
In Vitro and In Silico Antiviral Activity of Di-Halogenated Compounds Derived from L-Tyrosine against Human Immunodeficiency Virus 1 (HIV-1)

 , , , , ,
, , , , , 
Abstract
:1. Introduction
2. Materials and Methods
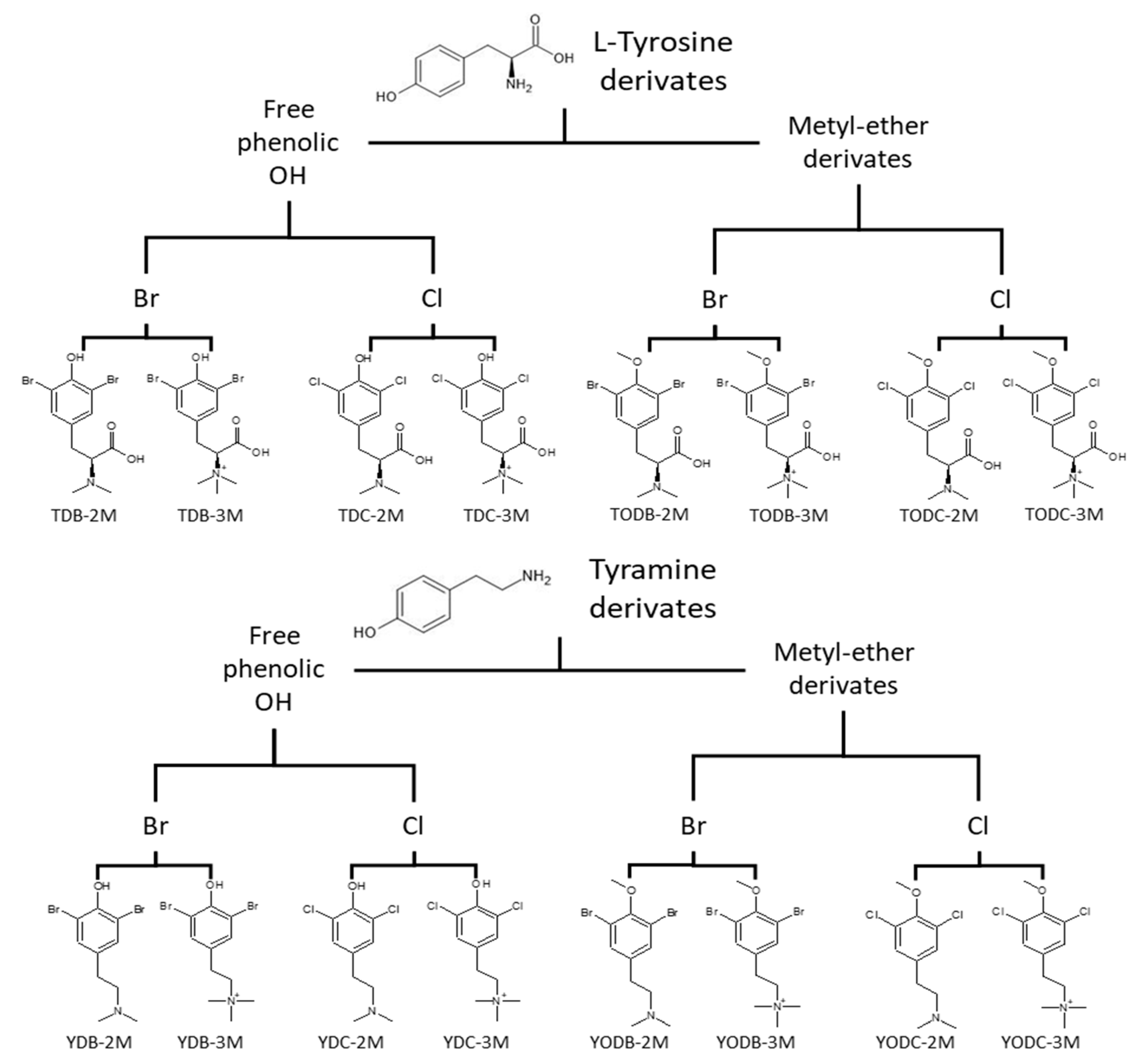
2.1. Di-Halogenated Compounds Derived from L-Tyrosine
2.2. Cells Lines and Culture Conditions
2.3. Virus Strains
2.4. Cytotoxicity Assay
2.5. Viability Assay
2.6. In Silico Toxicological Modeling/ADMET Profiles
2.7. Anti-HIV Activity in Infected TZM-bl Cells
2.8. VSV-Pseudotyped HIV-1 Infection Assay and Quantification of the Percentage of Infected Cells Using Flow Cytometry
2.9. Anti-HIV Activity of Di-Halogenated Compounds Derived from L-Tyrosine on HIV Infection of PBMC
2.10. Quantification of HIV-1 p24 Antigen Using ELISA
2.11. Molecular Docking Simulation
2.12. Molecular Dynamics Simulations
2.13. Statistical Analysis
3. Results
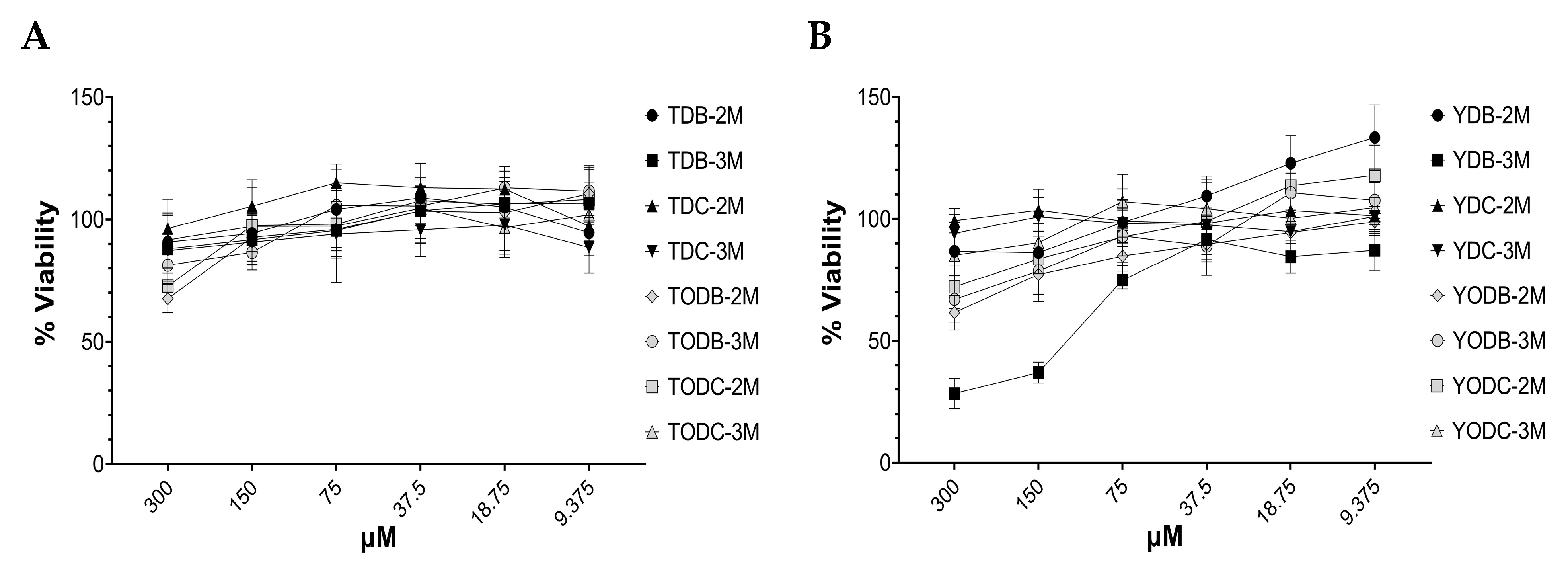
3.1. Di-Halogenated Compounds Derived from L-Tyrosine Have No Significant Cytotoxic Effect on TZM-bl Cells
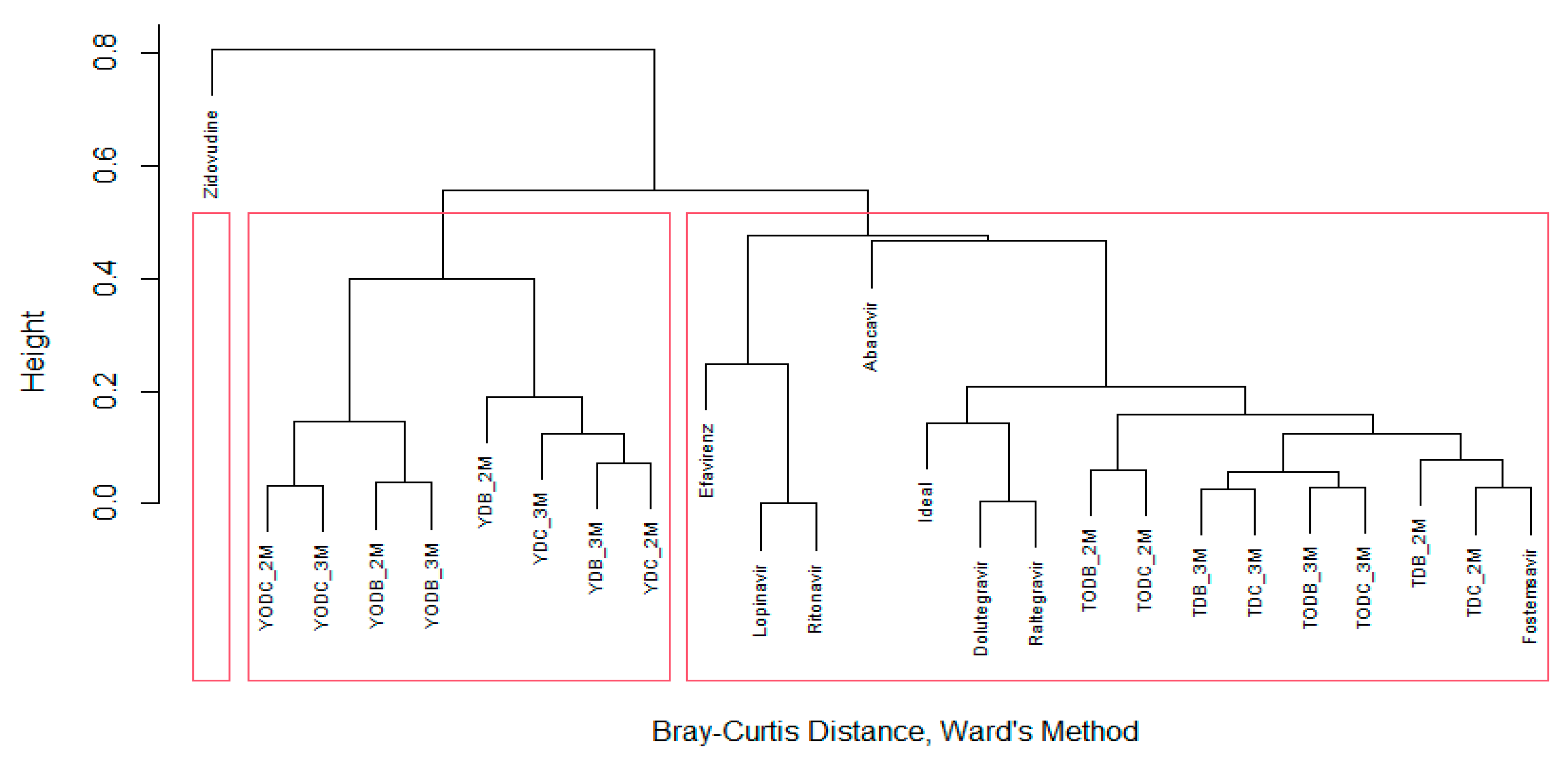
3.2. In Silico Prediction Indicates That Di-Halogenated Compounds Are Not Highly Toxic
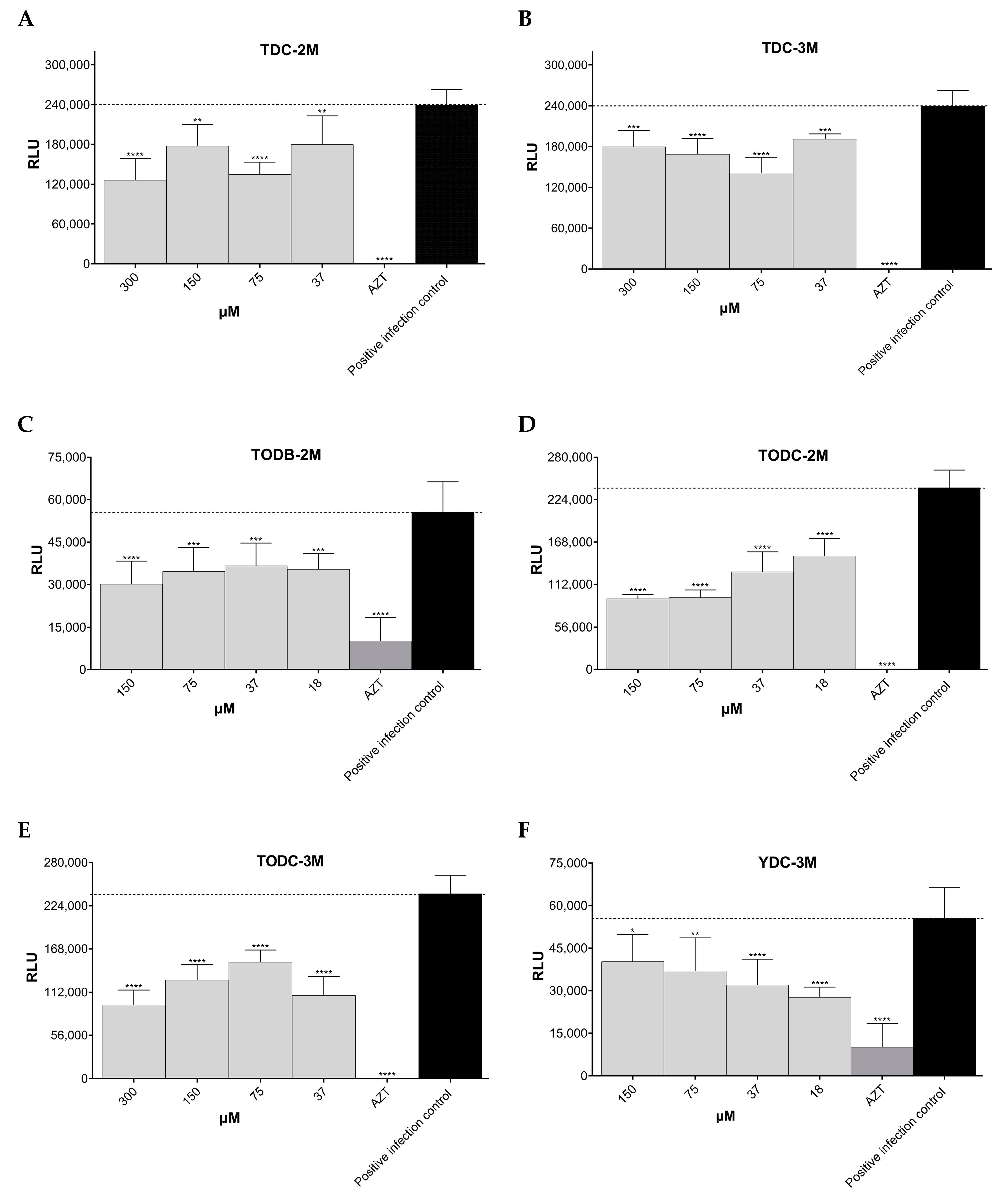
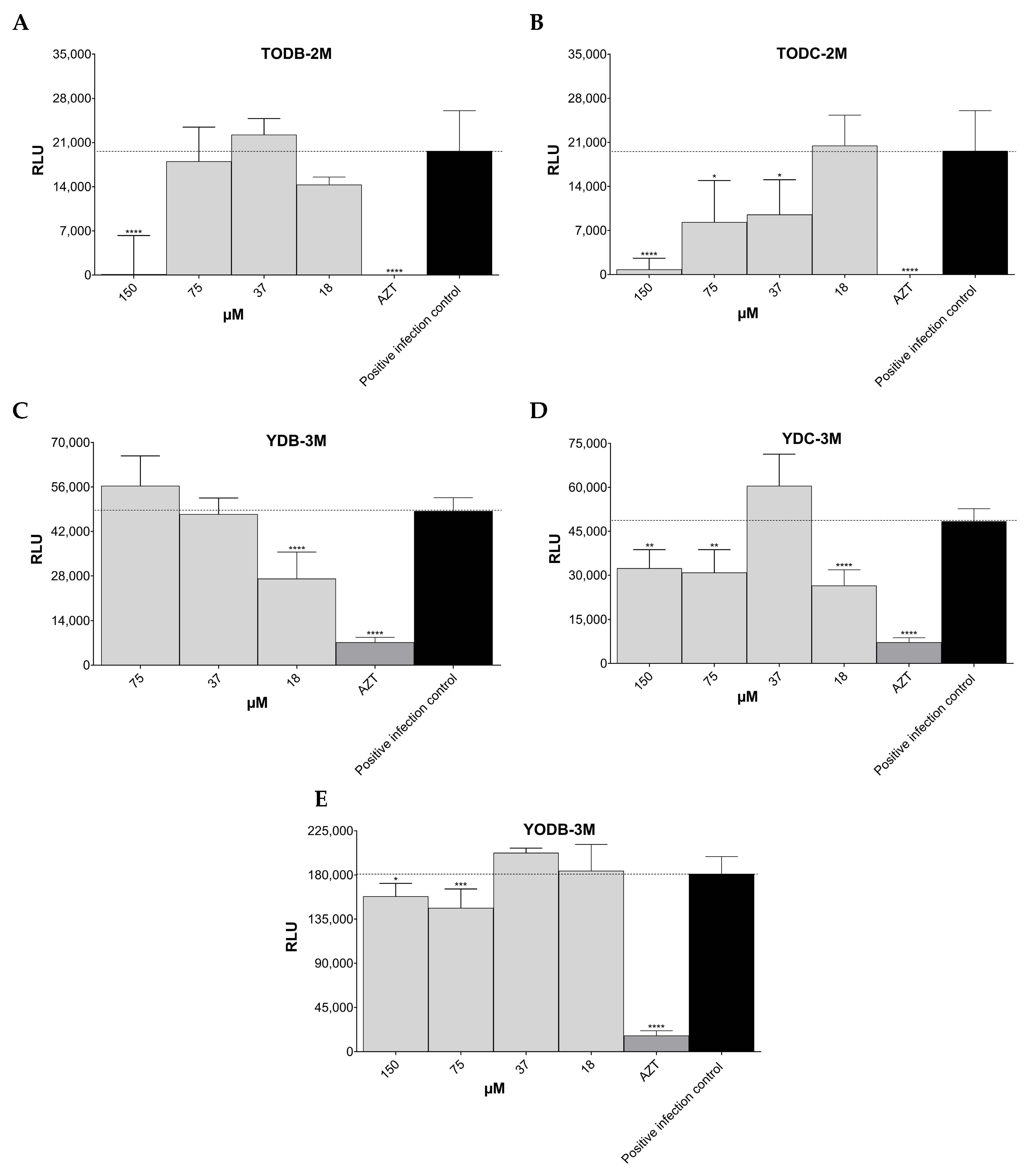
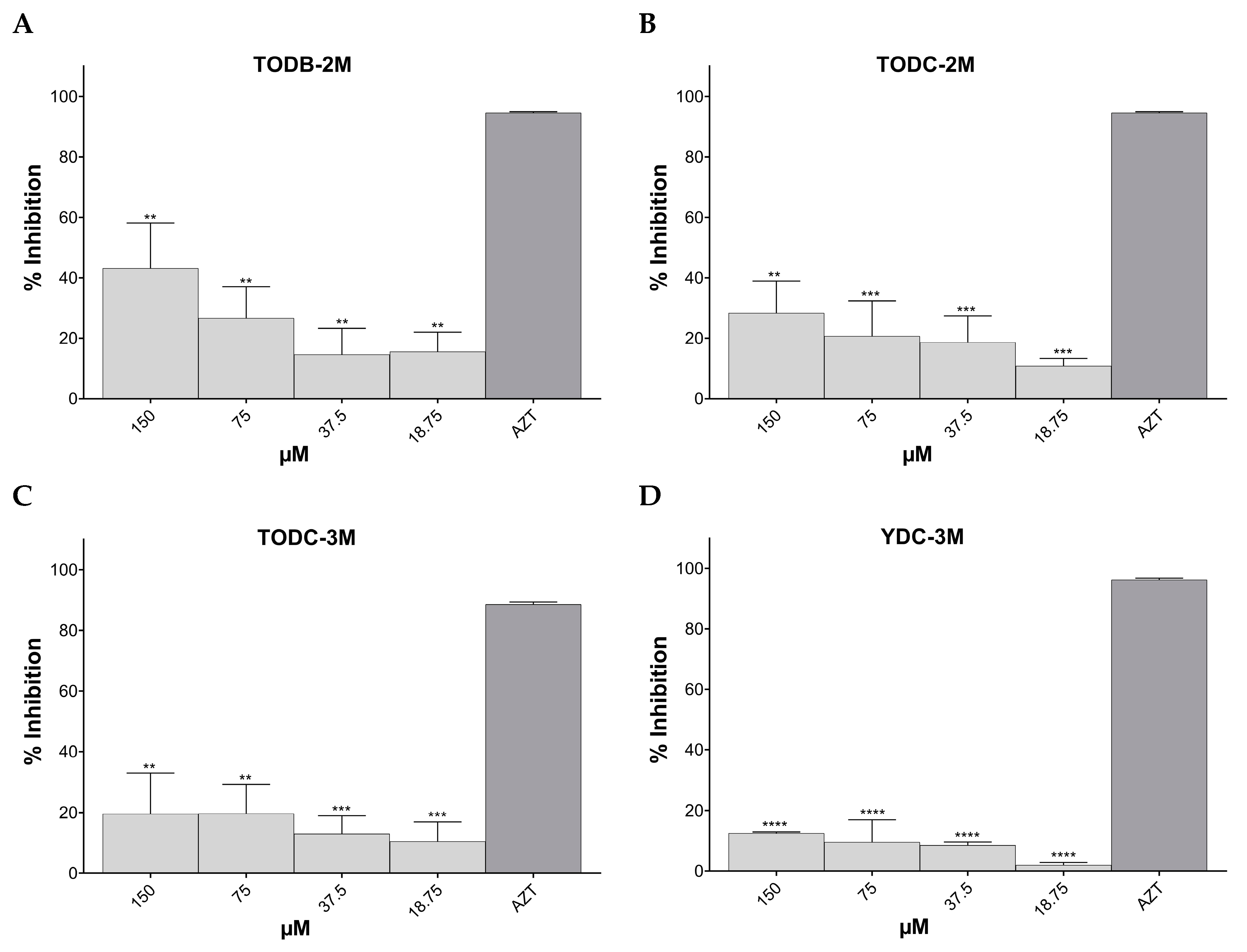
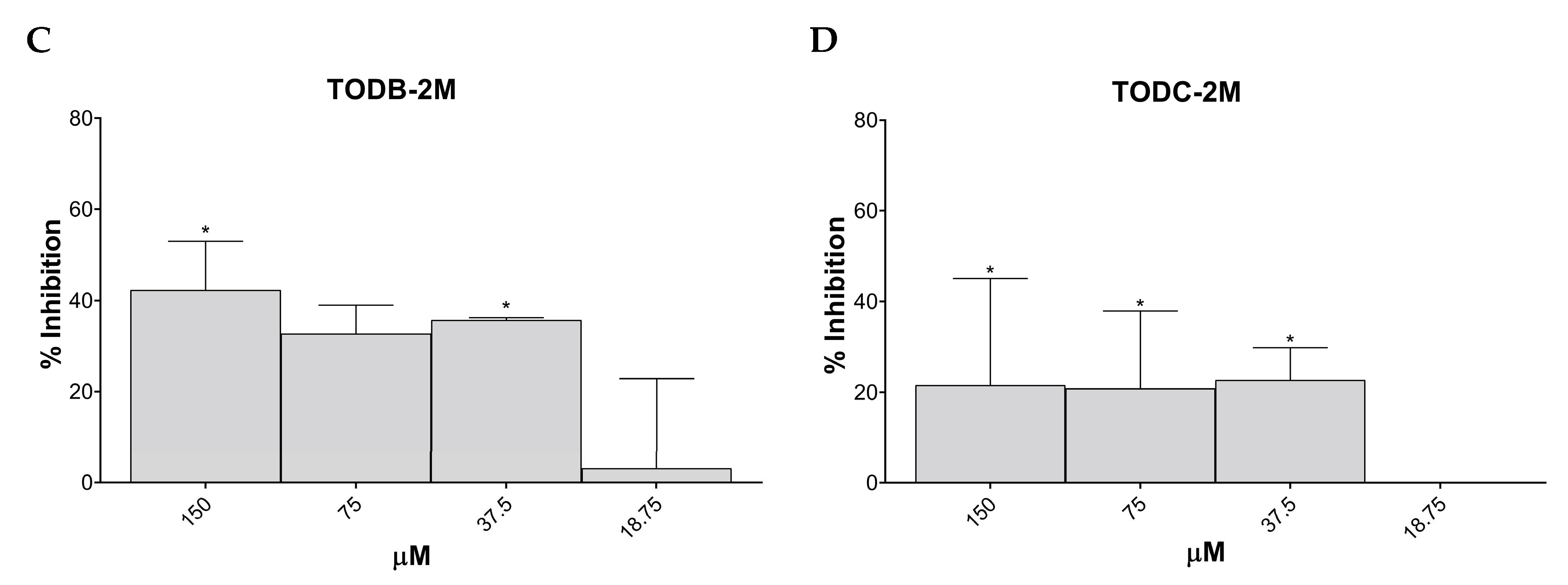
3.3. Some Di-Halogenated Compounds Inhibit Replication of HIV-1 BaL (R5 Strain)
3.4. Some Di-Halogenated Compounds Inhibit Replication of HIV-1IIIB (X4 Strain)
3.5. TODB-2M and Other Di-Halogenated Compounds Inhibit Replication of HIV-GFP-VSV-G
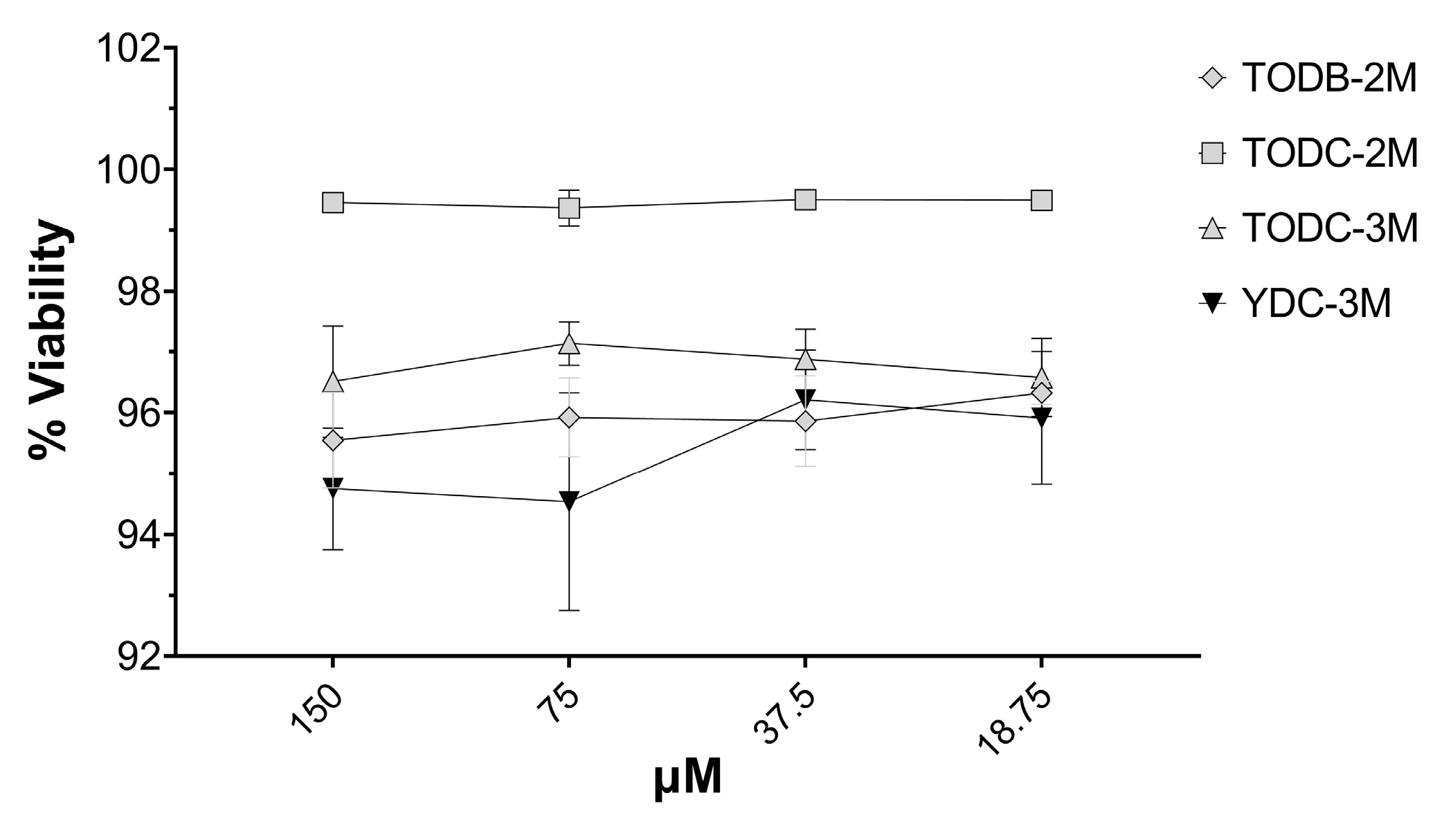
3.6. The Most Promising Compounds Have no Impact on the Cell Viability of PBMC
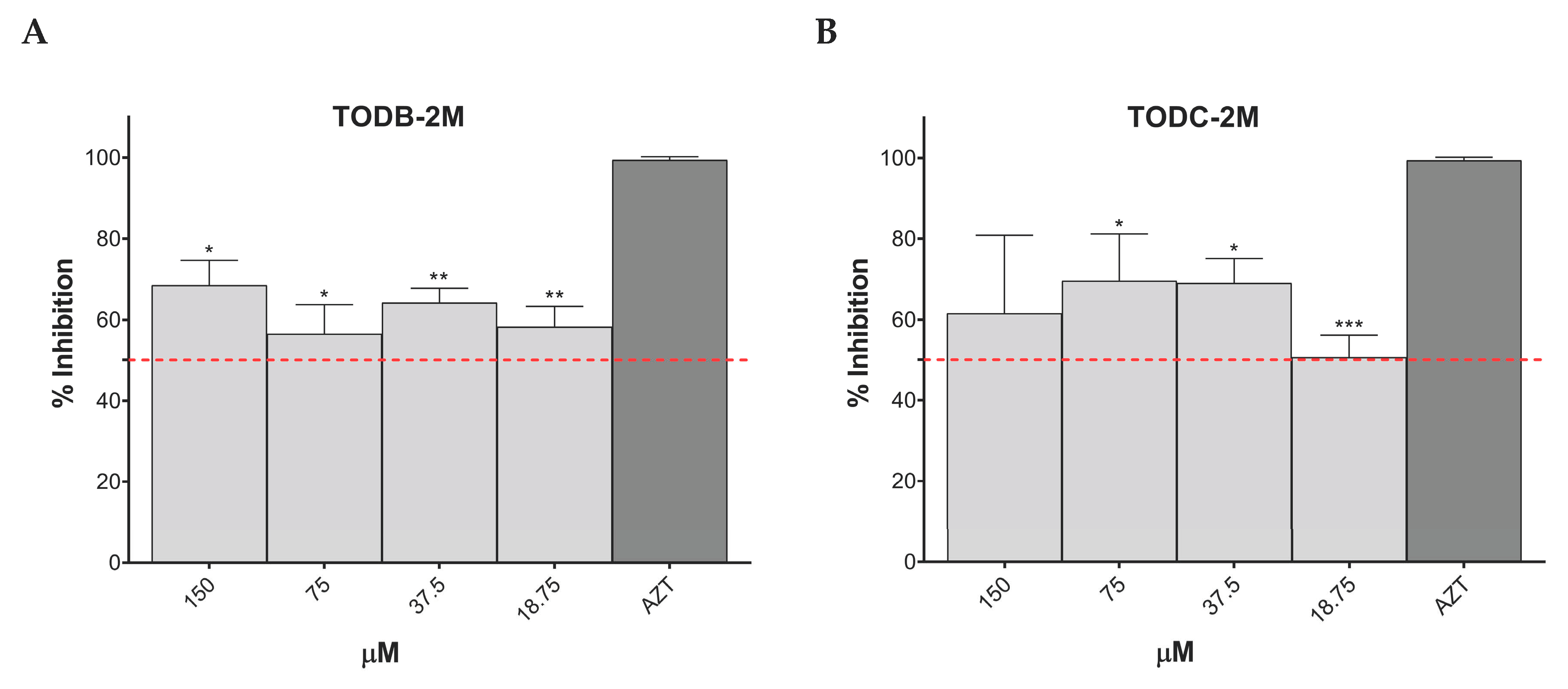
3.7. TODB-2M and TODC-2M Compounds Inhibit Replication of HIV-1 BaL on PBMC
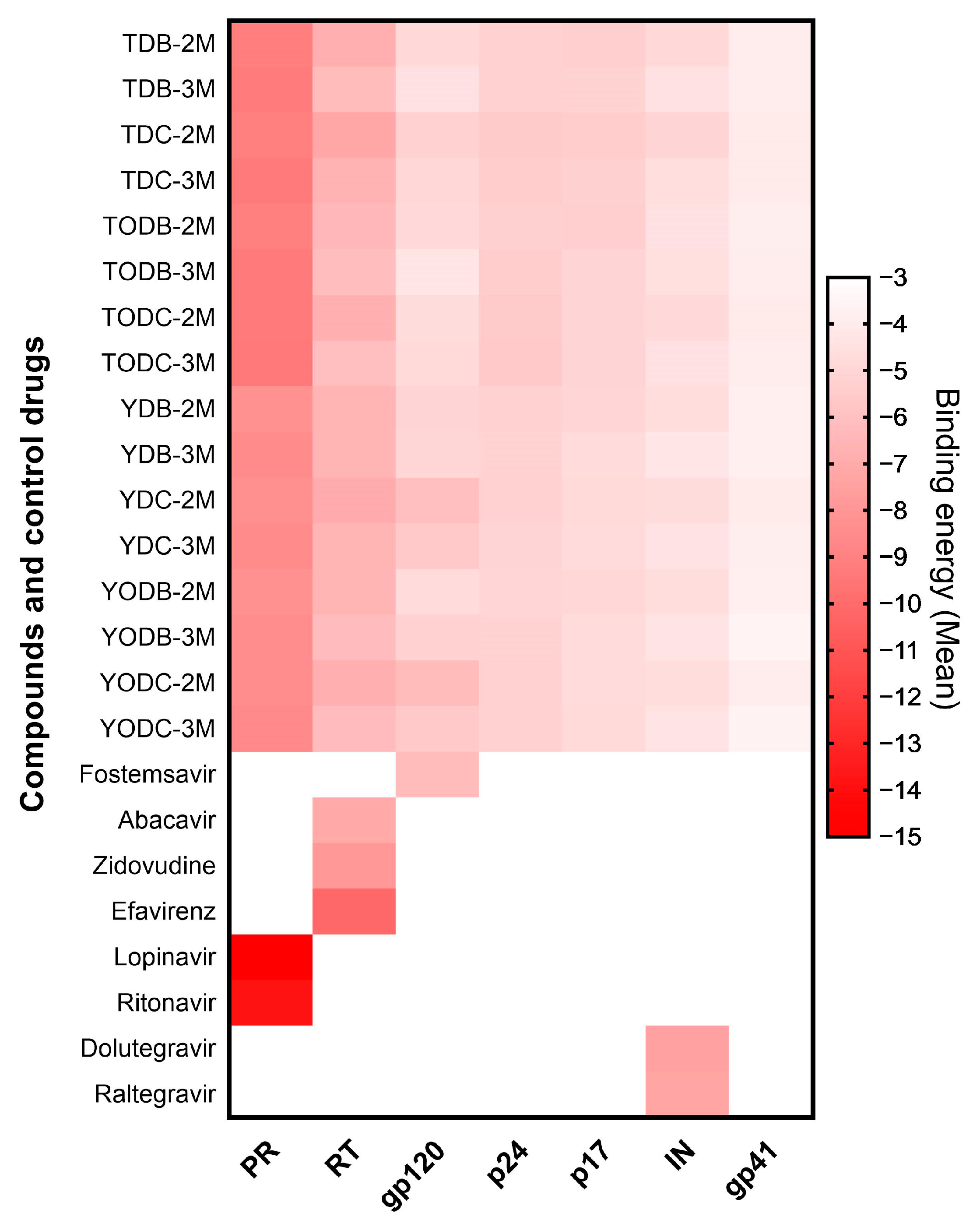
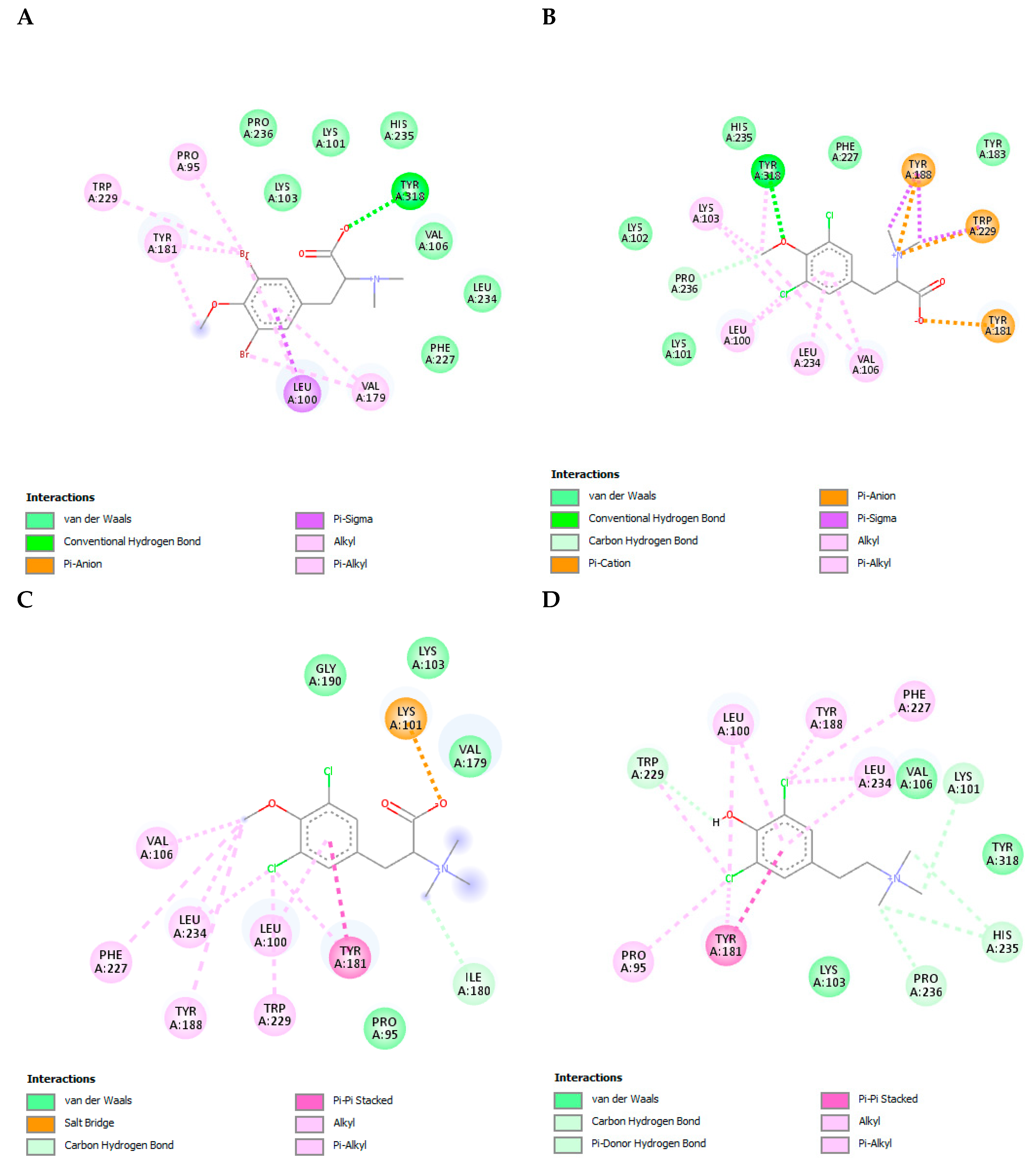
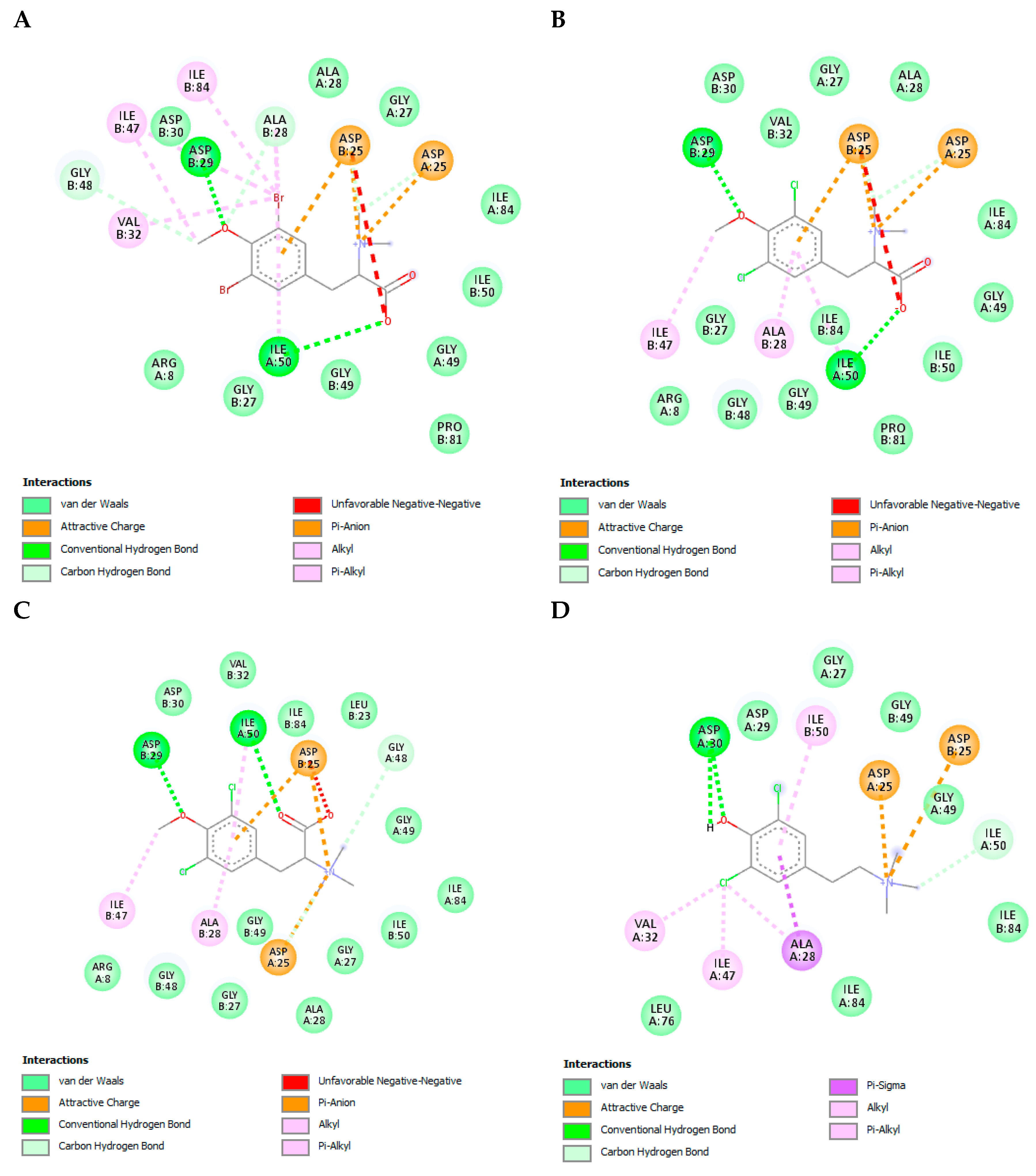
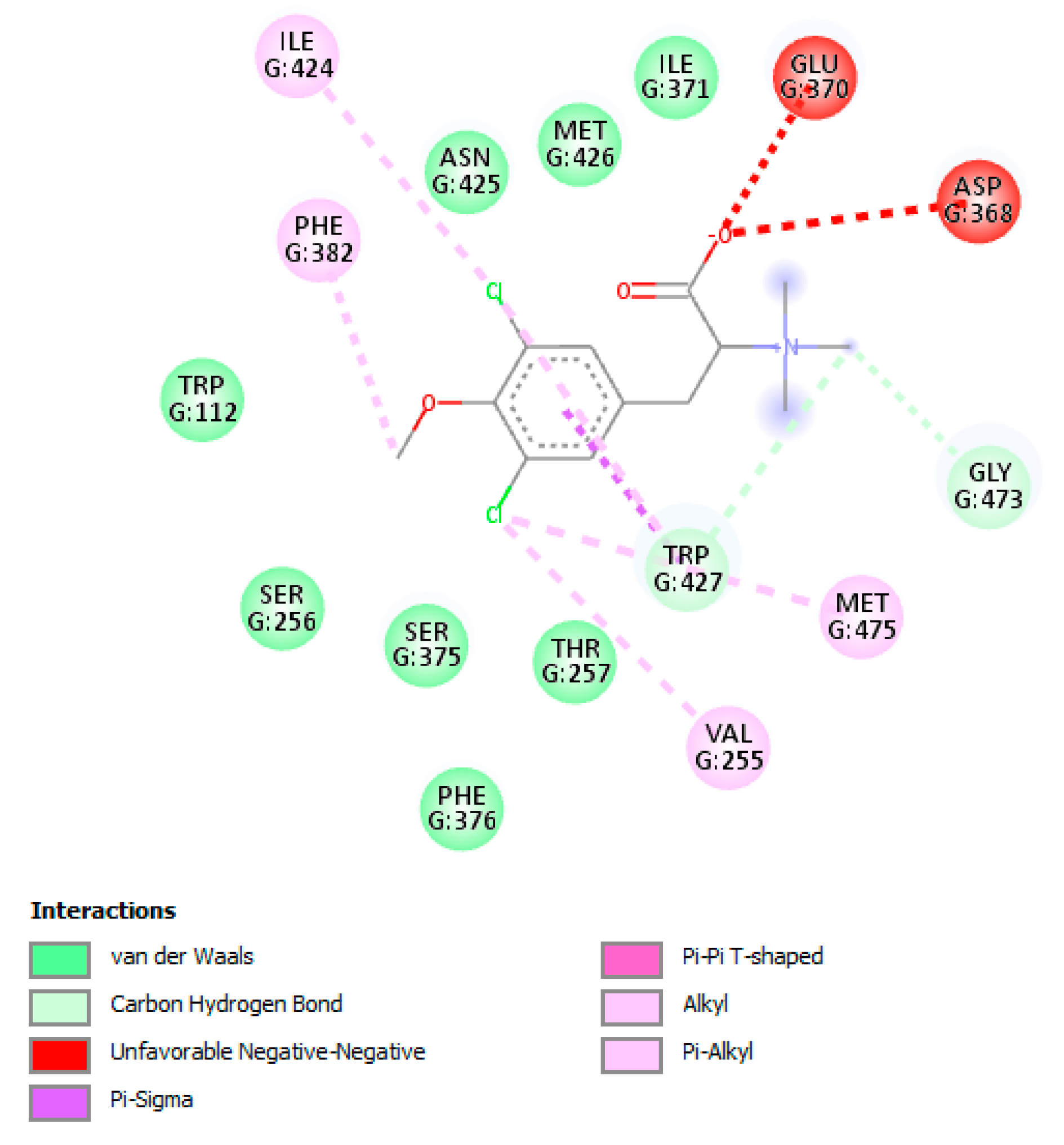
3.8. The L-Tyrosine Derivatives Exhibit Favorable Binding Energies with the Tested Proteins
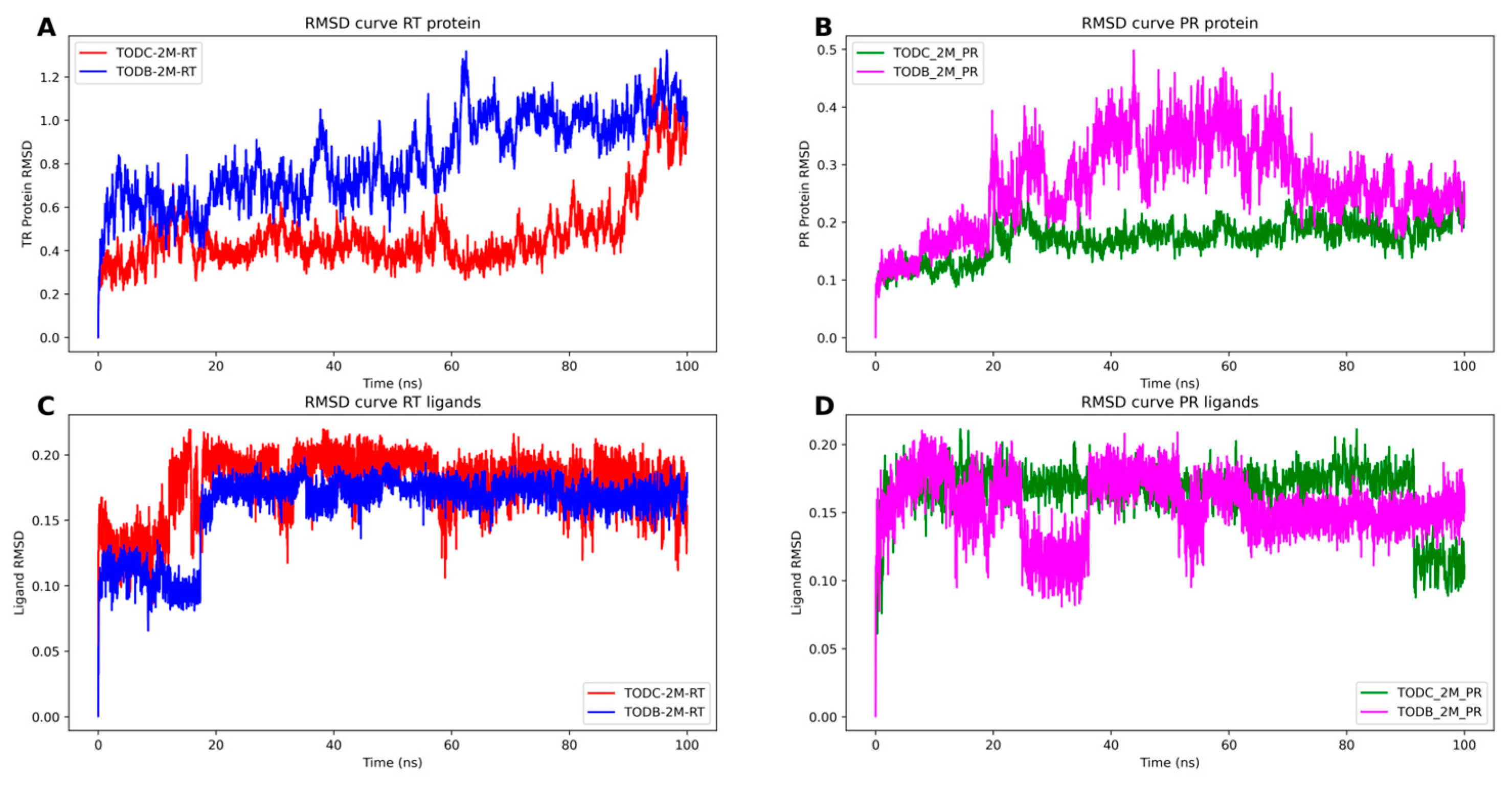
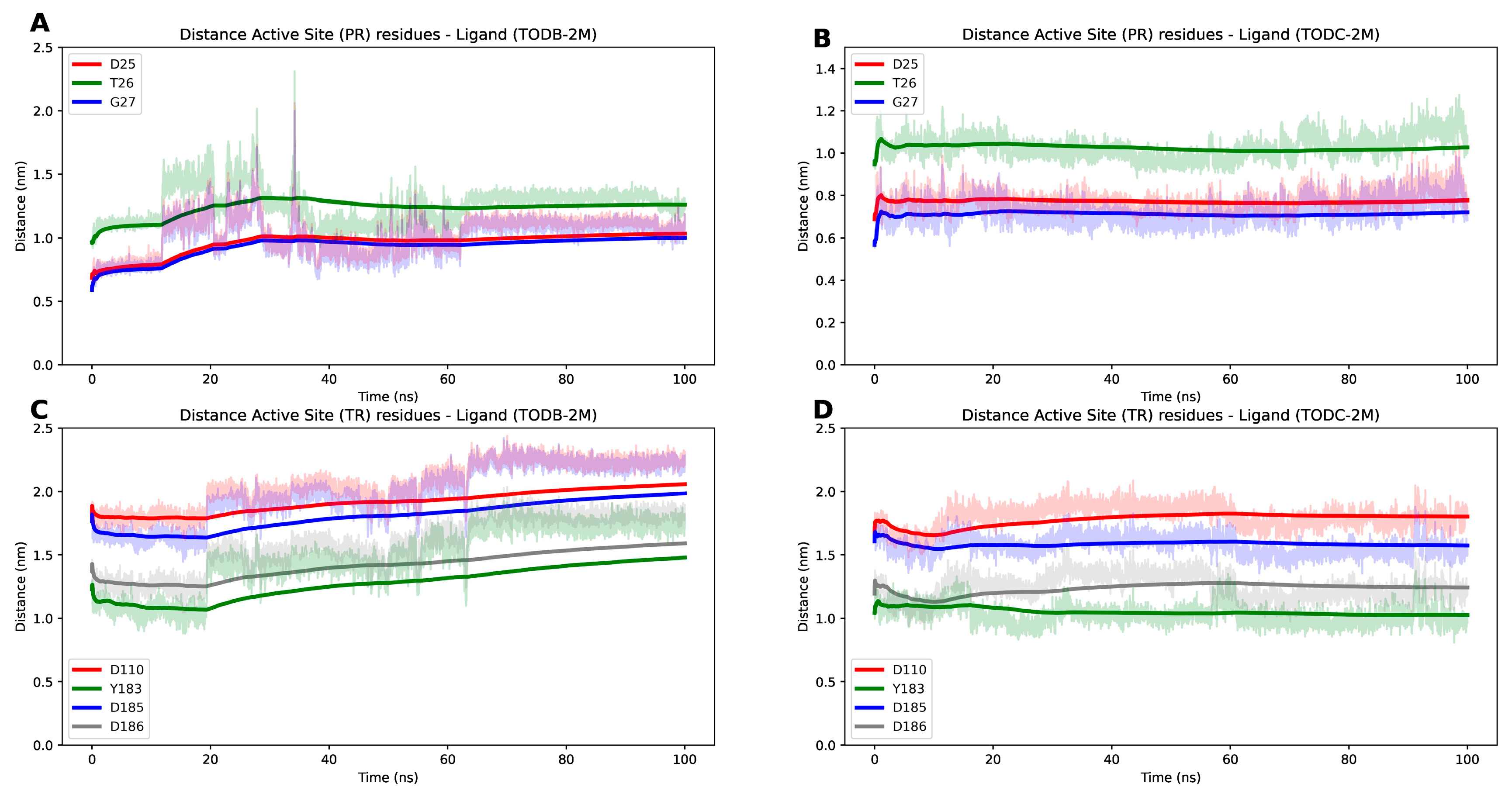
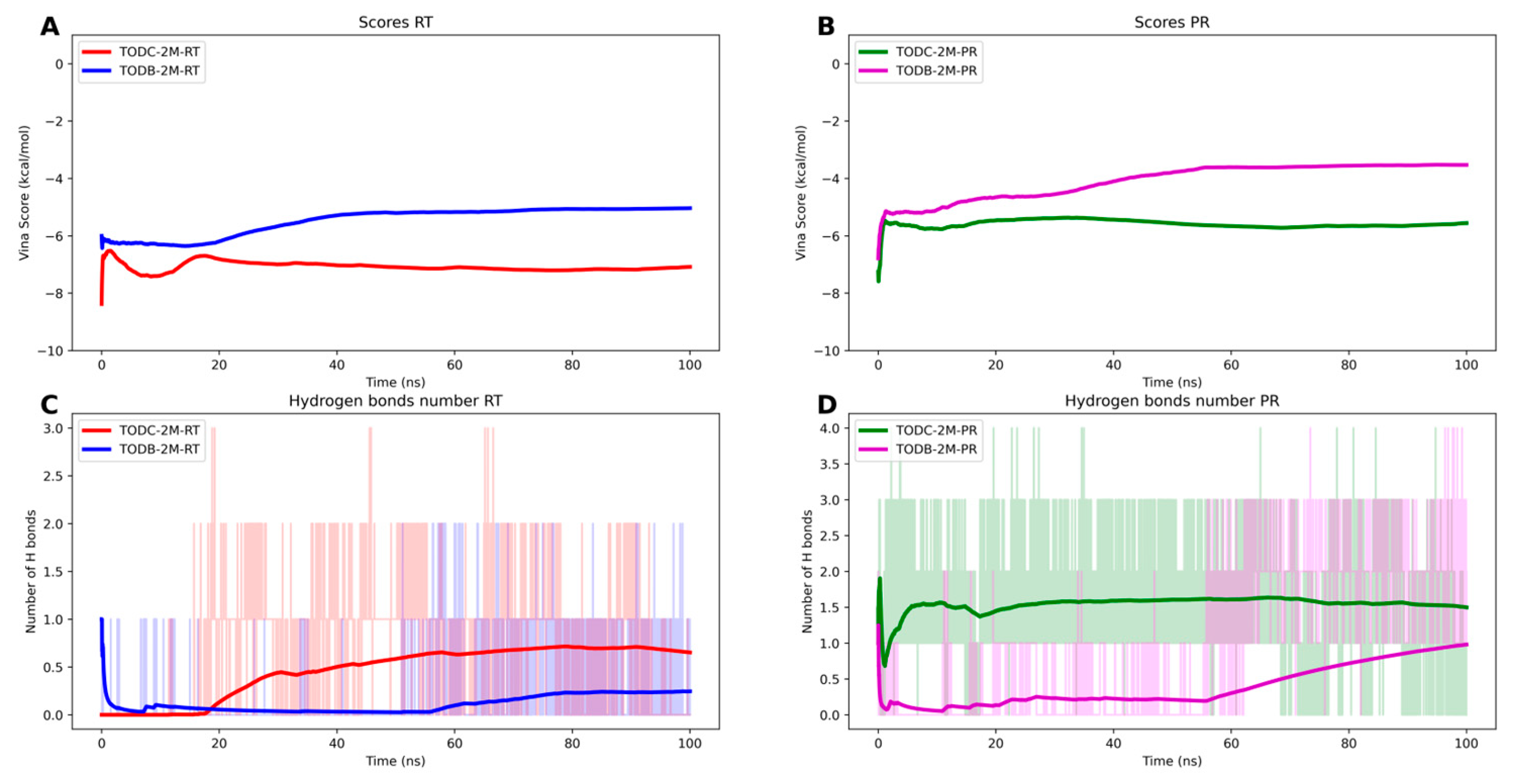
3.9. The L-Tyrosine Derivatives Exhibit Stable Interaction with the Tested Proteins during Molecular Dynamics Simulations
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- UNAIDS. Global HIV & AIDS Statistics—Fact Sheet; UNAIDS: Geneva, Switzerland, 2023; Available online: https://www.unaids.org/en/resources/fact-sheet (accessed on 24 July 2023).
- Arentoft, A.; Troxell, K.; Alvarez, K.; Aghvinian, M.; Rivera Mindt, M.; Cherner, M.; Van Dyk, K.; Razani, J.; Roxas, M.; Gavilanes, M. HIV Antiretroviral Medication Neuropenetrance and Neurocognitive Outcomes in HIV+ Adults: A Review of the Literature Examining the Central Nervous System Penetration Effectiveness Score. Viruses 2022, 14, 1151. [Google Scholar] [CrossRef]
- Hughes, Y.; Tomlins, L.; Usherwood, T. Prescribing for patients taking antiretroviral therapy. Aust. Prescr. 2022, 45, 80. [Google Scholar] [CrossRef]
- Melhuish, A.; Lewthwaite, P. Natural history of HIV and AIDS. Medicine 2022, 50, 356–361. [Google Scholar] [CrossRef]
- Nachega, B.J.; Marconi, C.V.; van Zyl, U.G.; Gardner, M.E.; Preiser, W.; Hong, Y.S.; Mills, J.E.; Gross, R. HIV treatment adherence, drug resistance, virologic failure: Evolving concepts. Infect. Disord. Drug Targets 2011, 11, 167–174. [Google Scholar] [CrossRef]
- Gardner, E.M.; Burman, W.J.; Steiner, J.F.; Anderson, P.L.; Bangsberg, D.R. Antiretroviral medication adherence and the development of class-specific antiretroviral resistance. AIDS 2009, 23, 1035–1046. [Google Scholar] [CrossRef]
- WHO. Fact Sheet: HIV Drug Resistance; World Health Organization: Geneva, Switzerland, 2021; Available online: https://www.who.int/news-room/fact-sheets/detail/hiv-drug-resistance (accessed on 24 July 2023).
- Calixto, J.B. The Role of Natural Products in Modern Drug Discovery; Anais da Academia Brasileira de Ciencias: Rio de Janeiro, RJ, Brasil, 2019. [Google Scholar]
- Serna-Arbeláez, M.S.; Florez-Sampedro, L.; Orozco, L.P.; Ramírez, K.; Galeano, E.; Zapata, W. Natural Products with Inhibitory Activity against Human Immunodeficiency Virus Type 1. Adv. Virol. 2021, 2021, 5552088. [Google Scholar] [CrossRef] [PubMed]
- de Medeiros, A.I.; Gandolfi, R.C.; Secatto, A.; Falcucci, R.M.; Faccioli, L.H.; Hajdu, E.; Peixinho, S.; Berlinck, R.G. 11-Oxoaerothionin isolated from the marine sponge Aplysina fistularis shows anti-inflammatory activity in LPS-stimulated macrophages. Immunopharmacol. Immunotoxicol. 2012, 34, 919–924. [Google Scholar] [CrossRef] [PubMed]
- Jang, J.H.; van Soest, R.W.; Fusetani, N.; Matsunaga, S. Pseudoceratins A and B, antifungal bicyclic bromotyrosine-derived metabolites from the marine sponge Pseudoceratina purpurea. J. Org. Chem. 2007, 72, 1211–1217. [Google Scholar] [CrossRef] [PubMed]
- Kon, Y.; Kubota, T.; Shibazaki, A.; Gonoi, T.; Kobayashi, J.I. Ceratinadins A-C, new bromotyrosine alkaloids from an Okinawan marine sponge Pseudoceratina sp. Bioorg. Med. Chem. Lett. 2010, 20, 4569–4572. [Google Scholar] [CrossRef]
- Acosta, A.L.; Rodríguez, A.D. 11-oxoaerothionin: A cytotoxic antitumor bromotyrosine-derived alkaloid from the Caribbean marine sponge Aplysina lacunosa. J. Nat. Prod. 1992, 55, 1007–1012. [Google Scholar] [CrossRef]
- Gómez-Archila, L.G.; Zapata, W.; Galeano, E.; Martínez, A.; Rugeles, M.T. Bromotyrosine derivatives from marine sponges inhibit the HIV-1 replication in vitro. Vitae 2014, 21, 114–125. [Google Scholar] [CrossRef]
- Pastrana Restrepo, M.; Galeano Jaramillo, E.; Martínez Martínez, A.; Robledo Restrepo, S. Synthesis and trypanocide activity of chloro-l-tyrosine and bromo-l-tyrosine derivatives. Med. Chem. Res. 2018, 27, 2454–2465. [Google Scholar] [CrossRef]
- Platt, E.J.; Wehrly, K.; Kuhmann, S.E.; Chesebro, B.; Kabat, D. Effects of CCR5 and CD4 cell surface concentrations on infections by macrophagetropic isolates of human immunodeficiency virus type 1. J. Virol. 1998, 72, 2855–2864. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.; Decker, J.M.; Liu, H.; Zhang, Z.; Arani, R.B.; Kilby, J.M.; Saag, M.S.; Wu, X.; Shaw, G.M.; Kappes, J.C. Emergence of resistant human immunodeficiency virus type 1 in patients receiving fusion inhibitor (T-20) monotherapy. Antimicrob. Agents Chemother. 2002, 46, 1896–1905. [Google Scholar] [CrossRef]
- Gartner, S.; Markovits, P.; Markovitz, D.M.; Kaplan, M.H.; Gallo, R.C.; Popovic, M. The role of mononuclear phagocytes in HTLV-III/LAV infection. Science 1986, 233, 215–219. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, H.; Kim, B.O.; Gattone, V.H.; Li, J.; Nath, A.; Blum, J.; He, J.J. CD4-independent infection of astrocytes by human immunodeficiency virus type 1: Requirement for the human mannose receptor. J. Virol. 2004, 78, 4120–4133. [Google Scholar] [CrossRef]
- Zapata, W.; Aguilar-Jiménez, W.; Feng, Z.; Weinberg, A.; Russo, A.; Potenza, N.; Estrada, H.; Rugeles, M.T. Identification of innate immune antiretroviral factors during in vivo and in vitro exposure to HIV-1. Microbes Infect. 2016, 18, 211–219. [Google Scholar] [CrossRef]
- Simulations Plus Inc. Lancaster, CA, USA. 2022. Available online: www.simulations-plus.com (accessed on 12 April 2022).
- Mächler, M.; Rousseeuw, P.; Struyf, A.; Hubert, M.; Hornik, K. Cluster Analysis Basics and Extensions, Version 2.1.4, 2022.
- Pasetto, S.; Pardi, V.; Murata, R.M. Anti-HIV-1 activity of flavonoid myricetin on HIV-1 infection in a dual-chamber in vitro model. PLoS ONE 2014, 9, e115323. [Google Scholar] [CrossRef]
- Avogadro Chemistry. Avogadro: An Open-Source Molecular Builder and Visualization Tool; @AvogadroChem: San Diego, CA, USA, 2022. [Google Scholar]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminformatics 2012, 4, 1–17. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef]
- Vajragupta, O.; Boonchoong, P.; Morris, G.M.; Olson, A.J. Active site binding modes of curcumin in HIV-1 protease and integrase. Bioorg. Med. Chem. Lett. 2005, 15, 3364–3368. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, L.; Engelman, A. Retroviral integrase proteins and HIV-1 DNA integration. J. Biol. Chem. 2012, 287, 40858–40866. [Google Scholar] [CrossRef] [PubMed]
- Sanner, M.F. Python: A programming language for software integration and development. J. Mol. Graph. Model. 1999, 17, 57–61. [Google Scholar]
- Lavi, A.; Ngan, C.H.; Movshovitz-Attias, D.; Bohnuud, T.; Yueh, C.; Beglov, D.; Schueler-Furman, O.; Kozakov, D. Detection of peptide-binding sites on protein surfaces: The first step toward the modeling and targeting of peptide-mediated interactions. Proteins 2013, 81, 2096–2105. [Google Scholar] [CrossRef] [PubMed]
- Brenke, R.; Kozakov, D.; Chuang, G.Y.; Beglov, D.; Hall, D.; Landon, M.R.; Mattos, C.; Vajda, S. Fragment-based identification of druggable ‘hot spots’ of proteins using Fourier domain correlation techniques. Bioinformatics 2009, 25, 621–627. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- BIOVIA. [Discovery Studio Visualizer v21.2.0.20298]; Systèmes, D., Ed.; Dassault Systèmes: San Diego, CA, USA, 2021. [Google Scholar]
- Hess, B.; Kutzner, C.; Van Der Spoel, D.; Lindahl, E. GROMACS 4: Algorithms for Highly Efficient, Load-Balanced, and Scalable Molecular Simulation. J. Chem. Theory Comput. 2008, 4, 435–447. [Google Scholar] [CrossRef]
- Vanommeslaeghe, K.; Hatcher, E.; Acharya, C.; Kundu, S.; Zhong, S.; Shim, J.; Darian, E.; Guvench, O.; Lopes, P.; Vorobyov, I.; et al. CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem. 2010, 31, 671–690. [Google Scholar] [CrossRef]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef]
- Parrinello, M.; Rahman, A. Crystal Structure and Pair Potentials: A Molecular-Dynamics Study. Phys. Rev. Lett. 1980, 45, 1196–1199. [Google Scholar] [CrossRef]
- London, R.E. HIV-1 Reverse Transcriptase: A Metamorphic Protein with Three Stable States. Structure 2019, 27, 420–426. [Google Scholar] [CrossRef] [PubMed]
- La Monica, G.; Lauria, A.; Bono, A.; Martorana, A. Off-Target-Based Design of Selective HIV-1 PROTEASE Inhibitors. Int. J. Mol. Sci. 2021, 22, 6070. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Chen, C. Understanding HIV-1 protease autoprocessing for novel therapeutic development. Future Med. Chem. 2013, 5, 1215–1229. [Google Scholar] [CrossRef] [PubMed]
- Tamamis, P.; Floudas, C.A. Molecular recognition of CCR5 by an HIV-1 gp120 V3 loop. PLoS ONE 2014, 9, e95767. [Google Scholar] [CrossRef] [PubMed]
- Ochoa, R.; Watowich, S.J.; Flórez, A.; Mesa, C.V.; Robledo, S.M.; Muskus, C. Drug search for leishmaniasis: A virtual screening approach by grid computing. J. Comput. Aided Mol. Des. 2016, 30, 541–552. [Google Scholar] [CrossRef] [PubMed]
- Ta, T.M.; Malik, S.; Anderson, E.M.; Jones, A.D.; Perchik, J.; Freylikh, M.; Sardo, L.; Klase, Z.A.; Izumi, T. Insights Into Persistent HIV-1 Infection and Functional Cure: Novel Capabilities and Strategies. Front. Microbiol. 2022, 13, 862270. [Google Scholar] [CrossRef] [PubMed]
- Loaiza-Cano, V.; Monsalve-Escudero, L.M.; Restrepo, M.P.; Quintero-Gil, D.C.; Pulido Muñoz, S.A.; Galeano, E.; Zapata, W.; Martinez-Gutierrez, M. In Vitro and In Silico Anti-Arboviral Activities of Dihalogenated Phenolic Derivates of L-Tyrosine. Molecules 2021, 26, 3430. [Google Scholar] [CrossRef] [PubMed]
- Van De Waterbeemd, H.; Gifford, E. ADMET in silico modelling: Towards prediction paradise? Nat. Rev. Drug Discov. 2003, 2, 192–204. [Google Scholar] [CrossRef]
- Pau, A.K.; George, J.M. Antiretroviral therapy: Current drugs. Infect. Dis. Clin. N. Am. 2014, 28, 371–402. [Google Scholar] [CrossRef]
- Clark, R.D.; Morris, D.N.; Chinigo, G.; Lawless, M.S.; Prudhomme, J.; Le Roch, K.G.; Lafuente, M.J.; Ferrer, S.; Gamo, F.J.; Gadwood, R.; et al. Design and tests of prospective property predictions for novel antimalarial 2-aminopropylaminoquinolones. J. Comput.-Aided Mol. Des. 2020, 34, 1117–1132. [Google Scholar] [CrossRef]
- Richard, K.; Williams, D.E.; De Silva, E.D.; Brockman, M.A.; Brumme, Z.L.; Andersen, R.J.; Tietjen, I. Identification of Novel HIV-1 Latency-Reversing Agents from a Library of Marine Natural Products. Viruses 2018, 10, 348. [Google Scholar] [CrossRef] [PubMed]
- Ross, S.A.; Weete, J.D.; Schinazi, R.F.; Wirtz, S.S.; Tharnish, P.; Scheuer, P.J.; Hamann, M.T. Mololipids, a new series of anti-HIV bromotyramine-derived compounds from a sponge of the order verongida. J. Nat. Prod. 2000, 63, 501–503. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.B.; Wu, P.; Zhang, X.L.; Xia, C.; Li, G.Q.; Ye, W.C.; Wang, G.C.; Li, Y.L. Phenolic Compounds from the Flowers of Bombax malabaricum and Their Antioxidant and Antiviral Activities. Molecules 2015, 20, 19947–19957. [Google Scholar] [CrossRef] [PubMed]
- El-Demerdash, A.; Hassan, A.; Abd El-Aziz, T.M.; Stockand, J.D.; Arafa, R.K. Marine Brominated Tyrosine Alkaloids as Promising Inhibitors of SARS-CoV-2. Molecules 2021, 26, 6171. [Google Scholar] [CrossRef]
- Lim, C.T.; Tan, K.W.; Wu, M.; Ulferts, R.; Armstrong, L.A.; Ozono, E.; Drury, L.S.; Milligan, J.C.; Zeisner, T.U.; Zeng, J.; et al. Identifying SARS-CoV-2 antiviral compounds by screening for small molecule inhibitors of Nsp3 papain-like protease. Biochem. J. 2021, 478, 2517–2531. [Google Scholar] [CrossRef]
- Fais, S.; Lapenta, C.; Santini, S.M.; Spada, M.; Parlato, S.; Logozzi, M.; Rizza, P.; Belardelli, F. Human immunodeficiency virus type 1 strains R5 and X4 induce different pathogenic effects in hu-PBL-SCID mice, depending on the state of activation/differentiation of human target cells at the time of primary infection. J. Virol. 1999, 73, 6453–6459. [Google Scholar] [CrossRef]
- Weinberger, A.D.; Perelson, A.S. Persistence and emergence of X4 virus in HIV infection. Math. Biosci. Eng. MBE 2011, 8, 605. [Google Scholar]
- Ichiba, T.; Scheuer, P.J.; Kelly-Borges, M. Three bromotyrosine derivatives, one terminating in an unprecedented diketocyclopentenylidene enamine. J. Org. Chem. 1993, 58, 4149–4150. [Google Scholar] [CrossRef]
- Gochfeld, D.J.; El Sayed, K.A.; Yousaf, M.; Hu, J.; Bartyzel, P.; Dunbar, D.C.; Wilkins, S.P.; Zjawiony, J.K.; Schinazi, R.F.; Schlueter Wirtz, S.; et al. Marine natural products as lead anti-HIV agents. Mini Rev. Med. Chem. 2003, 3, 401–424. [Google Scholar] [CrossRef]
- Wu, Z.Y.; Liu, N.; Qin, B.; Huang, L.; Yu, F.; Qian, K.; Morris-Natschke, S.L.; Jiang, S.; Chen, C.H.; Lee, K.H.; et al. Optimization of the antiviral potency and lipophilicity of halogenated 2,6-diarylpyridinamines as a novel class of HIV-1 NNRTIS. ChemMedChem 2014, 9, 1546–1555. [Google Scholar] [CrossRef]
- Curran, A.; Ribera, E. From old to new nucleoside reverse transcriptase inhibitors: Changes in body fat composition, metabolic parameters and mitochondrial toxicity after the switch from thymidine analogs to tenofovir or abacavir. Expert. Opin. Drug Saf. 2011, 10, 389–406. [Google Scholar] [CrossRef]
- Ferreira, L.G.; Dos Santos, R.N.; Oliva, G.; Andricopulo, A.D. Molecular docking and structure-based drug design strategies. Molecules 2015, 20, 13384–13421. [Google Scholar] [CrossRef] [PubMed]
- Pinzi, L.; Rastelli, G. Molecular Docking: Shifting Paradigms in Drug Discovery. Int. J. Mol. Sci. 2019, 20, 4331. [Google Scholar] [CrossRef] [PubMed]
- Razzaghi-Asl, N.; Sepehri, S.; Ebadi, A.; Miri, R.; Shahabipour, S. Effect of Biomolecular Conformation on Docking Simulation: A Case Study on a Potent HIV-1 Protease Inhibitor. Iran. J. Pharm. Res. 2015, 14, 785–802. [Google Scholar] [PubMed]
- Bollini, M.; Domaoal, R.A.; Thakur, V.V.; Gallardo-Macias, R.; Spasov, K.A.; Anderson, K.S.; Jorgensen, W.L. Computationally-guided optimization of a docking hit to yield catechol diethers as potent anti-HIV agents. J. Med. Chem. 2011, 54, 8582–8591. [Google Scholar] [CrossRef]
- Amblard, F.; Patel, D.; Michailidis, E.; Coats, S.J.; Kasthuri, M.; Biteau, N.; Tber, Z.; Ehteshami, M.; Schinazi, R.F. HIV nucleoside reverse transcriptase inhibitors. Eur. J. Med. Chem. 2022, 240, 114554. [Google Scholar] [CrossRef]
- Ortega, J.T.; Suárez, A.I.; Serrano, M.L.; Baptista, J.; Pujol, F.H.; Rangel, H.R. The role of the glycosyl moiety of myricetin derivatives in anti-HIV-1 activity in vitro. AIDS Res. Ther. 2017, 14, 57. [Google Scholar] [CrossRef]
- Smerdon, S.J.; Jäger, J.; Wang, J.; Kohlstaedt, L.A.; Chirino, A.J.; Friedman, J.M.; Rice, P.A.; Steitz, T.A. Structure of the binding site for nonnucleoside inhibitors of the reverse transcriptase of human immunodeficiency virus type 1. Proc. Natl. Acad. Sci. USA 1994, 91, 3911–3915. [Google Scholar] [CrossRef]
- Kohlstaedt, L.A.; Wang, J.; Friedman, J.M.; Rice, P.A.; Steitz, T.A. Crystal structure at 3.5 A resolution of HIV-1 reverse transcriptase complexed with an inhibitor. Science 1992, 256, 1783–1790. [Google Scholar] [CrossRef]
- Hertje, M.; Zhou, M.; Dietrich, U. Inhibition of HIV-1 Entry: Multiple Keys to Close the Door. ChemMedChem 2010, 5, 1825–1835. [Google Scholar] [CrossRef]
- Governa, P.; Manetti, F. Recent research results have converted gp120 binders to a therapeutic option for the treatment of HIV-1 infection. A medicinal chemistry point of view. Eur. J. Med. Chem. 2022, 229, 114078. [Google Scholar] [CrossRef] [PubMed]
- Madrid, M.; Lukin, J.A.; Madura, J.D.; Ding, J.; Arnold, E. Molecular dynamics of HIV-1 reverse transcriptase indicates increased flexibility upon DNA binding. Proteins 2001, 45, 176–182. [Google Scholar] [CrossRef] [PubMed]
- Rana, N.; Singh, A.K.; Shuaib, M.; Gupta, S.; Habiballah, M.M.; Alkhanani, M.F.; Haque, S.; Reshi, M.S.; Kumar, S. Drug Resistance Mechanism of M46I-Mutation-Induced Saquinavir Resistance in HIV-1 Protease Using Molecular Dynamics Simulation and Binding Energy Calculation. Viruses 2022, 14, 697. [Google Scholar] [CrossRef] [PubMed]
- Padariya, M.; Kalathiya, U.; Baginski, M. Molecular basis and potential activity of HIV-1 reverse transcriptase toward trimethylamine-based compounds. Biotechnol. Appl. Biochem. 2017, 64, 810–826. [Google Scholar] [CrossRef]
- Zondagh, J.; Balakrishnan, V.; Achilonu, I.; Dirr, H.W.; Sayed, Y. Molecular dynamics and ligand docking of a hinge region variant of South African HIV-1 subtype C protease. J. Mol. Graph. Model. 2018, 82, 1–11. [Google Scholar] [CrossRef]
- Yu, Y.X.; Liu, W.T.; Li, H.Y.; Wang, W.; Sun, H.B.; Zhang, L.L.; Wu, S.L. Decoding molecular mechanism underlying binding of drugs to HIV-1 protease with molecular dynamics simulations and MM-GBSA calculations. SAR QSAR Environ. Res. 2021, 32, 889–915. [Google Scholar] [CrossRef]
- Louis, J.M.; Ishima, R.; Torchia, D.A.; Weber, I.T. HIV-1 protease: Structure, dynamics, and inhibition. Adv. Pharmacol. 2007, 55, 261–298. [Google Scholar]
- Tiefenbrunn, T.; Forli, S.; Happer, M.; Gonzalez, A.; Tsai, Y.; Soltis, M.; Elder, J.H.; Olson, A.J.; Stout, C.D. Crystallographic fragment-based drug discovery: Use of a brominated fragment library targeting HIV protease. Chem. Biol. Drug Des. 2014, 83, 141–148. [Google Scholar] [CrossRef]
- Ho, P.S. Biomolecular halogen bonds. Top. Curr. Chem. 2015, 358, 241–276. [Google Scholar]
- Shinada, N.K.; de Brevern, A.G.; Schmidtke, P. Halogens in Protein-Ligand Binding Mechanism: A Structural Perspective. J. Med. Chem. 2019, 62, 9341–9356. [Google Scholar] [CrossRef]

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | PDB Code | Resolution (Å) | Sequence Length | Chain | Active Site Amino Acids |
|---|---|---|---|---|---|
| gp120 | 4J6R | 1.64 | 359 | A | Trp112, Thr257, Asp368, Glu370, Ser375, Phe382, Ile424, Met426, Trp427, Met434, Met475 |
| gp41 | 3VTQ | 1.53 | 37 | A | Leu565, Leu566, Thr569, Val570, Trp571, Gly572, Ile573, Lys574, Leu576, Gln577 |
| RT | 4G1Q | 1.51 | 557 | A | Asp110, Tyr183, Asp185, Asp186, Leu100, Lys101, Lys103, Val106, Thr107, Val108, Val179, Tyr181, Tyr188, Val189, Gly190, Phe227, Tyr318, Glu138 |
| Protease | 5YOK | 0.85 | 100 | A-B | Asp25, Thr26, Gly27 |
| Integrase | 1QS4 | 2.1 | 154 | A | Asp64, Asp116, Glu152 |
| p24 | 2XDE | 1.4 | 145 | A | Phe32, Glu35, Val36, Asn57, Val59, Glu60, His62, Gln63, Ala65, Gln67, Lys70, Thr107, Met118, Thr119 |
| p17 | 4JMU | 2.0 | 112 | A | Leu21, Arg22, Pro23, Tyr29, Lys32, His33, Trp36, Ser77, Leu88, Asn80, Thr81, Thr81, Thr97, Lys98, Leu101 |
| TDB-2M | TDB-3M | TDC-2M | TDC-3M | TODB-2M | TODB-3M | TODC-2M | TODC-3M | YDB-2M | YDB-3M | YDC-2M | YDC-3M | YODB-2M | YODB-3M | YODC-2M | YODC-3M | ABC | DTG | EFV | FTR | LPV | RAL | RTV | AZT | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Chromosome aberrations | 0 | 1 | 0 | 1 | 0 | 1 | 0 | 1 | 1 | 1 | 1 | 1 | 0 | 0 | 1 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 1 |
| Skin sensitization | 1 | 1 | 0 | 0 | 1 | 1 | 0 | 0 | 1 | 1 | 1 | 0 | 1 | 1 | 0 | 0 | 1 | 1 | 1 | 0 | 0 | 1 | 0 | 1 |
| Respiratory sensitization | 0 | 0 | 0 | 0 | 1 | 0 | 1 | 0 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 0 | 1 | 0 | 0 | 0 | 0 | 0 |
| Neurotoxicity | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 1 | 1 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 |
| Cardiac toxicity | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Estrogen receptor tox | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 |
| Androgen receptor tox | 0 | 0 | 0 | 0 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 0 | 1 | 0 | 1 | 0 | 0 | 1 | 0 | 1 | 0 | 1 | 0 |
| ALP increase | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 1 | 0 | 1 | 0 | 1 | 0 | 1 | 1 |
| GGT increase | 1 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 1 | 1 |
| LDH increase | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 1 | 1 |
| SGOT increase | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 1 | 0 | 0 | 1 | 0 | 1 |
| SGPT increase | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 1 | 0 | 1 | 0 | 1 | 1 |
| Reproductive tox | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 1 | 0 | 1 | 1 |
| Accumulated tox | 3 | 3 | 1 | 2 | 5 | 4 | 3 | 3 | 7 | 5 | 6 | 6 | 6 | 6 | 4 | 4 | 6 | 2 | 8 | 1 | 6 | 2 | 6 | 8 |
| ADMET_Risk | 0.0 | 0.9 | 0.0 | 1.0 | 0.0 | 0.7 | 0.9 | 1.4 | 0.0 | 0.3 | 2.0 | 2.8 | 0.5 | 0.4 | 2.3 | 1.8 | 1.4 | 0.0 | 4.7 | 5.0 | 7.5 | 2.5 | 9.7 | 5.4 |
| Compound | CC50 (µM) | IC50 (µM) | SI |
|---|---|---|---|
| TDC-2M | ~174.4 | 140.7 | ~1.24 |
| TDC-3M | >300 | NA | NA |
| TODB-2M | 164.9 | 113.5 | 1.45 |
| TODC-2M | >300 | 67.42 | >4.45 |
| TODC-3M | 74.9 | NA | NA |
| YDC-3M | 39.88 | 37.7 | 1.07 |
| Compound | CC50 (µM) | IC50 (µM) | SI |
|---|---|---|---|
| TODB-2M | 164.9 | ~82.68 | ~1.99 |
| TODC-2M | >300 | ~39.15 | ~7.66 |
| YDB-3M | ~100.9 | 3.09 | ~32.65 |
| YDC-3M | 39.88 | 22.31 | 1.65 |
| YODB-3M | 78.7 | NA | NA |
| Compound | CC50 (µM) | IC50 (µM) | SI |
|---|---|---|---|
| TODB-2M | 164.9 | 141.8 | 1.16 |
| TODC-2M | >300 | NA | NA |
| YDC-3M | 39.88 | NA | NA |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Serna-Arbeláez, M.S.; García-Cárcamo, V.; Rincón-Tabares, D.S.; Guerra, D.; Loaiza-Cano, V.; Martinez-Gutierrez, M.; Pereañez, J.A.; Pastrana-Restrepo, M.; Galeano, E.; Zapata, W. In Vitro and In Silico Antiviral Activity of Di-Halogenated Compounds Derived from L-Tyrosine against Human Immunodeficiency Virus 1 (HIV-1). Curr. Issues Mol. Biol. 2023, 45, 8173-8200. https://doi.org/10.3390/cimb45100516
Serna-Arbeláez MS, García-Cárcamo V, Rincón-Tabares DS, Guerra D, Loaiza-Cano V, Martinez-Gutierrez M, Pereañez JA, Pastrana-Restrepo M, Galeano E, Zapata W. In Vitro and In Silico Antiviral Activity of Di-Halogenated Compounds Derived from L-Tyrosine against Human Immunodeficiency Virus 1 (HIV-1). Current Issues in Molecular Biology. 2023; 45(10):8173-8200. https://doi.org/10.3390/cimb45100516
Chicago/Turabian StyleSerna-Arbeláez, Maria S., Valentina García-Cárcamo, Daniel S. Rincón-Tabares, Diego Guerra, Vanessa Loaiza-Cano, Marlen Martinez-Gutierrez, Jaime A. Pereañez, Manuel Pastrana-Restrepo, Elkin Galeano, and Wildeman Zapata. 2023. "In Vitro and In Silico Antiviral Activity of Di-Halogenated Compounds Derived from L-Tyrosine against Human Immunodeficiency Virus 1 (HIV-1)" Current Issues in Molecular Biology 45, no. 10: 8173-8200. https://doi.org/10.3390/cimb45100516
APA StyleSerna-Arbeláez, M. S., García-Cárcamo, V., Rincón-Tabares, D. S., Guerra, D., Loaiza-Cano, V., Martinez-Gutierrez, M., Pereañez, J. A., Pastrana-Restrepo, M., Galeano, E., & Zapata, W. (2023). In Vitro and In Silico Antiviral Activity of Di-Halogenated Compounds Derived from L-Tyrosine against Human Immunodeficiency Virus 1 (HIV-1). Current Issues in Molecular Biology, 45(10), 8173-8200. https://doi.org/10.3390/cimb45100516