1. Introduction
Chitins originate from crustaceans such as shrimp, crab, insects, mushrooms, green algae, and silkworms. They are one of the most abundant renewable biopolymers on earth and can be obtained inexpensively from marine sources with more than 10 GT on an annual basis [
1]. The molecular structure of chitins consists of
N-acetylated glucosamine (GlcNAc) and 2-amino-2-
d-glucose (
d-glucosamine, GlcN), and chitin contains a high proportion of GlcNAc [
2]. However, their poor solubility limits usage of this natural resource. One possible means of addressing this challenge involves chitosan, which is the product of chitin deacetylation under alkaline conditions. Chitosan is a linear polysaccharide containing various proportions of GlcN [
3]. It has applications in nutritional [
4], agricultural [
5], and medical areas [
6,
7,
8]. However, its poor solubility in aqueous solutions and its high viscosity are problematic for those applications.
Chitooligosaccharides (COS) are hydrolyzed products of chitosan, and previous studies indicate that they may be produced from chitosan using either chemical and physical methods, as well as enzymatic hydrolysis [
9,
10]. COS contains 2–20 glucosamine residues which are attached through β-
d-(1-4) glycoside linkages, similar to chitin and chitosan (
Figure 1). The family of COS compounds have received much attention because of their small molecular weight, good aqueous solubility, and diverse biological activity [
11]. Applications of interest include antibacterial efficacy [
12], antitumor activity [
13], moderation of cholesterol levels [
14,
15,
16], and weight loss [
17,
18].
The degree of deacetylation (DD, %) is defined as the molar fraction of GlcN in the copolymers (chitosan) composed of GlcNAc and GlcN [
19]. The DD value of a COS sample is one of the most important factor in assessing its applications in the medical, nutritional, sewage treatment, and biotechnological fields [
20]. It can also be related back to the specific biological and structural properties and functions of chitin or chitosan, and it should be clear that chitosan is the deacetylated form of chitin and it must be characterized by a degree of acetylation when a degree of deacetylation is valid for chitin, the initial form of the polymer nearly fully acetylated.
For chitin, chitosan, and COS, measurement of the DD is crucial in determination of their chemical structures, physical properties, and interrelation. It is also essential in providing a basis from which the functions and applications can be predicted and optimized [
2,
21]. Therefore, a suitable method of obtaining reliable DD values is essential for diverse researchers working in those fields.
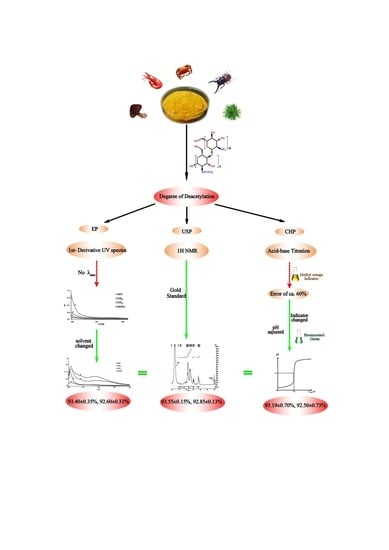
To date, several techniques have been developed and applied for determination of DD in chitosan [
19,
20,
22,
23,
24,
25,
26,
27,
28,
29,
30,
31,
32,
33,
34]. These have different levels of precision and accuracy. The currently accepted methods are specific to the concerned organization: U.S. Pharmacopoeia uses
1H NMR spectroscopy [
35],
European Pharmacopoeia has approved the first-order derivative UV spectrophotometric method [
36], and Chinese Pharmacopoeia prefers an acid-base titrametric procedure using methyl orange as the indicator [
37]. However, a standard method for determination of DD in COS for use by researchers, companies, and even state organizations is still lacking. Arguably, this predicament has resulted from variation in chitosan properties.
For COS, a method for DD determination based on
1H NMR spectroscopy has been reported [
26,
29,
32]; however, its large expense limits its application in developing countries. Unfortunately, the alternative procedures for DD determination in chitosan, which have been endorsed by the Chinese Pharmacopoeia and European Pharmacopoeia (vide supra), are highly inaccurate for evaluation of the DD in COS.
Our research team has long been engaged in the study of COS [
38] and has developed COS products such as the COS tablet [
39] and the COS capsule [
40]. We found that the weight loss function of COS is equal to that of Orlistat, the only Over the Counter (OTC) weight loss pill currently on the market, and that its side effects are significantly lower than those of Orlistat [
18]. The DD value of a COS sample is the one of the most important factor in assessing its quality, and the methods for determination of chitosan content recommended in the Chinese Pharmacopoeia and the European Pharmacopoeia are not applicable for evaluation of the extent of DD in COS. A need therefore exists for a method for DD determination in COS which is easy to perform, inexpensive, widely adaptable, and highly accurate. Our research group has addressed this challenge by pioneering the development of two new methods which are applicable to COS having several different molecular weight averages, and herein we report the results of those investigations for DD determination in COS using: (a) the acid-base titration with bromocresol green indicator; and (b) the first-order derivative of a UV spectrometric trace. The parameters we selected to optimize for each method were the indicator p
Ka and color change (titrametric method) and the solvent (UV method). Furthermore, the accuracy of both new methods in samples having relatively high and low molecular weights (COS
A and COS
B) were assessed, and those results, through comparison to those from the
1H NMR spectroscopic method, were verified.
3. Materials and Methods
3.1. Materials
Commercial samples of COS used in this study were obtained from Qingdao (Shangdong, China) and had average molecular weights of 1000 Da and 3000 Da, respectively. The DD of the commercial COS were >90% in glucosamine hydrochloride. The N-acetylglucosamine used in this study was purchased from Sigma Aldrich (St. Louis, MO, USA) and had a purity greater than 99%. The deuterium oxide used had an isotopic purity of 99.8%, and was obtained from Shanghai Macklin Biochemical Co., Ltd. (Tianjin, China). Bromocresol green and methyl orange were purchased from Tianjin Damao Chemical Reagent Factory (Tianjin, China) and methyl orange from Tianjin Zhiyuan Chemical Reagent Co., Ltd. (Tianjin, China), respectively. Sodium hydroxide pellets (NaOH), hydrochloric acid (HCl), and potassium hydroxide pellets were obtained from Tianjin Kermel Chemical Reagent Co., Ltd. (Tianjin, China). All other reagents and solvents were of analytical grade and used without further purification. All water used in the extraction and analysis was distilled and deionized.
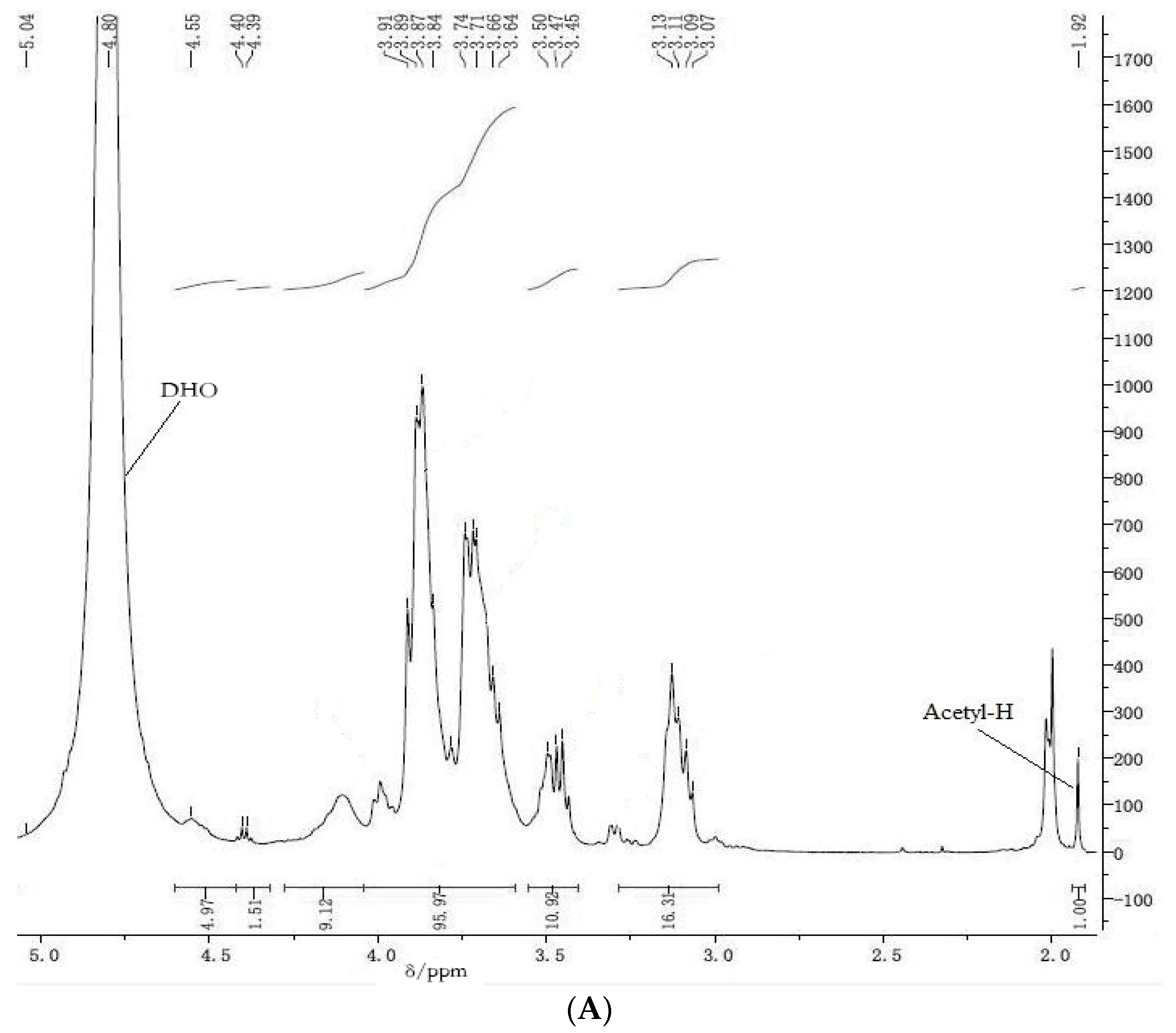
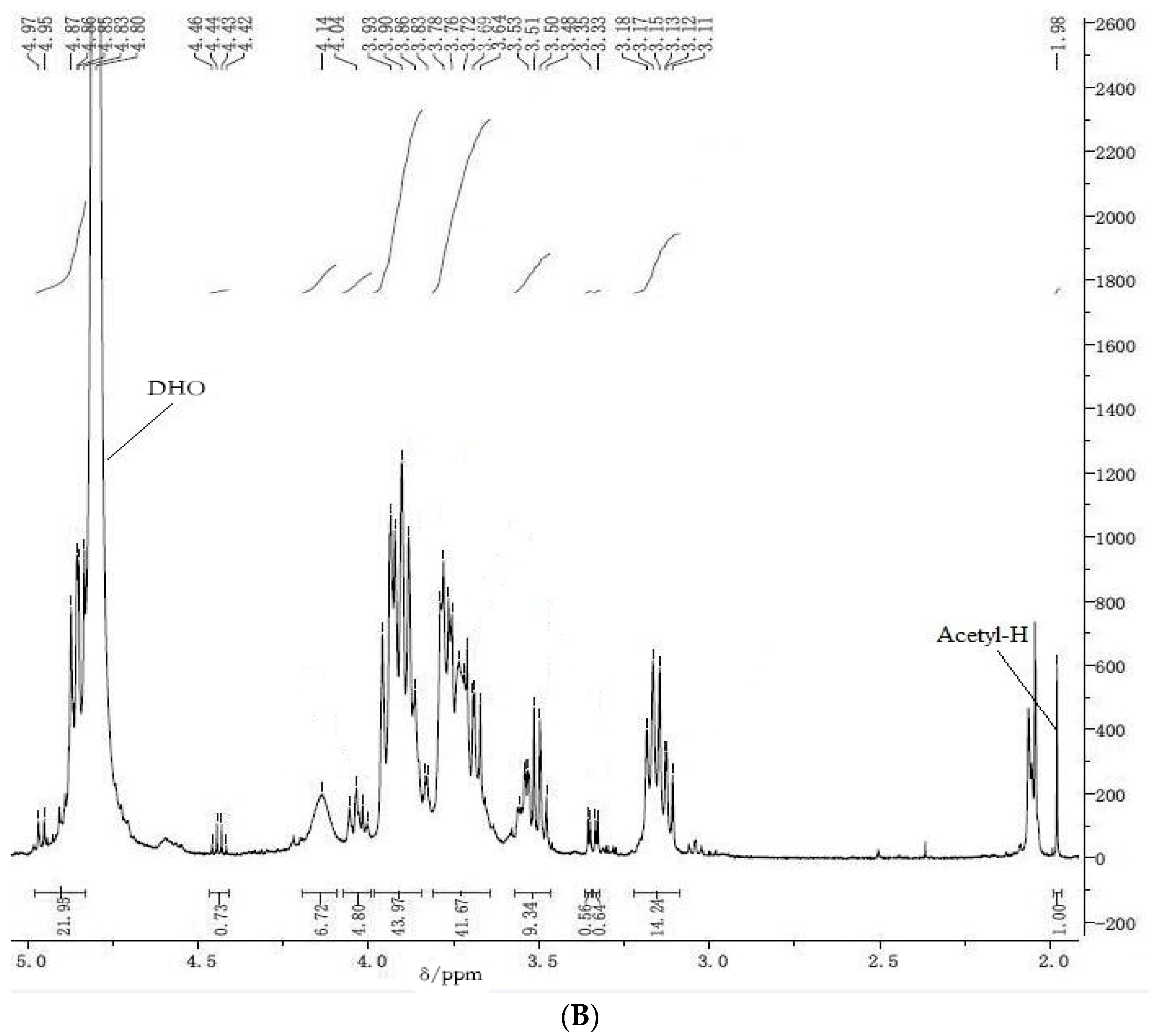
3.2. 1H NMR Spectroscopy
The following procedure, from the U.S. Pharmacopoeia, was used for
1H NMR spectroscopic determination of chitosan content. Approximately 20–30 mg COS samples were dissolved in 1 mL deuteroxide. Aliquots of 0.5–1.0 mL of the sample solution were transferred to a standard 8-mm NMR spinning tube. A spectral window of 8012 Hz, 297 K, and an acquisition time of 4.08 s were used for all measurements.
1H NMR spectra were recorded on a Bruker 500 MHz spectrometer (Bruker, Billerica, MA, USA) at 297 K. The quantitation method involved scanning the concerned sample over δ 0–5 ppm; every sample was evaluated three times. The average area of the segment in the δ 3–6 ppm range was recorded as
A1, and represented the seven protons adjacent to the oxygen atoms within the sugar molecule ring structure. The average areas below the resonance traces from the CH
3 protons on the acetyl groups appeared at ca. δ 2 ppm and were recorded as
A2 according to the equations listed below. The
1H NMR spectrum of a typical COS sample shows resonances at ca. 1.9–2.1 ppm from CH
3 of the
N-acetyl group in GlcNAc. The shifts of the protons attached to positions C
2–C
6 on the sugar ring appear at ca. 2.6–4.2 ppm [
4]. The DD of COS samples was determined according to Equation (1) [
35]:
where
A1 are the protons integral values of positions C
2–C
6 on the sugar ring and
A2 are the protons integral values of the three
N-acetyl protons of GlcNAc.
3.3. Determination of DD in COS Using the First-Order Derivative of UV Spectra
The UV spectra method of analysis is widely used in the analysis of drugs in pharmaceutical formulations due to its good sensitivity and cost effectiveness. Over the last three decades, derivative spectrophotometry has been extensively used in the determination of drugs in multicomponents having overlapping spectra, which eliminates interference from the formulation matrix by using the zero-crossing techniques. For GlcNAc, the absorption maximum occurs at λ
max = 204 nm in 0.3 M hydrochloric acid solution. GlcN also has an absorption which interferes with the determination of GlcNAc at 204 nm. However, the first-order derivative UV spectroscopic method compensates for this phenomenon. The procedure described in the European Pharmacopoeia for determination of chitosan DD in COS, which uses a first-order derivative UV spectroscopic method, is described as follows. According to this method, ca. 0.1 g of accurately weighed samples of COS are dried overnight at 45 °C at 1 atm pressure. The samples are then dissolved in 50.0 mL 0.3 M HCl solution with vigorous stirring. For analysis, aliquots of 1.0 mL of this solution are diluted to 10.0 mL with hydrochloric acid solution. As necessary, further dilutions of the analyte solution are performed. In this study, the maximum wavelength for analyzed solutions was obtained at 200–400 nm at a slit width of 2 nm, a scanning speed of 20 nm·min
−1, a time constant of 4 s, and a chart speed of 10 cm·min
−1. Cuvettes with 10 mm path length were used in the UV spectrophotometer (Presee T10CS, Beijing, China). Then, the analyte solution absorbance under the model of spectrum scanning over the 200–215 nm range was recorded, and the absorbance was transformed into differential calculation at a time. The absorbance of GlcNAc in the COS was determined by measuring the first derivative signal at 200–215 nm. All measurements were blanked against the same HCl solution. The masses of GlcNAc in the COS analyte solutions were determined according to the calibration curve. The DD was calculated according to Equation (2) [
36]:
where
Csample is the concentration of COS in the analyte solution (μg/mL);
C is the concentration of GlcNAc in the analyte solution, as determined from the calibration curve (μg/mL); 203 is the molecular mass of GlcNAc fragment in COS polymer, according to C
8H
13NO
5; and 42 is the difference in molecular masses between the GlcNAc and GlcN fragments.
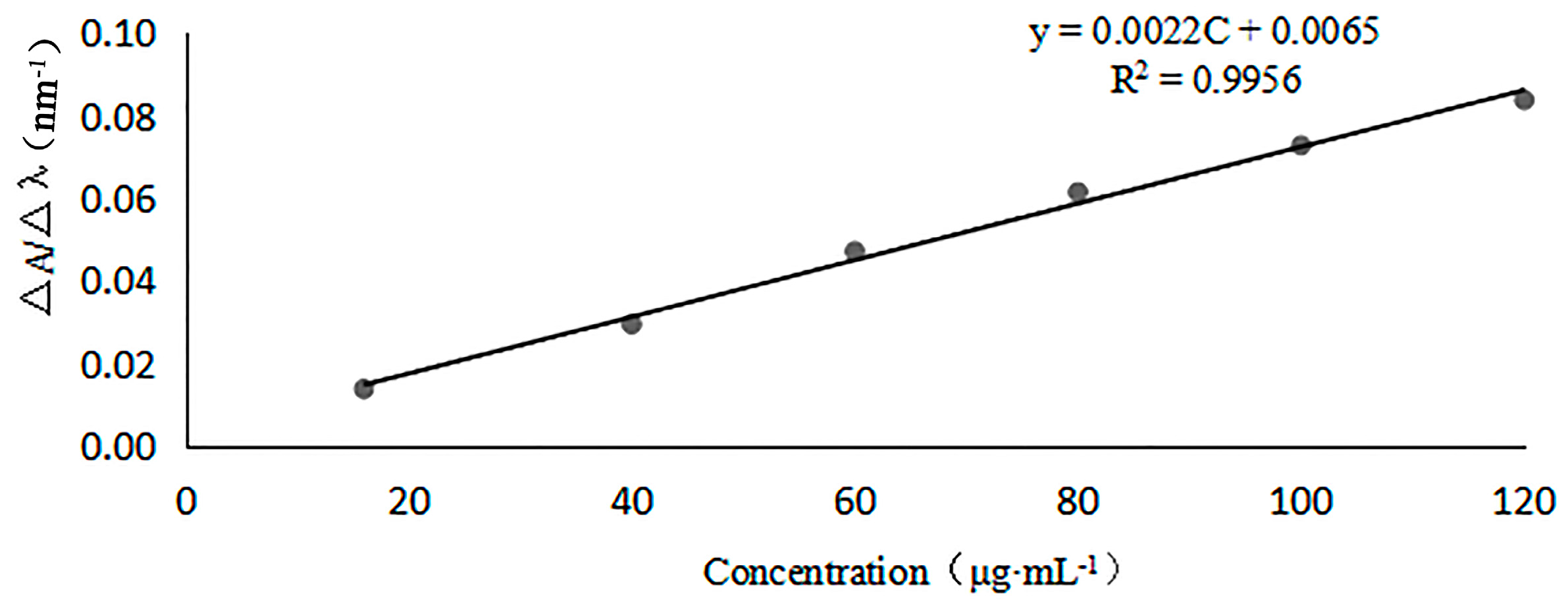
3.3.1. Linearity of First-Order Derivative UV Spectra
For linearity, six different standard solutions of GlcNAc, having concentrations of 16.0, 32.0, 64.0, 80.0, 100.0, and 120.0 μg/mL, were prepared in 0.3 M HCl solution. The first derivative of the UV absorption curve at λ = 200–215 nm of each solution was measured, and absorbance of GlcNAc under the dominant wavelength of 204 nm and a baseline wavelength of 202 nm were recorded so that amplitude ∆A/∆λ (∆A = A204nm − A202nm, ∆λ = 2 nm) and GlcNAc concentrations were obtained. Then, a calibration curve was made. The LOD and LOQ were determined by 3.3 σ/s and 10 σ/s criteria, respectively, where σ is the standard deviation of the analytical signal and s is the slope of the corresponding calibration curve.
3.3.2. Accuracy of First-Order Derivative UV Spectra
Accuracy of the first-order derivative UV spectroscopy was determined by calculating the recoveries of GlcNAc by the standard addition method, in which pre-analyzed samples (100 μg/mL) were taken, and GlcNAc was added at three different levels, i.e., 80%, 100%, and 120%. The total amount of GlcNAc was estimated by using the proposed methods in triplicate for every level. The percent recovery of the added GlcNAc was calculated as percent recovery = (Dt − Ds)/Da) × 100, where Dt is the total sample concentration measured after standard addition; Ds is the sample concentration in the formulation mixture; Da is the sample concentration added.
3.3.3. Precision of First-Order Derivative UV Spectra
To evaluate the first-order derivative UV spectra method precision, different concentrations were prepared from sample stock solution and analyzed three times within the same day and on three separate days to evaluate within-day and between-day precision, respectively. Under the first-order derivative model, the amplitude ∆A/∆λ (∆A = A204nm − A202nm, ∆λ = 2 nm) of COS samples is examined, and the equivalent amount of GlcNAc for the COS samples is analyzed through the calibration curve. The DD was calculated according to Equation (2). From the obtained data, the percent relative standard deviation (% RSD) was calculated.
3.4. Acid-Base Titrametric Determination of DD in COS Using Methyl Orange Indicator
The procedure described in the Chinese Pharmacopoeia for determination of chitosan DD in COS, using acid-base titration and methyl orange indictor, was followed. According to this method, ca. 0.5 g of accurately weighed samples of COS were dried overnight at 45 °C at 1 atm pressure. Afterwards, the samples were dissolved in 32.0 mL distilled and deionized water by stirring with a gradient mixer (TH-500A, Shanghai Chemical Co. Ltd., Shanghai, China). To the resulting solution, 18.00 mL of 0.3 M aqueous HCl solution (standardized to 0.314 M) was added. The volume of added aqueous HCl solution was noted as
VHCl, followed by addition of 2–3 drops of 1% M methyl orange indictor. Afterwards, the solution was back-titrated with 0.15 M aqueous NaOH (standardized to 0.146 M), until transition of the color from red to yellow had occurred. The volume of added standard aqueous NaOH solution was noted as
VNaOH. Calculation of DD was according to Equation (3) as follows [
37]:
where
CHCl is the concentration of standardized aqueous HCl solution (M);
CNaOH is the concentration of standardized aqueous NaOH solution (M);
VHCl is the volume of standardized aqueous HCl solution (mL);
VNaOH is the volume of standard NaOH aqueous solution (mL);
G is the weight of COS sample (g);
W is the aqueous concentration of COS sample (%); 0.016 is the equivalent weight of amino groups corresponding to 1 mL 0.1 M HCl (g); and 9.94% is the theoretical equivalent mass of amino groups, expressed as percentage.
3.5. Acid-Base Titrametric Determination of DD in COS Using Bromocresol Green Indicator
According to this procedure, ca. 0.5 g of accurately weighed samples of COS were dried overnight at 45 °C at 1 atm pressure. The samples were dissolved in 32.0 mL distilled and deionized water by stirring with a gradient mixer. The pH of the COS solutions were adjusted to 8.0 and monitored using a pH meter. An 18.00 mL aliquot of 0.3 M aqueous HCl solution (standardized to 0.314 M) was added to the solution. The volume of the aqueous HCl solution added was noted as VHCl, and then 2–3 drops of 1% bromocresol green indicator solution in ethyl alcohol were added. Afterwards, and while stirring, the solution was back-titrated with 0.15 M (standardized to 0.146 M) aqueous sodium hydroxide solution until transition of the indicator color from yellow to blue-green had occurred. The volume of added standardized NaOH aqueous solution was noted as VNaOH. DD was calculated according to Equation (3).
3.6. Moisture Determination
For determination of moisture levels, ca. 0.3–0.5 g sample of COS was weighed on the sample plate of an infrared moisture determination instrument (Sartorius, M7, Berlin, Germany). The sample was shaken for homogeneous distribution across the plate, moisture was determined until the scale heating test no longer moved, and the results were recorded.








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}