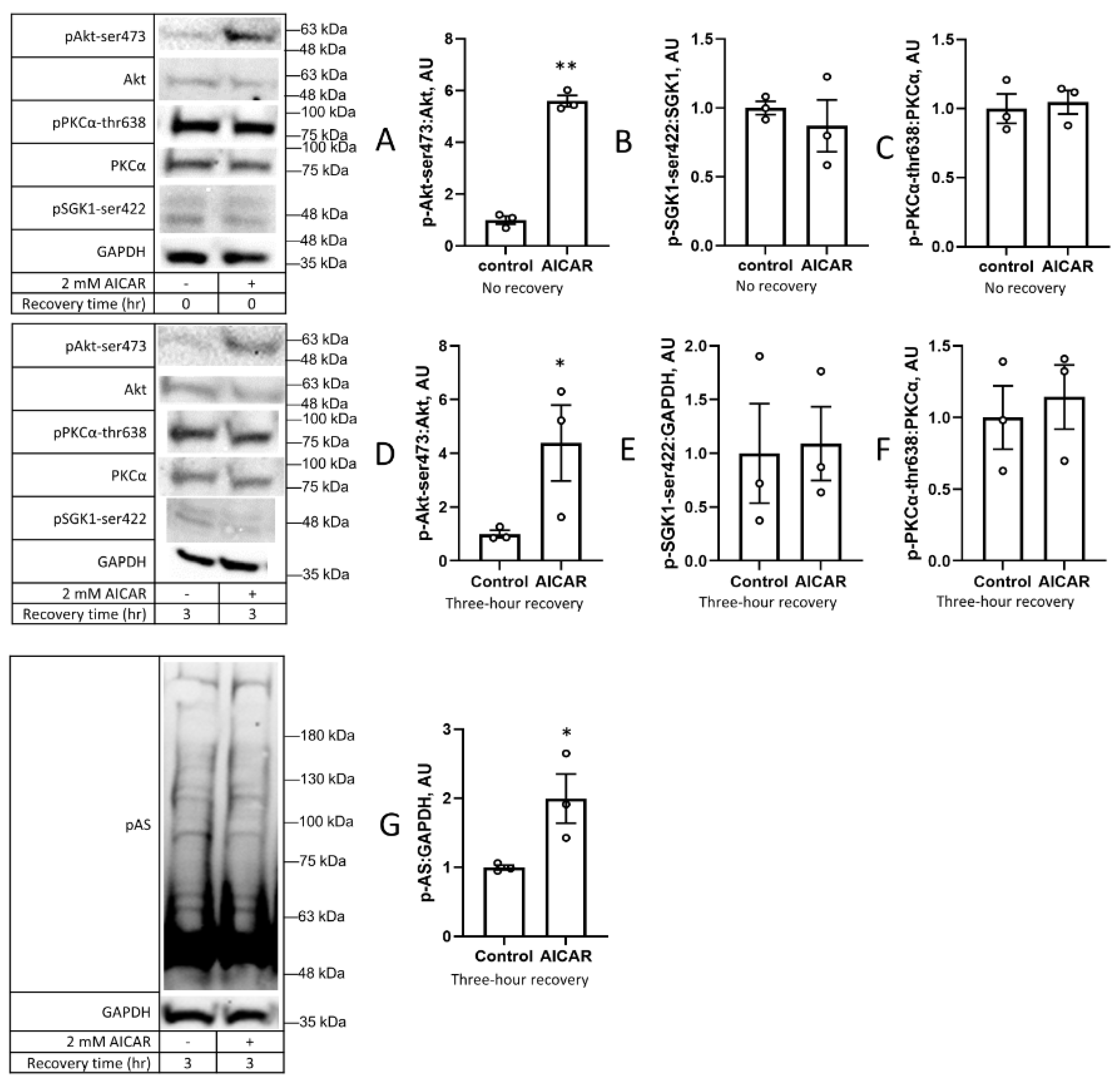
3. Results
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Corton, J.M.; Gillespie, J.G.; Hawley, S.A.; Hardie, D.G. 5-aminoimidazole-4-carboxamide ribonucleoside. A specific method for activating AMP-activated protein kinase in intact cells? Eur. J. Biochem. 1995, 229, 558–565. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.C.; Hardie, D.G. AMPK: Sensing Glucose as well as Cellular Energy Status. Cell Metab. 2018, 27, 299–313. [Google Scholar] [CrossRef]
- Merrill, G.F.; Kurth, E.J.; Hardie, D.G.; Winder, W.W. AICA riboside increases AMP-activated protein kinase, fatty acid oxidation, and glucose uptake in rat muscle. Am. J. Physiol. Metab. 1997, 273, E1107–E1112. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, T.; Hirshman, M.F.; Kurth, E.J.; Winder, W.W.; Goodyear, L.J. Evidence for 5’ AMP-activated protein kinase mediation of the effect of muscle contraction on glucose transport. Diabetes 1998, 47, 1369–1373. [Google Scholar] [CrossRef] [PubMed]
- Bolster, D.R.; Crozier, S.J.; Kimball, S.R.; Jefferson, L.S. AMP-activated protein kinase suppresses protein synthesis in rat skeletal muscle through down-regulated mammalian target of rapamycin (mTOR) signaling. J. Biol. Chem. 2002, 277, 23977–23980. [Google Scholar] [CrossRef] [PubMed]
- Višnjić, D.; Lalić, H.; Dembitz, V.; Tomić, B.; Smoljo, T. AICAr, a Widely Used AMPK Activator with Important AMPK-Independent Effects: A Systematic Review. Cells 2021, 10, 1095. [Google Scholar] [CrossRef] [PubMed]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef] [PubMed]
- Dibble, C.C.; Cantley, L.C. Regulation of mTORC1 by PI3K signaling. Trends Cell Biol. 2015, 25, 545–555. [Google Scholar] [CrossRef]
- Schiaffino, S.; Reggiani, C.; Akimoto, T.; Blaauw, B. Molecular Mechanisms of Skeletal Muscle Hypertrophy. J. Neuromuscul. Dis. 2021, 8, 169–183. [Google Scholar] [CrossRef]
- Sarbassov, D.D.; Guertin, D.A.; Ali, S.M.; Sabatini, D.M. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 2005, 307, 1098–1101. [Google Scholar] [CrossRef]
- Hresko, R.C.; Mueckler, M. mTOR.RICTOR is the Ser473 kinase for Akt/protein kinase B in 3T3-L1 adipocytes. J. Biol. Chem. 2005, 280, 40406–40416. [Google Scholar] [CrossRef]
- Fisher, J.S.; Gao, J.; Han, D.H.; Holloszy, J.O.; Nolte, L.A. Activation of AMP kinase enhances sensitivity of muscle glucose transport to insulin. Am. J. Physiol. Endocrinol. Metab. 2002, 282, E18–E23. [Google Scholar] [CrossRef] [PubMed]
- Baar, K.; Esser, K. Phosphorylation of p70(S6k) correlates with increased skeletal muscle mass following resistance exercise. Am. J. Physiol. Physiol. 1999, 276, C120–C127. [Google Scholar] [CrossRef] [PubMed]
- Sriwijitkamol, A.; Coletta, D.K.; Wajcberg, E.; Balbontin, G.B.; Reyna, S.M.; Barrientes, J.; Eagan, P.A.; Jenkinson, C.P.; Cersosimo, E.; DeFronzo, R.A.; et al. Effect of Acute Exercise on AMPK Signaling in Skeletal Muscle of Subjects with Type 2 Diabetes: A Time-Course and Dose-Response Study. Diabetes 2007, 56, 836–848. [Google Scholar] [CrossRef]
- Schweitzer, G.G.; Arias, E.B.; Cartee, G.D. Sustained postexercise increases in AS160 Thr642 and Ser588 phosphorylation in skeletal muscle without sustained increases in kinase phosphorylation. J. Appl. Physiol. 2012, 113, 1852–1861. [Google Scholar] [CrossRef]
- Somwar, R.; Kim, D.Y.; Sweeney, G.; Huang, C.; Niu, W.; Lador, C.; Ramlal, T.; Klip, A. GLUT4 translocation precedes the stimulation of glucose uptake by insulin in muscle cells: Potential activation of GLUT4 via p38 mitogen-activated protein kinase. Biochem. J. 2001, 359 Pt 3, 639–649. [Google Scholar] [CrossRef] [PubMed]
- Eccardt, A.M.; Pelzel, R.J.; Mattathil, L.; Moon, Y.A.; Mannino, M.H.; Janowiak, B.E.; Fisher, J.S. A peroxidase mimetic protects skeletal muscle cells from peroxide challenge and stimulates insulin signaling. Am. J. Physiol. Cell Physiol. 2020, 318, C1214–C1225. [Google Scholar] [CrossRef]
- Smith, J.L.; Patil, P.B.; Fisher, J.S. AICAR and hyperosmotic stress increase insulin-stimulated glucose transport. J. Appl. Physiol. 2005, 99, 877–883. [Google Scholar] [CrossRef] [PubMed]
- Passonneau, J.V.; Lowry, O.H.; Lowry, O.H. Enzymatic Analysis: A Practical Guide; Humana Press: Totowa, NJ, USA, 1993. [Google Scholar]
- Kohan, A.B.; Talukdar, I.; Walsh, C.M.; Salati, L.M. A role for AMPK in the inhibition of glucose-6-phosphate dehydrogenase by polyunsaturated fatty acids. Biochem. Biophys. Res. Commun. 2009, 388, 117–121. [Google Scholar] [CrossRef] [PubMed]
- Heden, T.D.; Chow, L.S.; Hughey, C.C.; Mashek, D.G. Regulation and role of glycophagy in skeletal muscle energy metabolism. Autophagy 2021, 18, 1078–1089. [Google Scholar] [CrossRef]
- Herzig, S.; Shaw, R.J. AMPK: Guardian of metabolism and mitochondrial homeostasis. Nat. Rev. Mol. Cell Biol. 2018, 19, 121–135. [Google Scholar] [CrossRef] [PubMed]
- Kazyken, D.; Magnuson, B.; Bodur, C.; Acosta-Jaquez, H.A.; Zhang, D.; Tong, X.; Barnes, T.M.; Steinl, G.K.; Patterson, N.E.; Altheim, C.H.; et al. AMPK directly activates mTORC2 to promote cell survival during acute energetic stress. Sci. Signal. 2019, 12, eaav3249. [Google Scholar] [CrossRef]
- Li, J.; Kim, S.G.; Blenis, J. Rapamycin: One Drug, Many Effects. Cell Metab. 2014, 19, 373–379. [Google Scholar] [CrossRef] [PubMed]
- Thoreen, C.C.; Kang, S.A.; Chang, J.W.; Liu, Q.; Zhang, J.; Gao, Y.; Reichling, L.J.; Sim, T.; Sabatini, D.M.; Gray, N.S. An ATP-competitive mammalian target of rapamycin inhibitor reveals rapamycin-resistant functions of mTORC1. J. Biol. Chem. 2009, 284, 8023–8032. [Google Scholar] [CrossRef]
- Lee, J.; Pilch, P.F. The insulin receptor: Structure, function, and signaling. Am. J. Physiol. Cell Physiol. 1994, 266, C319–C334. [Google Scholar] [CrossRef]
- Vanhaesebroeck, B.; Alessi, D.R. The PI3K–PDK1 connection: More than just a road to PKB. Biochem. J. 2000, 346, 561–576. [Google Scholar] [CrossRef]
- Vadlakonda, L.; Dash, A.; Pasupuleti, M.; Anil Kumar, K.; Reddanna, P. The Paradox of Akt-mTOR Interactions. Front. Oncol. 2013, 3, 165. [Google Scholar] [CrossRef]
- Sakamoto, K.; Holman, G.D. Emerging role for AS160/TBC1D4 and TBC1D1 in the regulation of GLUT4 traffic. Am. J. Physiol.-Endocrinol. Metab. 2008, 295, E29–E37. [Google Scholar] [CrossRef]
- Kumar, A.; Harris, T.E.; Keller, S.R.; Choi, K.M.; Magnuson, M.A.; Lawrence, J.C. Muscle-Specific Deletion of Rictor Impairs Insulin-Stimulated Glucose Transport and Enhances Basal Glycogen Synthase Activity. Mol. Cell. Biol. 2008, 28, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Sano, H.; Kane, S.; Sano, E.; Mîinea, C.P.; Asara, J.M.; Lane, W.S.; Garner, C.W.; Lienhard, G.E. Insulin-stimulated Phosphorylation of a Rab GTPase-activating Protein Regulates GLUT4 Translocation. J. Biol. Chem. 2003, 278, 14599–14602. [Google Scholar] [CrossRef]
- Howlett, K.F.; Mathews, A.; Garnham, A.; Sakamoto, K. The effect of exercise and insulin on AS160 phosphorylation and 14-3-3 binding capacity in human skeletal muscle. Am. J. Physiol.-Endocrinol. Metab. 2008, 294, E401–E407. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Xu, W.; Li, G.; Cui, W. Weighing In on mTOR Complex 2 Signaling: The Expanding Role in Cell Metabolism. Oxidative Med. Cell. Longev. 2018, 2018, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Sarbassov, D.D.; Ali, S.M.; Kim, D.H.; Guertin, D.A.; Latek, R.R.; Erdjument-Bromage, H.; Tempst, P.; Sabatini, D.M. Rictor, a Novel Binding Partner of mTOR, Defines a Rapamycin-Insensitive and Raptor-Independent Pathway that Regulates the Cytoskeleton. Curr. Biol. 2004, 14, 1296–1302. [Google Scholar] [CrossRef]
- Qin, X.; Jiang, B.; Zhang, Y. 4E-BP1, a multifactor regulated multifunctional protein. Cell Cycle 2016, 15, 781–786. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Bautista, S.J.; Boras, I.; Vissa, A.; Mecica, N.; Yip, C.M.; Kim, P.K.; Antonescu, C.N. mTOR complex 1 controls the nuclear localization and function of glycogen synthase kinase 3β. J. Biol. Chem. 2018, 293, 14723–14739. [Google Scholar] [CrossRef] [PubMed]
- Møller, A.B.; Vendelbo, M.H.; Christensen, B.; Clasen, B.F.; Bak, A.M.; Jørgensen, J.O.L.; Møller, N.; Jessen, N. Physical exercise increases autophagic signaling through ULK1 in human skeletal muscle. J. Appl. Physiol. 2015, 118, 971–979. [Google Scholar] [CrossRef]
- Dowling, R.J.O.; Topisirovic, I.; Fonseca, B.D.; Sonenberg, N. Dissecting the role of mTOR: Lessons from mTOR inhibitors. Biochim. Biophys. Acta BBA—Proteins Proteom. 2010, 1804, 433–439. [Google Scholar] [CrossRef]
- Khandia, R.; Dadar, M.; Munjal, A.; Dhama, K.; Karthik, K.; Tiwari, R.; Yatoo, M.I.; Iqbal, H.M.; Singh, K.P.; Joshi, S.K.; et al. A Comprehensive Review of Autophagy and Its Various Roles in Infectious, Non-Infectious, and Lifestyle Diseases: Current Knowledge and Prospects for Disease Prevention, Novel Drug Design, and Therapy. Cells 2019, 8, 674. [Google Scholar] [CrossRef]
- Ha, J.; Guan, K.L.; Kim, J. AMPK and autophagy in glucose/glycogen metabolism. Mol. Asp. Med. 2015, 46, 46–62. [Google Scholar] [CrossRef]
- Mizushima, N. The role of mammalian autophagy in protein metabolism. Proc. Jpn. Acad. Ser. B 2007, 83, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Li, T.Y.; Sun, Y.; Liang, Y.; Liu, Q.; Shi, Y.; Zhang, C.-S.; Zhang, C.; Song, L.; Zhang, P.; Zhang, X.; et al. ULK1/2 Constitute a Bifurcate Node Controlling Glucose Metabolic Fluxes in Addition to Autophagy. Mol. Cell 2016, 62, 359–370. [Google Scholar] [CrossRef]
- Zachari, M.; Ganley, I.G. The mammalian ULK1 complex and autophagy initiation. Essays Biochem. 2017, 61, 585–596. [Google Scholar] [CrossRef] [PubMed]
- Backer, J.M. The regulation and function of Class III PI3Ks: Novel roles for Vps34. Biochem. J. 2008, 410, 1–17. [Google Scholar] [CrossRef]
- Dan, H.C.; Antonia, R.J.; Baldwin, A.S. PI3K/Akt promotes feedforward mTORC2 activation through IKKα. Oncotarget 2016, 7, 21064–21075. [Google Scholar] [CrossRef]
- Wei, Y.; Zhou, J.; Yu, H.; Jin, X. AKT phosphorylation sites of Ser473 and Thr308 regulate AKT degradation. Biosci. Biotechnol. Biochem. 2019, 83, 429–435. [Google Scholar] [CrossRef]
- Feldman, M.E.; Apsel, B.; Uotila, A.; Loewith, R.; Knight, Z.A.; Ruggero, D.; Shokat, K.M. Active-Site Inhibitors of mTOR Target Rapamycin-Resistant Outputs of mTORC1 and mTORC2. PLoS Biol. 2009, 7, e1000038. [Google Scholar] [CrossRef] [PubMed]
- García-Martínez, J.M.; Alessi, D.R. mTOR complex 2 (mTORC2) controls hydrophobic motif phosphorylation and activation of serum- and glucocorticoid-induced protein kinase 1 (SGK1). Biochem. J. 2008, 416, 375–385. [Google Scholar] [CrossRef]
- Apsel, B.; Blair, J.A.; Gonzalez, B.; Nazif, T.M.; E Feldman, M.; Aizenstein, B.; Hoffman, R.; Williams, R.L.; Shokat, K.M.; A Knight, Z. Targeted polypharmacology: Discovery of dual inhibitors of tyrosine and phosphoinositide kinases. Nat. Chem. Biol. 2008, 4, 691–699. [Google Scholar] [CrossRef]
- Janes, M.R.; Limon, J.J.; So, L.; Chen, J.; Lim, R.J.; A Chavez, M.; Vu, C.; Lilly, M.B.; Mallya, S.; Ong, S.T.; et al. Effective and selective targeting of leukemia cells using a TORC1/2 kinase inhibitor. Nat. Med. 2010, 16, 205–213. [Google Scholar] [CrossRef]
- Dong, L.Q.; Liu, F. PDK2: The missing piece in the receptor tyrosine kinase signaling pathway puzzle. Am. J. Physiol. Metab. 2005, 289, E187–E196. [Google Scholar] [CrossRef] [PubMed]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dedert, C.J.; Bagdady, K.R.; Fisher, J.S. Prior Treatment with AICAR Causes the Selective Phosphorylation of mTOR Substrates in C2C12 Cells. Curr. Issues Mol. Biol. 2023, 45, 8040-8052. https://doi.org/10.3390/cimb45100508
Dedert CJ, Bagdady KR, Fisher JS. Prior Treatment with AICAR Causes the Selective Phosphorylation of mTOR Substrates in C2C12 Cells. Current Issues in Molecular Biology. 2023; 45(10):8040-8052. https://doi.org/10.3390/cimb45100508
Chicago/Turabian StyleDedert, Cass J., Kazimir R. Bagdady, and Jonathan S. Fisher. 2023. "Prior Treatment with AICAR Causes the Selective Phosphorylation of mTOR Substrates in C2C12 Cells" Current Issues in Molecular Biology 45, no. 10: 8040-8052. https://doi.org/10.3390/cimb45100508
APA StyleDedert, C. J., Bagdady, K. R., & Fisher, J. S. (2023). Prior Treatment with AICAR Causes the Selective Phosphorylation of mTOR Substrates in C2C12 Cells. Current Issues in Molecular Biology, 45(10), 8040-8052. https://doi.org/10.3390/cimb45100508