On the Quest for In Vitro Platelet Production by Re-Tailoring the Concepts of Megakaryocyte Differentiation

 , ,
, ,
Abstract
:1. Background: Platelet Transfusion over the Years
2. In Vitro Megakaryocyte (and Platelet) Production: The Story Thus Far
2.1. Thrombopoietin and other Factors Influencing Megakaryopoiesis
2.2. The Source Material and Its Developmental Stage
3. Human Cell Sources to Produce In Vitro Megakaryocytes and Platelets
3.1. Hematopoietic Stem Cells and Precursors
3.2. Cell Reprogramming
3.3. Non-Hematopoietic Sources
4. The Culture Engineering
4.1. Bioreactors
4.2. Using the Lungs as In Vivo Bioreactors
4.3. Artificial Platelets and Applications beyond Transfusion Medicine
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Deutsch, V.R.; Tomer, A. Megakaryocyte development and platelet production. Br. J. Haematol. 2006, 134, 453–466. [Google Scholar] [CrossRef] [PubMed]
- Slichter, S.J. Platelet transfusion therapy. Hematol. Oncol. Clin. North. Am. 2007, 21, 697–729. [Google Scholar] [CrossRef]
- Blajchman, M.A.; Slichter, S.J.; Heddle, N.M.; Murphy, M.F. New Strategies for the Optimal Use of Platelet Transfusions. Hematology 2008, 2008, 198–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wandt, H.; Schaefer-Eckart, K.; Wendelin, K.; Pilz, B.; Wilhelm, M.; Thalheimer, M.; Mahlknecht, U.; Ho, A.; Schaich, M.; Kramer, M.; et al. Therapeutic platelet transfusion versus routine prophylactic transfusion in patients with haematological malignancies: An open-label, multicentre, randomised study. Lancet 2012, 380, 1309–1316. [Google Scholar] [CrossRef]
- Estcourt, L.J. Why has demand for platelet components increased? A review. Transfus. Med. 2014, 24, 260–268. [Google Scholar] [CrossRef] [PubMed]
- Yazer, M.H.; Shaz, B.; Seheult, J.N.; Apelseth, T.O.; De Korte, D.; Devin, G.; Devine, D.; Doncaster, C.; Field, S.; Flanagan, P.; et al. Trends in platelet distributions from 2008 to 2017: A survey of twelve national and regional blood collectors. Vox Sang. 2020. [Google Scholar] [CrossRef]
- Storch, E.K.; Custer, B.S.; Jacobs, M.R.; Menitove, J.E.; Mintz, P.D. Review of current transfusion therapy and blood banking practices. Blood Rev. 2019, 38, 100593. [Google Scholar] [CrossRef]
- Williamson, L.M.; Devine, D.V. Challenges in the management of the blood supply. Lancet 2013, 381, 1866–1875. [Google Scholar] [CrossRef]
- Flint, A.W.; McQuilten, Z.K.; Irwin, G.; Rushford, K.; Haysom, H.E.; Wood, E.M. Is Platelet Expiring Out of Date? A Systematic Review. Transfus. Med. Rev. 2020, 34, 42–50. [Google Scholar] [CrossRef]
- Allain, J.P.; Bianco, C.; Blajchman, M.A.; Brecher, M.E.; Busch, M.; Leiby, D.; Lin, L.; Stramer, S. Protecting the blood supply from emerging pathogens: The role of pathogen inactivation. Transfus. Med. Rev. 2005, 19, 110–126. [Google Scholar] [CrossRef]
- Strassel, C.; Gachet, C.; Lanza, F. On the way to in vitro platelet production. Transfus. Clin. Biol. 2018, 25, 220–227. [Google Scholar] [CrossRef]
- Salunkhe, V.; Van der Meer, P.F.; De Korte, D.; Seghatchian, J.; Gutierrez, L. Development of blood transfusion product pathogen reduction treatments: A review of methods, current applications and demands. Transfus. Apher. Sci. 2015, 52, 19–34. [Google Scholar] [CrossRef]
- Jimenez-Marco, T.; Garcia-Recio, M.; Girona-Llobera, E. Our experience in riboflavin and ultraviolet light pathogen reduction technology for platelets: From platelet production to patient care. Transfusion 2018, 58, 1881–1889. [Google Scholar] [CrossRef] [Green Version]
- Devine, D.V.; Serrano, K. The platelet storage lesion. Clin. Lab. Med. 2010, 30, 475–487. [Google Scholar] [CrossRef]
- Zeddies, S.; De Cuyper, I.M.; Van der Meer, P.F.; Daal, B.B.; De Korte, D.; Gutierrez, L.; Thijssen-Timmer, D.C. Pathogen reduction treatment using riboflavin and ultraviolet light impairs platelet reactivity toward specific agonists in vitro. Transfusion 2014, 54, 2292–2300. [Google Scholar] [CrossRef]
- Garraud, O.; Lozano, M. Pathogen inactivation/reduction technologies for platelet transfusion: Where do we stand? Transfus. Clin. Biol. 2018, 25, 165–171. [Google Scholar] [CrossRef]
- Magron, A.; Laugier, J.; Provost, P.; Boilard, E. Pathogen reduction technologies: The pros and cons for platelet transfusion. Platelets 2018, 29, 2–8. [Google Scholar] [CrossRef]
- Slichter, S.J.; Davis, K.; Enright, H.; Braine, H.; Gernsheimer, T.; Kao, K.-J.; Kickler, T.; Lee, E.; McFarland, J.; McCullough, J.; et al. Factors affecting posttransfusion platelet increments, platelet refractoriness, and platelet transfusion intervals in thrombocytopenic patients. Blood 2005, 105, 4106–4114. [Google Scholar] [CrossRef] [Green Version]
- Johnson, L.; Tan, S.; Wood, B.; Davis, A.; Marks, D.C. Refrigeration and cryopreservation of platelets differentially affect platelet metabolism and function: A comparison with conventional platelet storage conditions. Transfusion 2016, 56, 1807–1818. [Google Scholar] [CrossRef]
- Desborough, M.; Estcourt, L.J.; Doree, C.; Trivella, M.; Hopewell, S.; Stanworth, S.J.; Murphy, M.F. Alternatives, and adjuncts, to prophylactic platelet transfusion for people with haematological malignancies undergoing intensive chemotherapy or stem cell transplantation. Cochrane Database Syst. Rev. 2016, Cd010982. [Google Scholar] [CrossRef] [Green Version]
- Martínez-Botía, P.; Acebes-Huerta, A.; Seghatchian, J.; Gutiérrez, L. In vitro platelet production for transfusion purposes: Where are we now? Transfus. Apher. Sci. 2020, 59, 102864. [Google Scholar] [CrossRef]
- Franco, A.T.; Corken, A.; Ware, J. Platelets at the interface of thrombosis, inflammation, and cancer. Blood 2015, 126, 582–588. [Google Scholar] [CrossRef] [Green Version]
- Kaushansky, K. Historical review: Megakaryopoiesis and thrombopoiesis. Blood 2008, 111, 981–986. [Google Scholar] [CrossRef]
- Ghanima, W.; Cooper, N.; Rodeghiero, F.; Godeau, B.; Bussel, J.B. Thrombopoietin receptor agonists: Ten years later. Haematologica 2019, 104, 1112–1123. [Google Scholar] [CrossRef] [Green Version]
- Choi, E.; Nichol, J.; Hokom, M.; Hornkohl, A.; Hunt, P. Platelets generated in vitro from proplatelet-displaying human megakaryocytes are functional. Blood 1995, 85, 402–413. [Google Scholar] [CrossRef] [Green Version]
- Kaushansky, K.; Lok, S.; Holly, R.D.; Broudy, V.C.; Lin, N.; Bailey, M.C.; Forstrom, J.W.; Buddle, M.M.; Oort, P.J.; Hagen, F.S.; et al. Promotion of megakaryocyte progenitor expansion and differentiation by the c-Mpl ligand thrombopoietin. Nature 1994, 369, 568–571. [Google Scholar] [CrossRef]
- Terskikh, A.V.; Miyamoto, T.; Chang, C.; Diatchenko, L.; Weissman, I.L. Gene expression analysis of purified hematopoietic stem cells and committed progenitors. Blood 2003, 102, 94–101. [Google Scholar] [CrossRef]
- Ninos, J.M.; Jefferies, L.C.; Cogle, C.R.; Kerr, W.G. The thrombopoietin receptor, c-Mpl, is a selective surface marker for human hematopoietic stem cells. J. Transl. Med. 2006, 4, 9. [Google Scholar] [CrossRef] [Green Version]
- Yoshihara, H.; Arai, F.; Hosokawa, K.; Hagiwara, T.; Takubo, K.; Nakamura, Y.; Gomei, Y.; Iwasaki, H.; Matsuoka, S.; Miyamoto, K.; et al. Thrombopoietin/MPL signaling regulates hematopoietic stem cell quiescence and interaction with the osteoblastic niche. Cell Stem Cell 2007, 1, 685–697. [Google Scholar] [CrossRef] [Green Version]
- Nakamura-Ishizu, A.; Matsumura, T.; Stumpf, P.S.; Umemoto, T.; Takizawa, H.; Takihara, Y.; O’Neil, A.; Majeed, A.; MacArthur, B.D.; Suda, T. Thrombopoietin Metabolically Primes Hematopoietic Stem Cells to Megakaryocyte-Lineage Differentiation. Cell Rep. 2018, 25, 1772–1785.e1776. [Google Scholar] [CrossRef] [Green Version]
- Nakamura-Ishizu, A.; Suda, T. Multifaceted roles of thrombopoietin in hematopoietic stem cell regulation. Ann. N. Y. Acad. Sci. 2019. [Google Scholar] [CrossRef]
- De Bruyn, C.; Delforge, A.; Martiat, P.; Bron, D. Ex vivo expansion of megakaryocyte progenitor cells: Cord blood versus mobilized peripheral blood. Stem Cells Dev. 2005, 14, 415–424. [Google Scholar] [CrossRef]
- Van den Oudenrijn, S.; Von dem Borne, A.E.; De Haas, M. Differences in megakaryocyte expansion potential between CD34(+) stem cells derived from cord blood, peripheral blood, and bone marrow from adults and children. Exp. Hematol. 2000, 28, 1054–1061. [Google Scholar] [CrossRef]
- Niswander, L.M.; Fegan, K.H.; Kingsley, P.D.; McGrath, K.E.; Palis, J. SDF-1 dynamically mediates megakaryocyte niche occupancy and thrombopoiesis at steady state and following radiation injury. Blood 2014, 124, 277–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avecilla, S.T.; Hattori, K.; Heissig, B.; Tejada, R.; Liao, F.; Shido, K.; Jin, D.K.; Dias, S.; Zhang, F.; Hartman, T.E.; et al. Chemokine-mediated interaction of hematopoietic progenitors with the bone marrow vascular niche is required for thrombopoiesis. Nat. Med. 2004, 10, 64–71. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Orban, M.; Lorenz, M.; Barocke, V.; Braun, D.; Urtz, N.; Schulz, C.; Von Bruhl, M.L.; Tirniceriu, A.; Gaertner, F.; et al. A novel role of sphingosine 1-phosphate receptor S1pr1 in mouse thrombopoiesis. J. Exp. Med. 2012, 209, 2165–2181. [Google Scholar] [CrossRef] [PubMed]
- Ng, A.P.; Kauppi, M.; Metcalf, D.; Hyland, C.D.; Josefsson, E.C.; Lebois, M.; Zhang, J.G.; Baldwin, T.M.; Di Rago, L.; Hilton, D.J.; et al. Mpl expression on megakaryocytes and platelets is dispensable for thrombopoiesis but essential to prevent myeloproliferation. Proc. Natl. Acad. Sci. USA 2014, 111, 5884–5889. [Google Scholar] [CrossRef] [Green Version]
- Assinger, A.; Schrottmaier, W.C.; Salzmann, M.; Rayes, J. Platelets in Sepsis: An Update on Experimental Models and Clinical Data. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef]
- Müller-Newen, G.; Stope, M.B.; Kraus, T.; Ziegler, P. Development of platelets during steady state and inflammation. J. Leukoc. Biol. 2017, 101, 1109–1117. [Google Scholar] [CrossRef]
- Reems, J.A.; Pineault, N.; Sun, S. In vitro megakaryocyte production and platelet biogenesis: State of the art. Transfus. Med. Rev. 2010, 24, 33–43. [Google Scholar] [CrossRef] [Green Version]
- Salunkhe, V.; Papadopoulos, P.; Gutiérrez, L. Culture of Megakaryocytes from Human Peripheral Blood Mononuclear Cells. Bioprotocol 2015, 5, e1639. [Google Scholar] [CrossRef]
- Kaushansky, K.; Broudy, V.C.; Lin, N.; Jorgensen, M.J.; McCarty, J.; Fox, N.; Zucker-Franklin, D.; Lofton-Day, C. Thrombopoietin, the Mp1 ligand, is essential for full megakaryocyte development. Proc. Natl. Acad. Sci. USA 1995, 92, 3234–3238. [Google Scholar] [CrossRef] [Green Version]
- Gainsford, T.; Nandurkar, H.; Metcalf, D.; Robb, L.; Begley, C.G.; Alexander, W.S. The residual megakaryocyte and platelet production in c-mpl-deficient mice is not dependent on the actions of interleukin-6, interleukin-11, or leukemia inhibitory factor. Blood 2000, 95, 528–534. [Google Scholar] [CrossRef]
- Ku, H.; Yonemura, Y.; Kaushansky, K.; Ogawa, M. Thrombopoietin, the ligand for the Mpl receptor, synergizes with steel factor and other early acting cytokines in supporting proliferation of primitive hematopoietic progenitors of mice. Blood 1996, 87, 4544–4551. [Google Scholar] [CrossRef]
- Couldwell, G.; Machlus, K.R. Modulation of megakaryopoiesis and platelet production during inflammation. Thromb. Res. 2019, 179, 114–120. [Google Scholar] [CrossRef]
- Nishimura, S.; Nagasaki, M.; Kunishima, S.; Sawaguchi, A.; Sakata, A.; Sakaguchi, H.; Ohmori, T.; Manabe, I.; Italiano, J.E., Jr.; Ryu, T.; et al. IL-1alpha induces thrombopoiesis through megakaryocyte rupture in response to acute platelet needs. J. Cell Biol. 2015, 209, 453–466. [Google Scholar] [CrossRef] [Green Version]
- Wilcox, D.A. Megakaryocyte- and megakaryocyte precursor–related gene therapies. Blood 2016, 127, 1260–1268. [Google Scholar] [CrossRef] [Green Version]
- Avanzi, M.P.; Mitchell, W.B. Ex vivo production of platelets from stem cells. Br. J. Haematol. 2014, 165, 237–247. [Google Scholar] [CrossRef]
- Davenport, P.; Liu, Z.J.; Sola-Visner, M. Changes in megakaryopoiesis over ontogeny and their implications in health and disease. Platelets 2020, 1–8. [Google Scholar] [CrossRef]
- Strassel, C.; Eckly, A.; Léon, C.; Moog, S.; Cazenave, J.P.; Gachet, C.; Lanza, F. Hirudin and heparin enable efficient megakaryocyte differentiation of mouse bone marrow progenitors. Exp. Cell Res. 2012, 318, 25–32. [Google Scholar] [CrossRef]
- Italiano, J.E., Jr.; Lecine, P.; Shivdasani, R.A.; Hartwig, J.H. Blood platelets are assembled principally at the ends of proplatelet processes produced by differentiated megakaryocytes. J. Cell Biol. 1999, 147, 1299–1312. [Google Scholar] [CrossRef]
- Bluteau, O.; Langlois, T.; Rivera-Munoz, P.; Favale, F.; Rameau, P.; Meurice, G.; Dessen, P.; Solary, E.; Raslova, H.; Mercher, T.; et al. Developmental changes in human megakaryopoiesis. J. Thromb. Haemost. 2013, 11, 1730–1741. [Google Scholar] [CrossRef]
- Liu, Z.J.; Italiano, J., Jr.; Ferrer-Marin, F.; Gutti, R.; Bailey, M.; Poterjoy, B.; Rimsza, L.; Sola-Visner, M. Developmental differences in megakaryocytopoiesis are associated with up-regulated TPO signaling through mTOR and elevated GATA-1 levels in neonatal megakaryocytes. Blood 2011, 117, 4106–4117. [Google Scholar] [CrossRef] [Green Version]
- Slayton, W.B.; Wainman, D.A.; Li, X.M.; Hu, Z.; Jotwani, A.; Cogle, C.R.; Walker, D.; Fisher, R.C.; Wingard, J.R.; Scott, E.W.; et al. Developmental differences in megakaryocyte maturation are determined by the microenvironment. Stem Cells 2005, 23, 1400–1408. [Google Scholar] [CrossRef]
- Margraf, A.; Nussbaum, C.; Sperandio, M. Ontogeny of platelet function. Blood Adv. 2019, 3, 692–703. [Google Scholar] [CrossRef] [Green Version]
- Caparros-Perez, E.; Teruel-Montoya, R.; Lopez-Andreo, M.J.; Llanos, M.C.; Rivera, J.; Palma-Barqueros, V.; Blanco, J.E.; Vicente, V.; Martinez, C.; Ferrer-Marin, F. Comprehensive comparison of neonate and adult human platelet transcriptomes. PLoS ONE 2017, 12, e0183042. [Google Scholar] [CrossRef] [Green Version]
- Tao, H.; Gaudry, L.; Rice, A.; Chong, B. Cord blood is better than bone marrow for generating megakaryocytic progenitor cells. Exp. Hematol. 1999, 27, 293–301. [Google Scholar] [CrossRef]
- Guerriero, R.; Testa, U.; Gabbianelli, M.; Mattia, G.; Montesoro, E.; Macioce, G.; Pace, A.; Ziegler, B.; Hassan, H.J.; Peschle, C. Unilineage megakaryocytic proliferation and differentiation of purified hematopoietic progenitors in serum-free liquid culture. Blood 1995, 86, 3725–3736. [Google Scholar] [CrossRef] [Green Version]
- De Bruyn, C.; Delforge, A.; Lagneaux, L.; Bron, D. Characterization of CD34+ subsets derived from bone marrow, umbilical cord blood and mobilized peripheral blood after stem cell factor and interleukin 3 stimulation. Bone Marrow Transplant. 2000, 25, 377–383. [Google Scholar] [CrossRef] [Green Version]
- Miyazaki, R.; Ogata, H.; Iguchi, T.; Sogo, S.; Kushida, T.; Ito, T.; Inaba, M.; Ikehara, S.; Kobayashi, Y. Comparative analyses of megakaryocytes derived from cord blood and bone marrow. Br. J. Haematol. 2000, 108, 602–609. [Google Scholar] [CrossRef]
- Gaur, M.; Kamata, T.; Wang, S.; Moran, B.; Shattil, S.J.; Leavitt, A.D. Megakaryocytes derived from human embryonic stem cells: A genetically tractable system to study megakaryocytopoiesis and integrin function. J. Thromb. Haemost. 2006, 4, 436–442. [Google Scholar] [CrossRef] [PubMed]
- Herbein, G.; Sovalat, H.; Wunder, E.; Baerenzung, M.; Bachorz, J.; Lewandowski, H.; Schweitzer, C.; Schmitt, C.; Kirn, A.; Hénon, P. Isolation and identification of two CD34+ cell subpopulations from normal human peripheral blood. Stem Cells 1994, 12, 187–197. [Google Scholar] [CrossRef] [PubMed]
- Panuganti, S.; Schlinker, A.C.; Lindholm, P.F.; Papoutsakis, E.T.; Miller, W.M. Three-stage ex vivo expansion of high-ploidy megakaryocytic cells: Toward large-scale platelet production. Tissue Eng. Part A 2013, 19, 998–1014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ivanovic, Z.; Duchez, P.; Morgan, D.A.; Hermitte, F.; Lafarge, X.; Chevaleyre, J.; Praloran, V.; Dazey, B.; Vezon, G.; Boiron, J.M. Whole-blood leuko-depletion filters as a source of CD 34+ progenitors potentially usable in cell therapy. Transfusion 2006, 46, 118–125. [Google Scholar] [CrossRef]
- Six, K.R.; Sicot, G.; Devloo, R.; Feys, H.B.; Baruch, D.; Compernolle, V. A comparison of haematopoietic stem cells from umbilical cord blood and peripheral blood for platelet production in a microfluidic device. Vox Sang. 2019, 114, 330–339. [Google Scholar] [CrossRef]
- Lambert, M.P.; Sullivan, S.K.; Fuentes, R.; French, D.L.; Poncz, M. Challenges and promises for the development of donor-independent platelet transfusions. Blood 2013, 121, 3319–3324. [Google Scholar] [CrossRef]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [Green Version]
- Takayama, N.; Eto, K. Pluripotent stem cells reveal the developmental biology of human megakaryocytes and provide a source of platelets for clinical application. Cell. Mol. Life Sci. 2012, 69, 3419–3428. [Google Scholar] [CrossRef] [Green Version]
- Focosi, D.; Amabile, G.; Di Ruscio, A.; Quaranta, P.; Tenen, D.G.; Pistello, M. Induced pluripotent stem cells in hematology: Current and future applications. Blood Cancer J. 2014, 4, e211. [Google Scholar] [CrossRef] [Green Version]
- Lee, A.S.; Tang, C.; Rao, M.S.; Weissman, I.L.; Wu, J.C. Tumorigenicity as a clinical hurdle for pluripotent stem cell therapies. Nat. Med. 2013, 19, 998–1004. [Google Scholar] [CrossRef] [Green Version]
- Peng, Y.; Martin, D.A.; Kenkel, J.; Zhang, K.; Ogden, C.A.; Elkon, K.B. Innate and adaptive immune response to apoptotic cells. J. Autoimmun. 2007, 29, 303–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamura, S.; Takayama, N.; Hirata, S.; Seo, H.; Endo, H.; Ochi, K.; Fujita, K.; Koike, T.; Harimoto, K.; Dohda, T.; et al. Expandable megakaryocyte cell lines enable clinically applicable generation of platelets from human induced pluripotent stem cells. Cell Stem Cell 2014, 14, 535–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takayama, N.; Nishikii, H.; Usui, J.; Tsukui, H.; Sawaguchi, A.; Hiroyama, T.; Eto, K.; Nakauchi, H. Generation of functional platelets from human embryonic stem cells in vitro via ES-sacs, VEGF-promoted structures that concentrate hematopoietic progenitors. Blood 2008, 111, 5298–5306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hansen, M.; Von Lindern, M.; Van den Akker, E.; Varga, E. Human-induced pluripotent stem cell-derived blood products: State of the art and future directions. FEBS Lett. 2019, 593, 3288–3303. [Google Scholar] [CrossRef]
- Figueiredo, C.; Goudeva, L.; Horn, P.A.; Eiz-Vesper, B.; Blasczyk, R.; Seltsam, A. Generation of HLA-deficient platelets from hematopoietic progenitor cells. Transfusion 2010, 50, 1690–1701. [Google Scholar] [CrossRef] [Green Version]
- Pick, M.; Azzola, L.; Osborne, E.; Stanley, E.G.; Elefanty, A.G. Generation of megakaryocytic progenitors from human embryonic stem cells in a feeder- and serum-free medium. PLoS ONE 2013, 8, e55530. [Google Scholar] [CrossRef] [Green Version]
- Feng, Q.; Shabrani, N.; Thon, J.N.; Huo, H.; Thiel, A.; Machlus, K.R.; Kim, K.; Brooks, J.; Li, F.; Luo, C.; et al. Scalable Generation of Universal Platelets from Human Induced Pluripotent Stem Cells. Stem Cell Rep. 2014, 3, 817–831. [Google Scholar] [CrossRef] [Green Version]
- Moreau, T.; Evans, A.L.; Vasquez, L.; Tijssen, M.R.; Yan, Y.; Trotter, M.W.; Howard, D.; Colzani, M.; Arumugam, M.; Wu, W.H.; et al. Large-scale production of megakaryocytes from human pluripotent stem cells by chemically defined forward programming. Nature Commun. 2016, 7, 11208. [Google Scholar] [CrossRef]
- Lis, R.; Karrasch, C.C.; Poulos, M.G.; Kunar, B.; Redmond, D.; Duran, J.G.B.; Badwe, C.R.; Schachterle, W.; Ginsberg, M.; Xiang, J.; et al. Conversion of adult endothelium to immunocompetent haematopoietic stem cells. Nature 2017, 545, 439–445. [Google Scholar] [CrossRef]
- Ono, Y.; Wang, Y.; Suzuki, H.; Okamoto, S.; Ikeda, Y.; Murata, M.; Poncz, M.; Matsubara, Y. Induction of functional platelets from mouse and human fibroblasts by p45NF-E2/Maf. Blood 2012, 120, 3812–3821. [Google Scholar] [CrossRef] [Green Version]
- Ono-Uruga, Y.; Tozawa, K.; Horiuchi, T.; Murata, M.; Okamoto, S.; Ikeda, Y.; Suda, T.; Matsubara, Y. Human adipose tissue-derived stromal cells can differentiate into megakaryocytes and platelets by secreting endogenous thrombopoietin. J. Thromb. Haemost. 2016, 14, 1285–1297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tozawa, K.; Ono-Uruga, Y.; Yazawa, M.; Mori, T.; Murata, M.; Okamoto, S.; Ikeda, Y.; Matsubara, Y. Megakaryocytes and platelets from a novel human adipose tissue-derived mesenchymal stem cell line. Blood 2019, 133, 633–643. [Google Scholar] [CrossRef] [PubMed]
- Pinho, S.; Frenette, P.S. Haematopoietic stem cell activity and interactions with the niche. Nat. Rev. Mol. Cell Biol. 2019, 20, 303–320. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Li, L. Understanding hematopoietic stem-cell microenvironments. Trends Biochem. Sci. 2006, 31, 589–595. [Google Scholar] [CrossRef] [PubMed]
- Gurkan, U.A.; Akkus, O. The mechanical environment of bone marrow: A review. Ann. Biomed. Eng. 2008, 36, 1978–1991. [Google Scholar] [CrossRef]
- Junt, T.; Schulze, H.; Chen, Z.; Massberg, S.; Goerge, T.; Krueger, A.; Wagner, D.D.; Graf, T.; Italiano, J.E., Jr.; Shivdasani, R.A.; et al. Dynamic visualization of thrombopoiesis within bone marrow. Science 2007, 317, 1767–1770. [Google Scholar] [CrossRef] [Green Version]
- Di Buduo, C.A.; Kaplan, D.L.; Balduini, A. In vitro generation of platelets: Where do we stand? Transfus. Clin. Biol. 2017, 24, 273–276. [Google Scholar] [CrossRef]
- Aguilar, A.; Pertuy, F.; Eckly, A.; Strassel, C.; Collin, D.; Gachet, C.; Lanza, F.; Leon, C. Importance of environmental stiffness for megakaryocyte differentiation and proplatelet formation. Blood 2016, 128, 2022–2032. [Google Scholar] [CrossRef] [Green Version]
- Schlinker, A.C.; Radwanski, K.; Wegener, C.; Min, K.; Miller, W.M. Separation of in-vitro-derived megakaryocytes and platelets using spinning-membrane filtration. Biotechnol. Bioeng. 2015, 112, 788–800. [Google Scholar] [CrossRef] [Green Version]
- Ingavle, G.; Shabrani, N.; Vaidya, A.; Kale, V. Mimicking megakaryopoiesis in vitro using biomaterials: Recent advances and future opportunities. Acta Biomater. 2019, 96, 99–110. [Google Scholar] [CrossRef]
- Thon, J.N.; Dykstra, B.J.; Beaulieu, L.M. Platelet bioreactor: Accelerated evolution of design and manufacture. Platelets 2017, 28, 472–477. [Google Scholar] [CrossRef] [PubMed]
- Lasky, L.C.; Sullenbarger, B. Manipulation of oxygenation and flow-induced shear stress can increase the in vitro yield of platelets from cord blood. Tissue Eng. Part. C Methods 2011, 17, 1081–1088. [Google Scholar] [CrossRef] [PubMed]
- Kropp, C.; Kempf, H.; Halloin, C.; Robles-Diaz, D.; Franke, A.; Scheper, T.; Kinast, K.; Knorpp, T.; Joos, T.O.; Haverich, A.; et al. Impact of Feeding Strategies on the Scalable Expansion of Human Pluripotent Stem Cells in Single-Use Stirred Tank Bioreactors. Stem. Cells Transl. Med. 2016, 5, 1289–1301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sullenbarger, B.; Bahng, J.H.; Gruner, R.; Kotov, N.; Lasky, L.C. Prolonged continuous in vitro human platelet production using three-dimensional scaffolds. Exp. Hematol. 2009, 37, 101–110. [Google Scholar] [CrossRef] [Green Version]
- Nakagawa, Y.; Nakamura, S.; Nakajima, M.; Endo, H.; Dohda, T.; Takayama, N.; Nakauchi, H.; Arai, F.; Fukuda, T.; Eto, K. Two differential flows in a bioreactor promoted platelet generation from human pluripotent stem cell-derived megakaryocytes. Exp. Hematol. 2013, 41, 742–748. [Google Scholar] [CrossRef] [Green Version]
- Di Buduo, C.A.; Wray, L.S.; Tozzi, L.; Malara, A.; Chen, Y.; Ghezzi, C.E.; Smoot, D.; Sfara, C.; Antonelli, A.; Spedden, E.; et al. Programmable 3D silk bone marrow niche for platelet generation ex vivo and modeling of megakaryopoiesis pathologies. Blood 2015, 125, 2254–2264. [Google Scholar] [CrossRef] [Green Version]
- Tozzi, L.; Laurent, P.A.; Di Buduo, C.A.; Mu, X.; Massaro, A.; Bretherton, R.; Stoppel, W.; Kaplan, D.L.; Balduini, A. Multi-channel silk sponge mimicking bone marrow vascular niche for platelet production. Biomaterials 2018, 178, 122–133. [Google Scholar] [CrossRef]
- Pineault, N.; Boucher, J.F.; Cayer, M.P.; Palmqvist, L.; Boyer, L.; Lemieux, R.; Proulx, C. Characterization of the effects and potential mechanisms leading to increased megakaryocytic differentiation under mild hyperthermia. Stem Cells Dev. 2008, 17, 483–493. [Google Scholar] [CrossRef]
- Dunois-Lardé, C.; Capron, C.; Fichelson, S.; Bauer, T.; Cramer-Bordé, E.; Baruch, D. Exposure of human megakaryocytes to high shear rates accelerates platelet production. Blood 2009, 114, 1875–1883. [Google Scholar] [CrossRef] [Green Version]
- Thon, J.N.; Mazutis, L.; Wu, S.; Sylman, J.L.; Ehrlicher, A.; Machlus, K.R.; Feng, Q.; Lu, S.; Lanza, R.; Neeves, K.B.; et al. Platelet bioreactor-on-a-chip. Blood 2014, 124, 1857–1867. [Google Scholar] [CrossRef] [Green Version]
- Blin, A.; Le Goff, A.; Magniez, A.; Poirault-Chassac, S.; Teste, B.; Sicot, G.; Nguyen, K.A.; Hamdi, F.S.; Reyssat, M.; Baruch, D. Microfluidic model of the platelet-generating organ: Beyond bone marrow biomimetics. Sci. Rep. 2016, 6, 21700. [Google Scholar] [CrossRef] [Green Version]
- Avanzi, M.P.; Oluwadara, O.E.; Cushing, M.M.; Mitchell, M.L.; Fischer, S.; Mitchell, W.B. A novel bioreactor and culture method drives high yields of platelets from stem cells. Transfusion 2016, 56, 170–178. [Google Scholar] [CrossRef] [PubMed]
- Martinez, A.F.; McMahon, R.D.; Horner, M.; Miller, W.M. A uniform-shear rate microfluidic bioreactor for real-time study of proplatelet formation and rapidly-released platelets. Biotechnol. Prog. 2017, 33, 1614–1629. [Google Scholar] [CrossRef] [PubMed]
- Ito, Y.; Nakamura, S.; Sugimoto, N.; Shigemori, T.; Kato, Y.; Ohno, M.; Sakuma, S.; Ito, K.; Kumon, H.; Hirose, H.; et al. Turbulence Activates Platelet Biogenesis to Enable Clinical Scale Ex Vivo Production. Cell 2018, 174, 636–648.e618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heazlewood, S.Y.; Nilsson, S.K.; Cartledge, K.; Be, C.L.; Vinson, A.; Gel, M.; Haylock, D.N. Progress in bio-manufacture of platelets for transfusion. Platelets 2017, 28, 649–656. [Google Scholar] [CrossRef]
- Do Sacramento, V.; Mallo, L.; Freund, M.; Eckly, A.; Hechler, B.; Mangin, P.; Lanza, F.; Gachet, C.; Strassel, C. Functional properties of human platelets derived in vitro from CD34+ cells. Sci. Rep. 2020, 10, 914. [Google Scholar] [CrossRef] [Green Version]
- Josefsson, E.C.; White, M.J.; Dowling, M.R.; Kile, B.T. Platelet life span and apoptosis. Methods Mol. Biol. 2012, 788, 59–71. [Google Scholar] [CrossRef]
- Lefrançais, E.; Ortiz-Muñoz, G.; Caudrillier, A.; Mallavia, B.; Liu, F.; Sayah, D.M.; Thornton, E.E.; Headley, M.B.; David, T.; Coughlin, S.R.; et al. The lung is a site of platelet biogenesis and a reservoir for haematopoietic progenitors. Nature 2017, 544, 105–109. [Google Scholar] [CrossRef]
- Fuentes, R.; Wang, Y.; Hirsch, J.; Wang, C.; Rauova, L.; Worthen, G.S.; Kowalska, M.A.; Poncz, M. Infusion of mature megakaryocytes into mice yields functional platelets. J. Clin. Invest. 2010, 120, 3917–3922. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Hayes, V.; Jarocha, D.; Sim, X.; Harper, D.C.; Fuentes, R.; Sullivan, S.K.; Gadue, P.; Chou, S.T.; Torok-Storb, B.J.; et al. Comparative analysis of human ex vivo-generated platelets vs megakaryocyte-generated platelets in mice: A cautionary tale. Blood 2015, 125, 3627–3636. [Google Scholar] [CrossRef] [Green Version]
- Eicke, D.; Baigger, A.; Schulze, K.; Latham, S.L.; Halloin, C.; Zweigerdt, R.; Guzman, C.A.; Blasczyk, R.; Figueiredo, C. Large-scale production of megakaryocytes in microcarrier-supported stirred suspension bioreactors. Sci. Rep. 2018, 8, 10146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monzen, S.; Osuda, K.; Miyazaki, Y.; Hayashi, N.; Takahashi, K.; Kashiwakura, I. Radiation sensitivities in the terminal stages of megakaryocytic maturation and platelet production. Radiat Res. 2009, 172, 314–320. [Google Scholar] [CrossRef] [PubMed]
- Xi, J.; Zhu, H.; Liu, D.; Nan, X.; Zheng, W.; Liu, K.; Shi, W.; Chen, L.; Lv, Y.; Yan, F.; et al. Infusion of megakaryocytic progenitor products generated from cord blood hematopoietic stem/progenitor cells: Results of the phase 1 study. PLoS ONE 2013, 8, e54941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Modery-Pawlowski, C.L.; Tian, L.L.; Pan, V.; McCrae, K.R.; Mitragotri, S.; Sen Gupta, A. Approaches to synthetic platelet analogs. Biomaterials 2013, 34, 526–541. [Google Scholar] [CrossRef]
- Barroso, J.; Osborne, B.; Teramura, G.; Pellham, E.; Fitzpatrick, M.; Biehl, R.; Yu, A.; Pehta, J.; Slichter, S.J. Safety evaluation of a lyophilized platelet-derived hemostatic product. Transfusion 2018, 58, 2969–2977. [Google Scholar] [CrossRef]
- Nasiri, S. Infusible platelet membrane as a platelet substitute for transfusion: An overview. Blood Transfus. 2013, 11, 337–342. [Google Scholar] [CrossRef]
- Levi, M.; Friederich, P.W.; Middleton, S.; De Groot, P.G.; Wu, Y.P.; Harris, R.; Biemond, B.J.; Heijnen, H.F.; Levin, J.; Ten Cate, J.W. Fibrinogen-coated albumin microcapsules reduce bleeding in severely thrombocytopenic rabbits. Nat. Med. 1999, 5, 107–111. [Google Scholar] [CrossRef]
- Coller, B.S.; Springer, K.T.; Beer, J.H.; Mohandas, N.; Scudder, L.E.; Norton, K.J.; West, S.M. Thromboerythrocytes. In vitro studies of a potential autologous, semi-artificial alternative to platelet transfusions. J. Clin. Invest. 1992, 89, 546–555. [Google Scholar] [CrossRef]
- Bertram, J.P.; Williams, C.A.; Robinson, R.; Segal, S.S.; Flynn, N.T.; Lavik, E.B. Intravenous hemostat: Nanotechnology to halt bleeding. Sci. Transl. Med. 2009, 1, 11ra22. [Google Scholar] [CrossRef] [Green Version]
- Hickman, D.A.; Pawlowski, C.L.; Shevitz, A.; Luc, N.F.; Kim, A.; Girish, A.; Marks, J.; Ganjoo, S.; Huang, S.; Niedoba, E.; et al. Intravenous synthetic platelet (SynthoPlate) nanoconstructs reduce bleeding and improve ’golden hour’ survival in a porcine model of traumatic arterial hemorrhage. Sci. Rep. 2018, 8, 3118. [Google Scholar] [CrossRef]
- Xu, P.; Zuo, H.; Chen, B.; Wang, R.; Ahmed, A.; Hu, Y.; Ouyang, J. Doxorubicin-loaded platelets as a smart drug delivery system: An improved therapy for lymphoma. Sci. Rep. 2017, 7, 42632. [Google Scholar] [CrossRef] [PubMed]
- Díaz, A.; Saxena, V.; González, J.; David, A.; Casañas, B.; Carpenter, C.; Batteas, J.D.; Colón, J.L.; Clearfield, A.; Hussain, M.D. Zirconium phosphate nano-platelets: A novel platform for drug delivery in cancer therapy. Chem. Commun. 2012, 48, 1754–1756. [Google Scholar] [CrossRef] [PubMed]
- Girish, A.; Hickman, D.A.; Banerjee, A.; Luc, N.; Ma, Y.; Miyazawa, K.; Sekhon, U.D.S.; Sun, M.; Huang, S.; Sen Gupta, A. Trauma-targeted delivery of tranexamic acid improves hemostasis and survival in rat liver hemorrhage model. J. Thromb. Haemost. 2019, 17, 1632–1644. [Google Scholar] [CrossRef] [PubMed]
- Morrell, C.N.; Aggrey, A.A.; Chapman, L.M.; Modjeski, K.L. Emerging roles for platelets as immune and inflammatory cells. Blood 2014, 123, 2759–2767. [Google Scholar] [CrossRef] [Green Version]
- Leblanc, R.; Peyruchaud, O. Metastasis: New functional implications of platelets and megakaryocytes. Blood 2016, 128, 24–31. [Google Scholar] [CrossRef]
- Nurden, A.T. Platelets, inflammation and tissue regeneration. Thromb. Haemost. 2011, 105 (Suppl. 1), S13–S33. [Google Scholar] [CrossRef]
- Rayes, J.; Watson, S.P.; Nieswandt, B. Functional significance of the platelet immune receptors GPVI and CLEC-2. J. Clin. Invest. 2019, 129, 12–23. [Google Scholar] [CrossRef] [Green Version]
- Lam, F.W.; Vijayan, K.V.; Rumbaut, R.E. Platelets and Their Interactions with Other Immune Cells. Compr. Physiol. 2015, 5, 1265–1280. [Google Scholar] [CrossRef] [Green Version]
- Acebes-Huerta, A.; Arias-Fernández, T.; Bernardo, Á.; Muñoz-Turrillas, M.C.; Fernández-Fuertes, J.; Seghatchian, J.; Gutiérrez, L. Platelet-derived bio-products: Classification update, applications, concerns and new perspectives. Transfus. Apher. Sci. 2020, 59, 102716. [Google Scholar] [CrossRef]
- Sim, X.; Poncz, M.; Gadue, P.; French, D.L. Understanding platelet generation from megakaryocytes: Implications for in vitro-derived platelets. Blood 2016, 127, 1227–1233. [Google Scholar] [CrossRef] [Green Version]



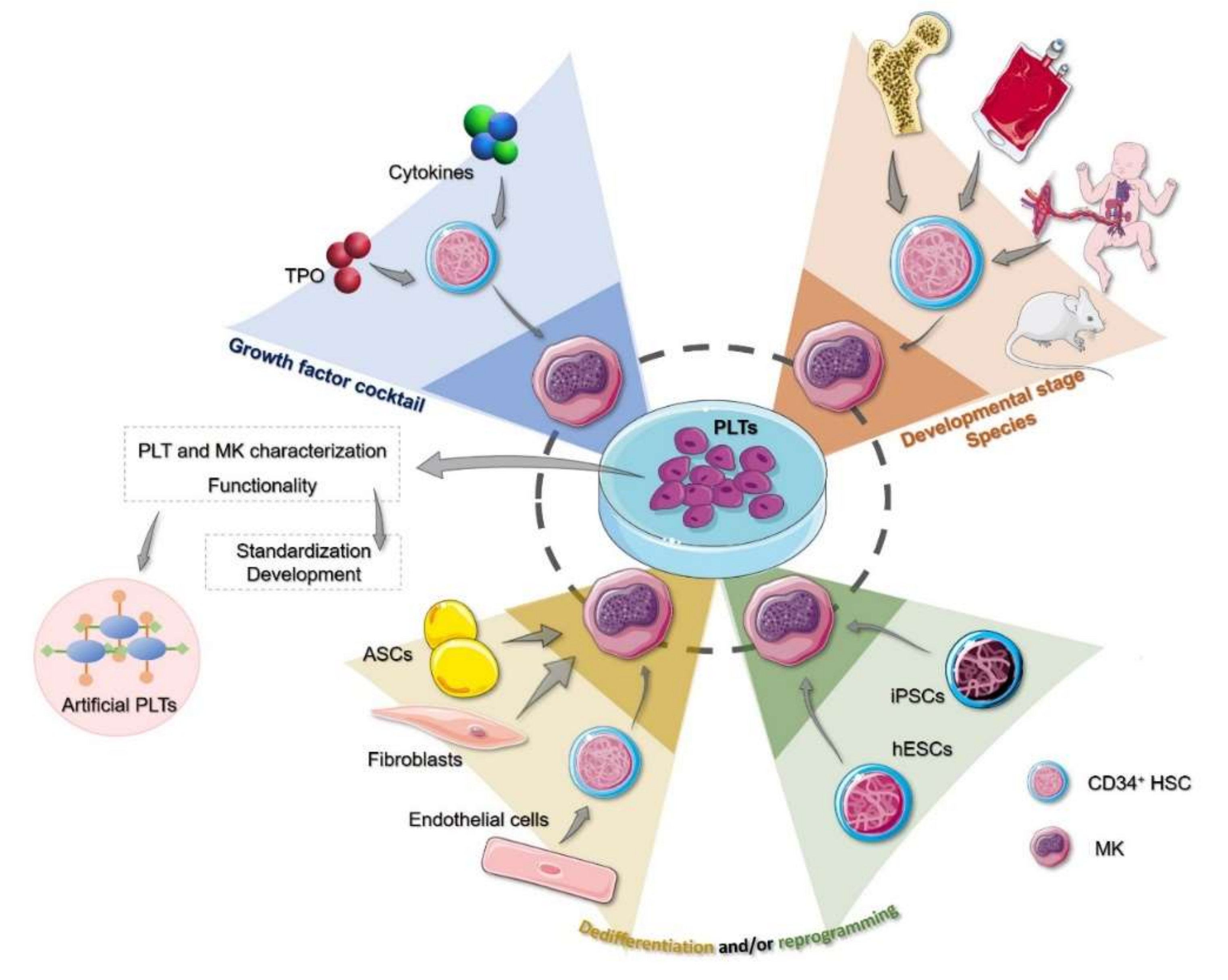{kind=link}
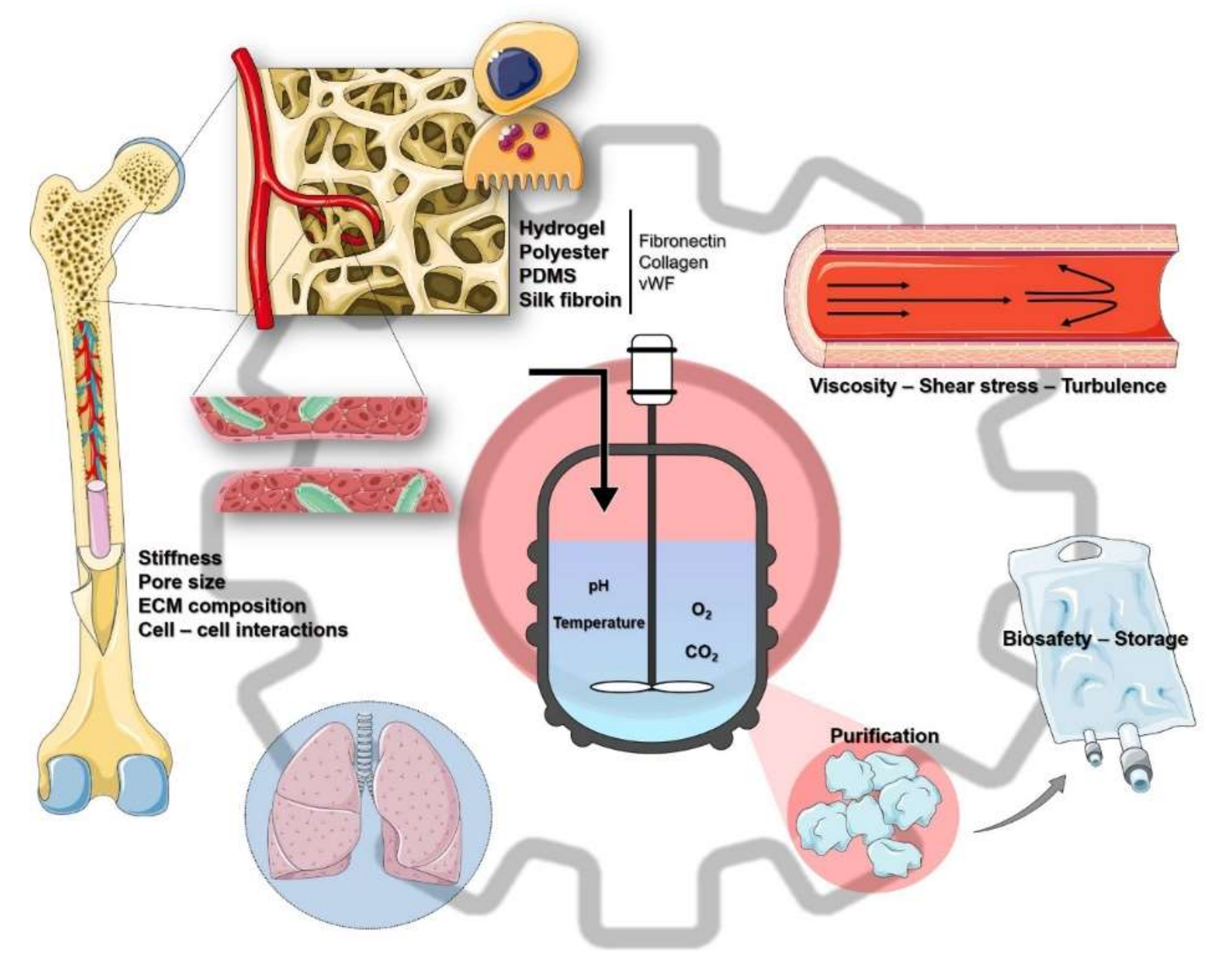{kind=link}
| Standardization Phase | Quality Control | |
|---|---|---|
| PLT characterization | Immunophenotyping–comprehensive receptor expression (flow cytometry) | Immunophenotyping selected receptors |
| Morphology (size, granularity; flow cytometry, e-microscopy) | Morphology (flow cytometry) | |
| Viability/Apoptosis tests (mitochondrial function, caspase activity) [107] | If relevant from standardization, mitochondrial function | |
| Proteomics | If relevant from standardization, selected proteins (WB, ELISA, Luminex) | |
| Transcriptomics | If relevant from standardization, selected transcripts (qRT-PCR) | |
| PLT function | Adhesion (comprehensive substrate panel) | The ideal would be to minimize functional tests, when the characterization reaches a certain quality level as derived from the standardization phase |
| Degranulation (comprehensive agonist panel) | ||
| Aggregation (comprehensive agonist panel) | Aggregation (selected agonists) | |
| Thrombi formation (perfusion assays in vitro) | ||
| In vivo transfusion assays |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martínez-Botía, P.; Acebes-Huerta, A.; Seghatchian, J.; Gutiérrez, L. On the Quest for In Vitro Platelet Production by Re-Tailoring the Concepts of Megakaryocyte Differentiation. Medicina 2020, 56, 671. https://doi.org/10.3390/medicina56120671
Martínez-Botía P, Acebes-Huerta A, Seghatchian J, Gutiérrez L. On the Quest for In Vitro Platelet Production by Re-Tailoring the Concepts of Megakaryocyte Differentiation. Medicina. 2020; 56(12):671. https://doi.org/10.3390/medicina56120671
Chicago/Turabian StyleMartínez-Botía, Patricia, Andrea Acebes-Huerta, Jerard Seghatchian, and Laura Gutiérrez. 2020. "On the Quest for In Vitro Platelet Production by Re-Tailoring the Concepts of Megakaryocyte Differentiation" Medicina 56, no. 12: 671. https://doi.org/10.3390/medicina56120671
APA StyleMartínez-Botía, P., Acebes-Huerta, A., Seghatchian, J., & Gutiérrez, L. (2020). On the Quest for In Vitro Platelet Production by Re-Tailoring the Concepts of Megakaryocyte Differentiation. Medicina, 56(12), 671. https://doi.org/10.3390/medicina56120671