
Biscembranoids and Cembranoids from the Soft Coral Sarcophyton elegans

Abstract
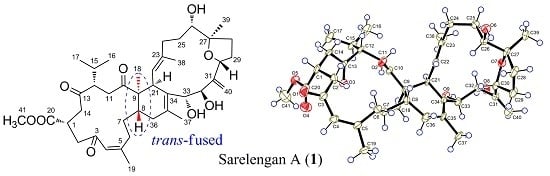
:
1. Introduction
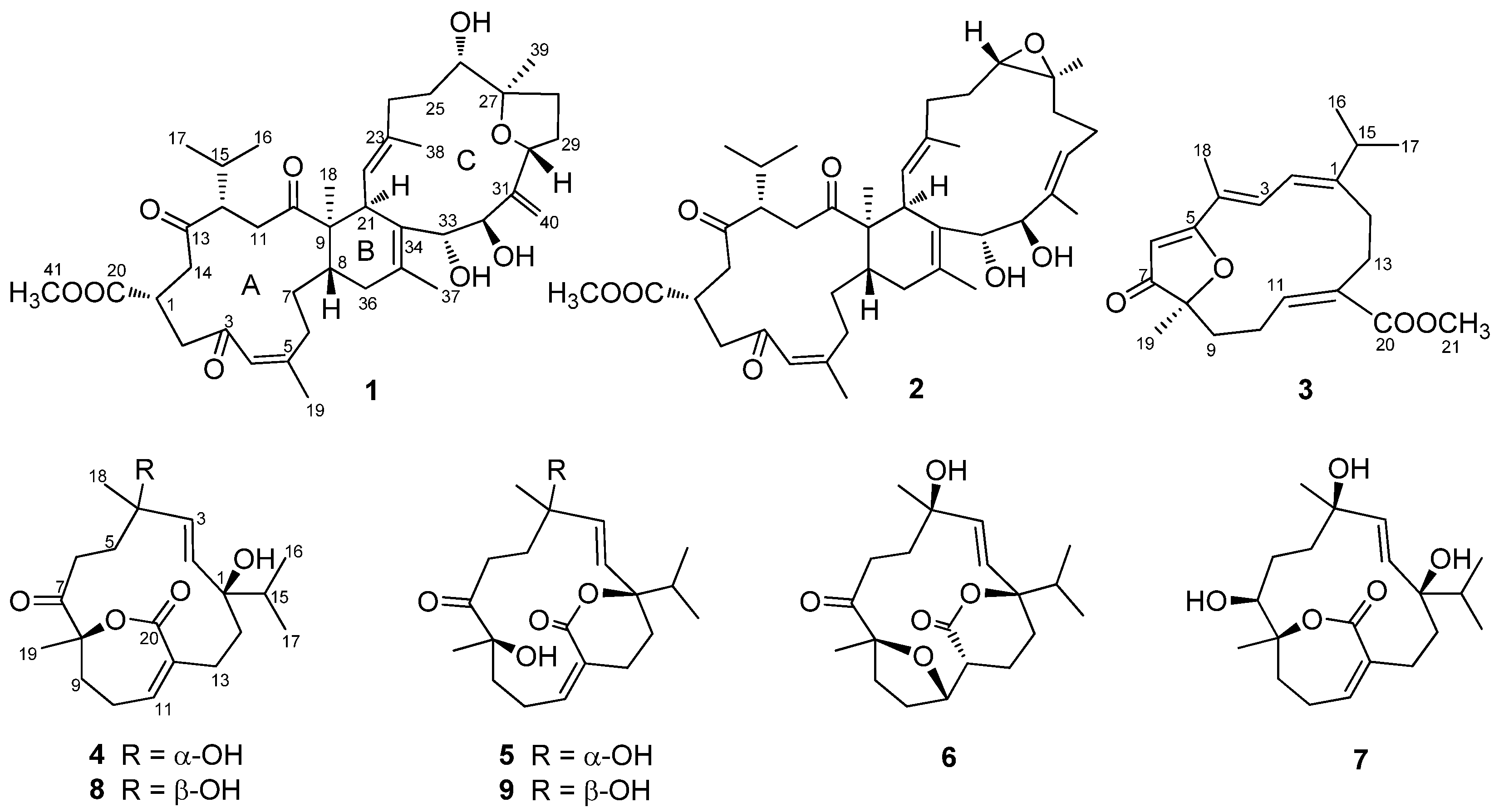
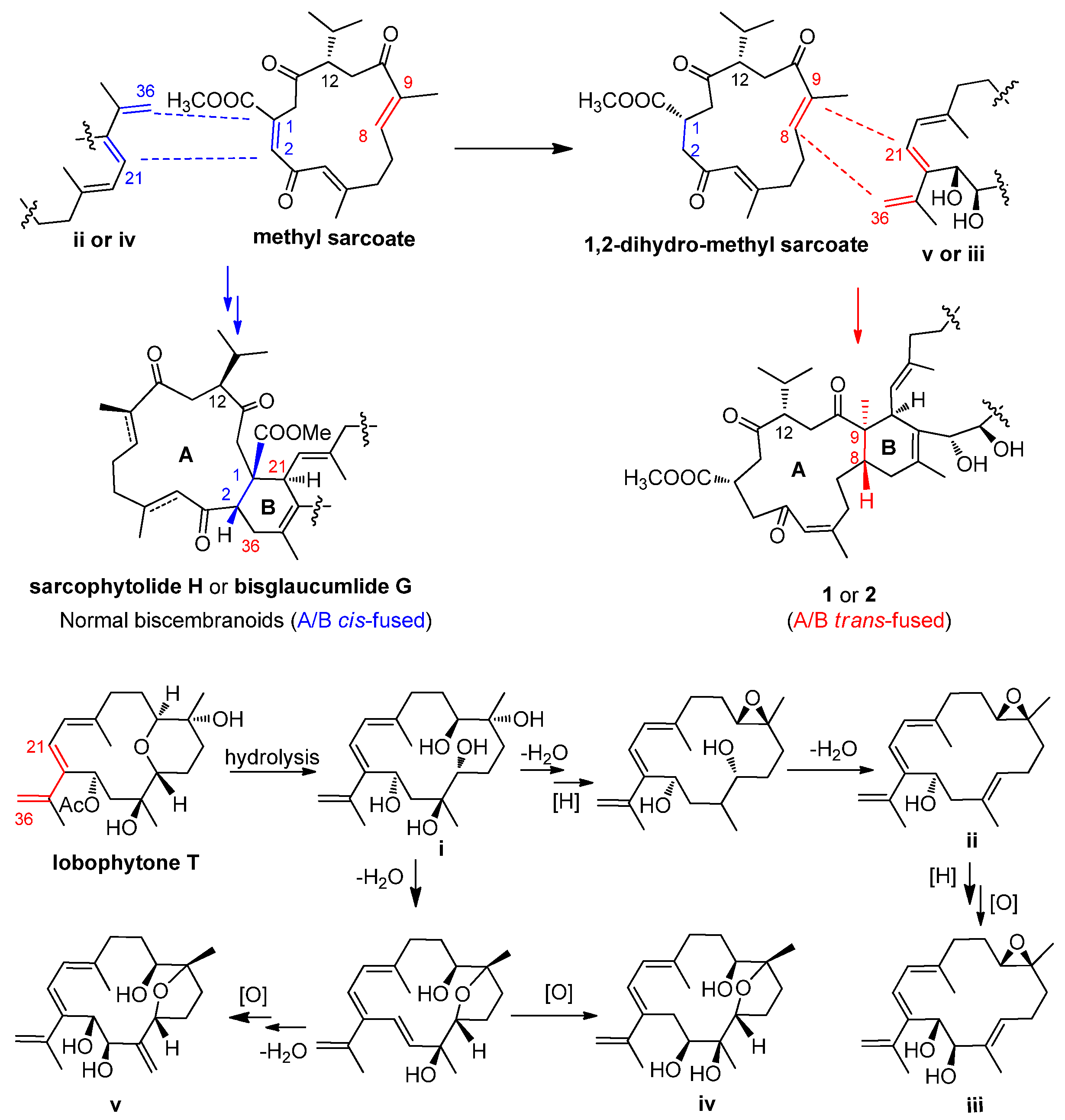
2. Results and Discussion
3. Materials and Methods
3.1. General Experimental Procedures
3.2. Animal Material
3.3. Extraction and Isolation
3.4. Chemical Transformation of 9 to 6
3.5. Cell Culture and Viability Assay
3.6. Measurement of NO Production
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Su, J.Y.; Long, K.H.; Pang, T.S.; He, C.H.; Clardy, J. The Structure of Methyl Isosartortuoate, a Novel Tetracyclic Tetraterpenoid from the Soft Coral Sarcophyton tortuosum. J. Am. Chem. Soc. 1986, 108, 177–178. [Google Scholar] [CrossRef]
- Kusumi, T.; Igari, M.; Ishitsuka, M.O.; Ichikawa, A.; Itezono, Y.; Nakayama, N.; Kakisawa, H. A Novel Chlorinated Biscembranoid from the Marine Soft Coral Sarcophyton glaucum. J. Org. Chem. 1990, 55, 6286–6289. [Google Scholar] [CrossRef]
- Leone, P.A.; Bowden, B.F.; Carroll, A.R.; Coll, J.C.; Meehan, G.V. Studies of Australian soft corals, XLIX. A new biscembranoid and its probable biosynthetic precursors from the soft coral Sarcophyton tortuosum. J. Nat. Prod. 1993, 56, 521–526. [Google Scholar] [CrossRef]
- Chen, B.W.; Chao, C.H.; Su, J.H.; Huang, C.Y.; Dai, C.F.; Wen, Z.H.; Sheu, J.H. A novel symmetric sulfur-containing biscembranoid from the Formosan soft coral Sinularia flexibilis. Tetrahedron Lett. 2010, 51, 5764–5766. [Google Scholar] [CrossRef]
- Yan, P.C.; Deng, Z.W.; van Ofwegen, L.; Proksch, P.; Lin, W.H. Lobophytones H–N, Biscembranoids from the Chinese Soft Coral Lobophytum pauciflorum. Chem. Pharm. Bull. 2010, 58, 1591–1595. [Google Scholar] [CrossRef] [PubMed]
- Zeng, L.M.; Lan, W.J.; Su, J.Y.; Zhang, G.W.; Feng, X.L.; Liang, Y.J.; Yang, X.P. Two New Cytotoxic Tetracyclic Tetraterpenoids from the Soft Coral Sarcophyton tortuosum. J. Nat. Prod. 2004, 67, 1915–1918. [Google Scholar] [CrossRef]
- Iwagawa, T.; Hashimoto, K.; Okamura, H.; Kurawaki, J.I.; Nakatani, M.; Hou, D.X.; Fujii, M.; Doe, M.; Morimoto, Y.; Takemura, K. Biscembranes from the Soft Coral Sarcophyton glaucum. J. Nat. Prod. 2006, 69, 1130–1133. [Google Scholar] [CrossRef] [PubMed]
- Bishara, A.; Rudi, A.; Benayahu, Y.; Kashman, Y. Three Biscembranoids and their Monomeric Counterpart Cembranoid, a Biogenetic Diels–Alder Precursor, from the Soft Coral Sarcophyton elegans. J. Nat. Prod. 2007, 70, 1951–1954. [Google Scholar] [CrossRef]
- Jia, R.; Guo, Y.W.; Chen, P.; Yang, Y.M.; Mollo, E.; Gavagnin, M.; Cimino, G. Biscembranoids and Their Probable Biogenetic Precursor from the Hainan Soft Coral Sarcophyton tortuosum. J. Nat. Prod. 2007, 70, 1158–1166. [Google Scholar] [CrossRef] [PubMed]
- Yan, P.C.; Lv, Y.; van Ofwegen, L.; Proksch, P.; Lin, W.H. Lobophytones A–G, New Isobiscembranoids from the Soft Coral Lobophytum pauciflorum. Org. Lett. 2010, 12, 2484–2487. [Google Scholar] [CrossRef]
- Sun, P.; Yu, Q.; Li, J.; Riccio, R.; Lauro, G.; Bifulco, G.; Kurtan, T.; Mandi, A.; Tang, H.; Li, T.J.; et al. Bissubvilides A and B, Cembrane–Capnosane Heterodimers from the Soft Coral Sarcophyton subviride. J. Nat. Prod. 2016, 79, 2552–2558. [Google Scholar] [CrossRef]
- Yan, P.C.; Deng, Z.W.; van Ofwegen, L.; Proksch, P.; Lin, W.H. Lobophytones U–Z1, Biscembranoids from the Chinese Soft Coral Lobophytum pauciflorum. Chem. Biodivers. 2011, 8, 1724–1734. [Google Scholar] [CrossRef] [PubMed]
- Xi, Z.F.; Bie, W.; Chen, W.; Liu, D.; van Ofwegen, L.; Proksch, P.; Lin, W.H. Sarcophytolides G–L, New Biscembranoids from the Soft Coral Sarcophyton elegans. Helv. Chim. Acta 2013, 96, 2218–2227. [Google Scholar] [CrossRef]
- Ichige, T.; Okano, Y.; Kanoh, N.; Nakata, M. Total Synthesis of Methyl Sarcophytoate. J. Am. Chem. Soc. 2007, 129, 9862–9863. [Google Scholar] [CrossRef] [PubMed]
- Ichige, T.; Okano, Y.; Kanoh, N.; Nakata, M. Total Synthesis of Methyl Sarcophytoate, a Marine Natural Biscembranoid. J. Org. Chem. 2009, 74, 230–243. [Google Scholar] [CrossRef] [PubMed]
- Xi, Z.F.; Bie, W.; Chen, W.; Liu, D.; van Ofwegen, L.; Proksch, P.; Lin, W.H. Sarcophyolides B–E, New Cembranoids from the Soft Coral Sarcophyton elegans. Mar. Drugs 2013, 11, 3186–3196. [Google Scholar] [CrossRef] [PubMed]
- Anjaneyulu, A.S. R.; Venugopal, M.J. R.V.; Sarada, P.; Rao, G.V.; Clardy, J.; Lobkovsky, E. Sarcophytin, A Novel Tetracyclic Diterpenoid From The Indian Ocean Soft Coral Sarcophyton elegans. Tetrahedron Lett. 1998, 39, 135–138. [Google Scholar] [CrossRef]
- Moldowan, J.M.; Tursch, B.M.; Djerassi, C. 24ξ-Methylcholestane-3β,5α,6β,25-tetrol 25-monoacetate, a novel polyhydroxylated steroid from an alcyonarian. Steroids 1974, 24, 387–398. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, J.S.; Liu, Q.; Tang, G.H.; Wang, H.S.; Fan, C.Q.; Yin, S. Bioactive Cembranoids from the South China Sea Soft Coral Sarcophyton elegans. Molecules 2015, 20, 13324–13335. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.Q.; Zhao, J.J.; Li, Z.Z.; Tang, G.H.; Zhao, Z.M.; Yin, S. Natural nitric oxide (NO) inhibitors from Chloranthus japonicus. Bioorg. Med. Chem. Lett. 2016, 26, 3163–3166. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.Y.; Cheng, B.; Zheng, Y.J.; Dong, Z.; Lin, S.L.; Tang, G.H.; Gu, Q.; Yin, S. Enantiomeric neolignans and sesquineolignans from Jatropha integerrima and their absolute configurations. RSC Adv. 2015, 5, 12202–12208. [Google Scholar] [CrossRef]
- Yan, P.C.; Deng, Z.W.; van Ofwegen, L.; Proksch, P.; Lin, W.H. Lobophytones O–T, New Biscembranoids and Cembranoid from Soft Coral Lobophytum pauciflorum. Mar. Drugs 2010, 8, 2837–2848. [Google Scholar] [CrossRef] [PubMed]
- Ichige, T.; Kamimura, S.; Mayumi, K.; Sakamoto, Y.; Terashita, S.; Ohteki, E.; Kanoh, N.; Nakata, M. Synthetic studies on biscembranoids: Asymmetric total synthesis of methyl sarcoate. Tetrahedron Lett. 2005, 46, 1263–1267. [Google Scholar] [CrossRef]
- Iwagawa, T.; Hashimoto, K.; Yokogawa, Y.; Okamura, H.; Nakatani, M.; Doe, M.; Morimoto, Y.; Takemura, K. Cytotoxic Biscembranes from the Soft Coral Sarcophyton glaucum. J. Nat. Prod. 2009, 72, 946–949. [Google Scholar] [CrossRef]
- McMurry, J.E.; Rico, J.G.; Shih, Y.N. Synthesis and stereochemistry of sarcophytol B: An anticancer cembranoid. Tetrahedron Lett. 1989, 30, 1173–1176. [Google Scholar] [CrossRef]
- Kurtan, T.; Jia, R.; Li, Y.; Pescitelli, G.; Guo, Y.W. Absolute Configuration of Highly Flexible Natural Products by the Solid-State ECD/TDDFT Method: Ximaolides and Sinulaparvalides. Eur. J. Org. Chem. 2012, 2012, 6722–6728. [Google Scholar] [CrossRef]
- Chimichi, S.; Boccalini, M.; Cosimelli, B.; Dall’Acqua, F.; Viola, G. New 5-(2-ethenylsubstituted)-3(2H)-furanones with in vitro antiproliferative activity. Tetrahedron 2003, 59, 5215–5223. [Google Scholar] [CrossRef]
- Liang, L.F.; Lan, L.F.; Taglialatela-Scafati, O.; Guo, Y.W. Sartrolides A–G and bissartrolide, new cembranolides from the South China Sea soft coral Sarcophyton trocheliophorum Marenzeller. Tetrahedron 2013, 69, 7381–7386. [Google Scholar] [CrossRef]

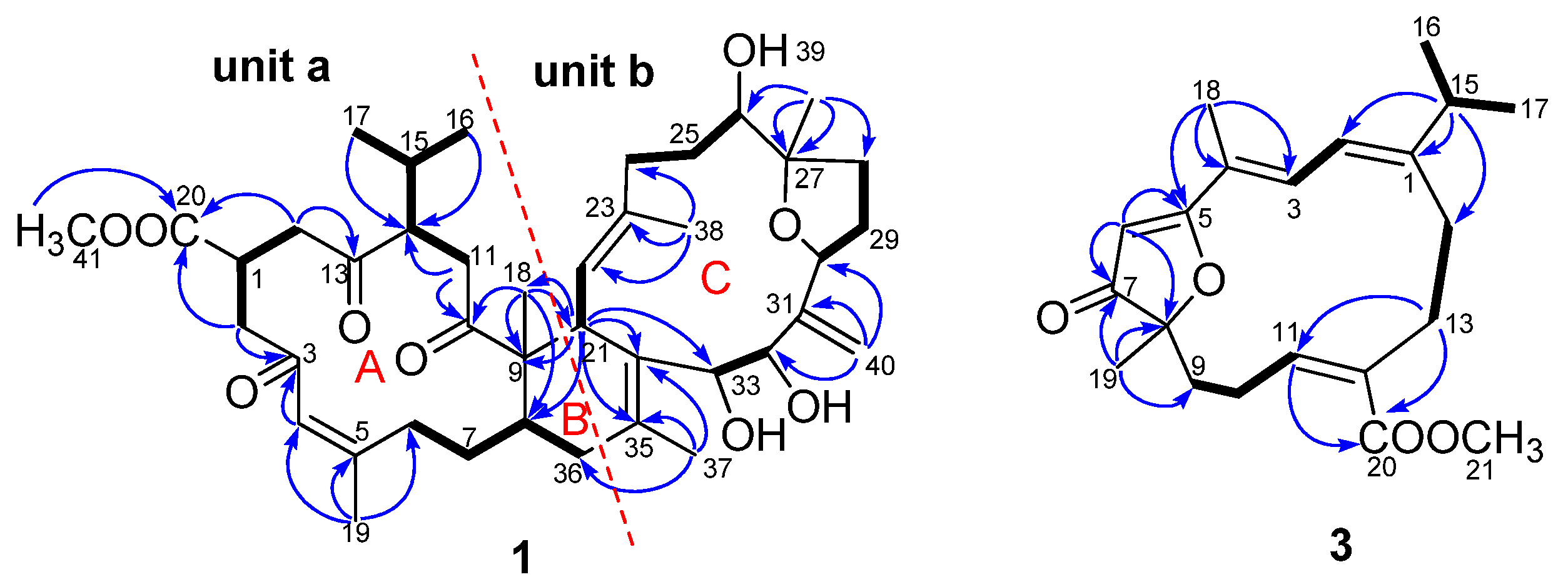
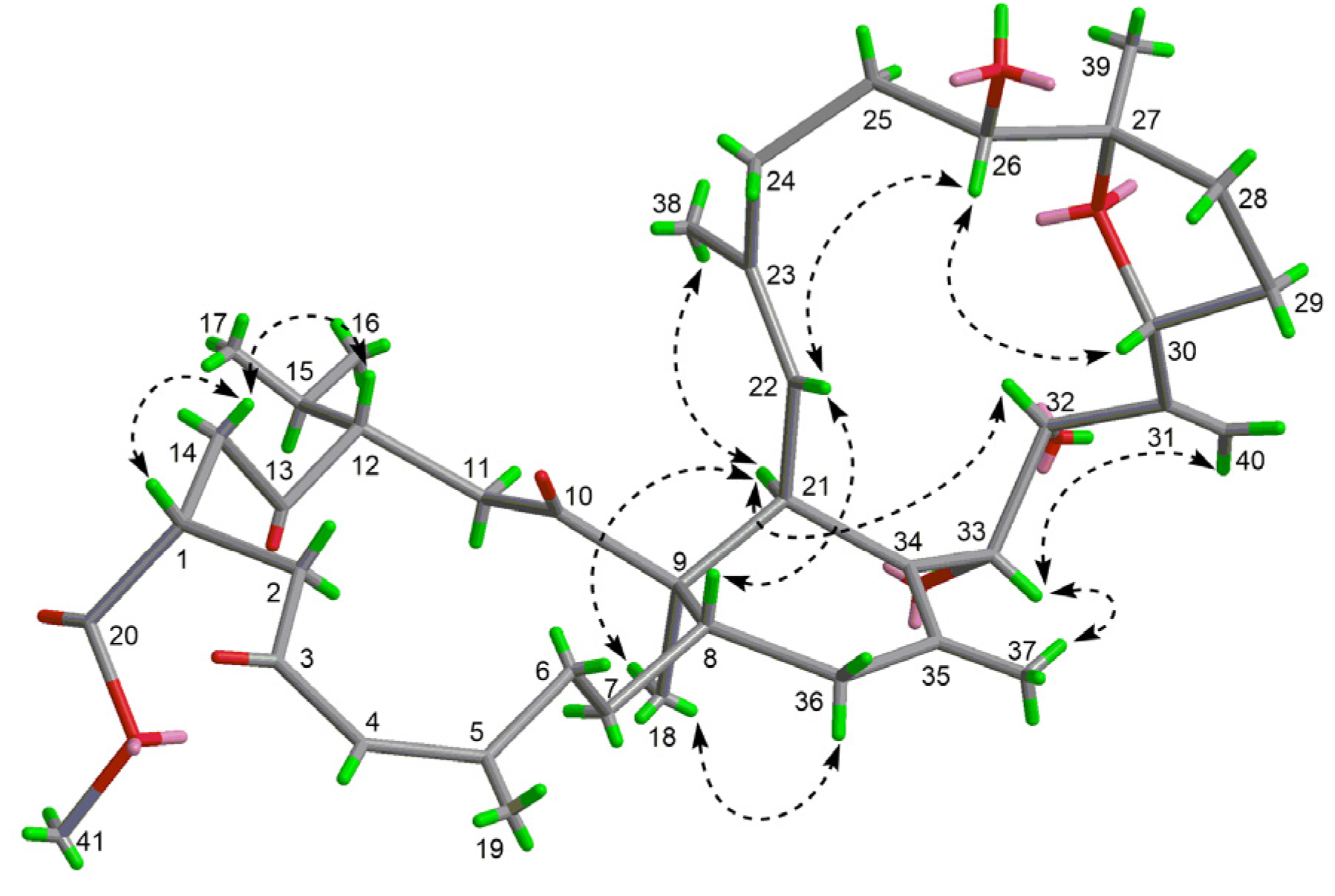
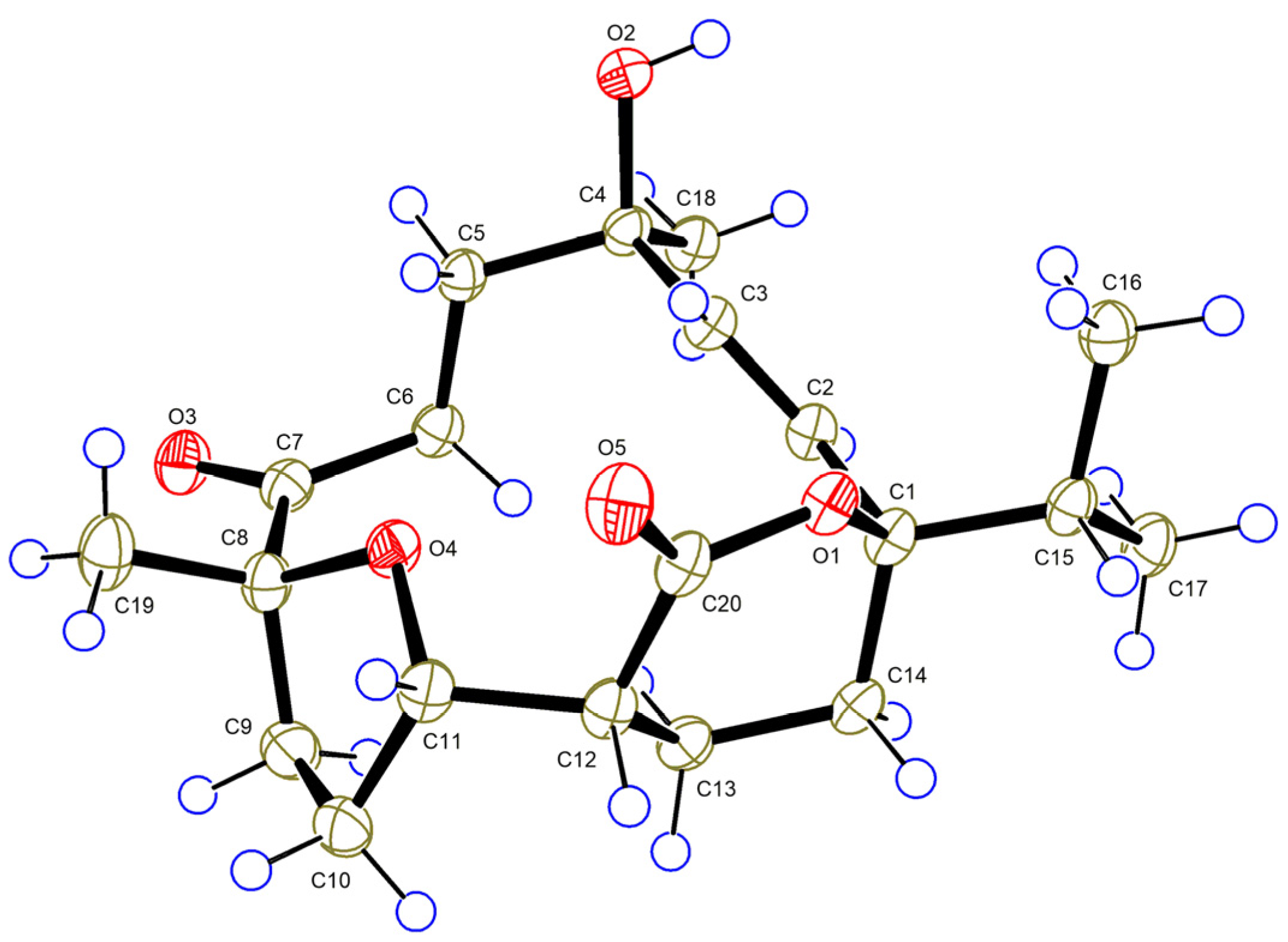
 ) and key HMBC (heteronuclear multiple-bond correlation spectroscopy) (
) and key HMBC (heteronuclear multiple-bond correlation spectroscopy) (  ) correlations of compounds 1 and 3.
) and key HMBC (heteronuclear multiple-bond correlation spectroscopy) ( ) correlations of compounds 1 and 3.
) correlations of compounds 1 and 3.
) and key HMBC (heteronuclear multiple-bond correlation spectroscopy) ( ) correlations of compounds 1 and 3.







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | 1 | 2 | ||
|---|---|---|---|---|
| δC, Type | δH, Multi. (J in Hz) | δC, Type | δH, Multi. (J in Hz) | |
| 1 | 36.7, CH | 3.15, m | 36.5, CH | 3.14, m |
| 2 | 43.0, CH2 | a 3.06, dd (17.6, 4.6) | 43.3, CH2 | a 3.07, dd (14.0, 4.4) |
| b 2.86, m | b 2.89, m | |||
| 3 | 203.4, C | 202.5, C | ||
| 4 | 127.4, CH | 5.96, d (1.2) | 126.7, CH | 6.03, d (1.1) |
| 5 | 154.1, C | 156.0, C | ||
| 6 | 34.1, CH2 | a 2.80, m | 33.2, CH2 | a 2.54, m |
| b 2.07, m | b 2.36, m | |||
| 7 | 36.7, CH2 | 1.05, m | 35.6, CH2 | 1.06, m |
| 8 | 33.2, CH | 2.30, m | 33.0, CH | 2.33, m |
| 9 | 55.9, C | 56.4, C | ||
| 10 | 215.5, C | 214.7, C | ||
| 11 | 41.1, CH2 | a 2.99, dd (18.0, 10.6) | 41.8, CH2 | 2.95, m |
| b 2.61, dd (18.0, 3.3) | ||||
| 12 | 54.8, CH | 2.51, m | 55.8, CH | 2.35, m |
| 13 | 215.4, C | 215.5, C | ||
| 14 | 44.1, CH2 | a 3.44, dd (18.1, 3.7) | 42.0, CH2 | a 3.18, dd (17.9, 3.8) |
| b 2.87, m | b 2.74, dd (17.9, 6.2) | |||
| 15 | 31.2, CH | 1.70, m | 31.4, CH | 1.72, m |
| 16 | 21.0, CH3 | 0.92, d (6.7) | 21.1, CH3 | 0.96, d (6.7) |
| 17 | 21.2, CH3 | 0.92, d (6.7) | 21.2, CH3 | 0.90, d (6.7) |
| 18 | 16.8, CH3 | 1.07, s | 16.5, CH3 | 1.13, s |
| 19 | 26.1, CH3 | 1.86, d (1.2) | 27.4, CH3 | 1.90, d (1.1) |
| 20 | 176.3, C | 176.6, C | ||
| 21 | 41.6, CH | 3.68, d (10.6) | 42.0, CH | 3.40, d (11.2) |
| 22 | 128.2, CH | 4.88, d (10.6) | 126.4, CH | 4.89, d (11.2) |
| 23 | 136.9, C | 133.1, C | ||
| 24 | 38.3, CH2 | 2.00, m | 35.5, CH2 | a 2.22, m |
| b 1.99, m | ||||
| 25 | 32.2, CH2 | a 1.79, m | 26.6, CH2 | 1.41, m |
| b 1.43, m | ||||
| 26 | 76.2, CH | 3.47, m | 64.0, CH | 2.90, m |
| 27 | 84.4, C | 61.2, C | ||
| 28 | 37.1, CH2 | a 2.08, m | 39.4, CH2 | a 1.99, m |
| b 1.69, m | b 1.01, m | |||
| 29 | 29.4, CH2 | 1.76, m | 24.3, CH2 | a 2.22, m |
| b 1.98, m | ||||
| 30 | 79.3, CH | 3.89, dd (10.2, 4.2) | 130.6, CH | 5.14, dd (9.1, 4.8) |
| 31 | 151.6, C | 135.6, C | ||
| 32 | 72.0, CH | 4.07, d (9.7) | 78.7, CH | 3.80, d (10.0) |
| 33 | 76.4, CH | 4.39, d (9.7) | 71.8, CH | 4.66, d (10.0) |
| 34 | 130.2, C | 130.7, C | ||
| 35 | 132.7, C | 132.7, C | ||
| 36 | 38.9, CH2 | a 2.45, m | 38.7, CH2 | a 2.55, m |
| b 1.62, m | b 1.65, m | |||
| 37 | 20.2, CH3 | 1.54, s | 19.6, CH3 | 1.71, s |
| 38 | 18.1, CH3 | 1.70, s | 18.9, CH3 | 1.59, s |
| 39 | 20.3, CH3 | 1.08, s | 16.3, CH3 | 1.30, s |
| 40 | 112.8, CH2 | a 5.47, s | 11.4, CH3 | 1.61, s |
| b 5.21, s | ||||
| 41 | 52.3, CH3 | 3.63, s | 52.3, CH3 | 3.64, s |
| No. | 3 a | 4 a | 5 a | 6 a | 7 b |
|---|---|---|---|---|---|
| 2 | 6.29, d (12.1) | 5.45, d (15.5) | 5.69, d (15.9) | 5.50, d (16.2) | 5.57, d (15.7) |
| 3 | 7.42, dd (12.1, 1.1) | 5.53, d (15.5) | 5.64, d (15.9) | 5.56, d (16.2) | 5.66, d (15.7) |
| 5 | a 1.93, m | a 1.75, m | 1.74, m | a 1.89, m | |
| b 1.79, m | b 1.59, ddd (13.4, 10.7, 7.0) | b 1.68, m | |||
| 6 | 5.64, s | 2.65, m | a 2.79, ddd (20.4, 10.9, 7.0) | a 2.65, m | 1.54, m |
| b 2.35, ddd (20.4, 10.7, 2.4) | b 2.35, m | ||||
| 7 | 4.37, m | ||||
| 9 | a 2.09, m | a 2.72, m | a 1.92, m | a 2.06, m | a 2.26, m |
| b 2.04, m | b 2.18, m | b 1.81, m | b 1.74, m | b 1.99, m | |
| 10 | a 2.74, m | 2.41, m | a 3.37, m | a 2.04, m | a 2.58, m |
| b 2.64, m | b 2.18, m | b 1.72, m | b 2.49, m | ||
| 11 | 5.99, t (6.4) | 6.09, m | 5.73, m | 4.87, m | 6.34, t (4.3) |
| 12 | 2.69, m | ||||
| 13 | a 2.73, m | 2.37, m | 2.46, m | 1.82, m | a 2.99, m |
| b 2.13, m | b 2.13, m | ||||
| 14 | 2.48, t (6.2) | 1.73, m | a 2.05, ddd (13.9, 6.6, 2.1) | a 2.04, m | a 2.00, m |
| b 1.77, m | b 1.82, m | b 1.73, m | |||
| 15 | 2.52, m | 1.72, m | 1.85, m | 1.89, m | 1.80, m |
| 16 | 1.12, d (6.8) | 0.92, d (6.8) | 0.96, d (6.7) | 0.98, d (6.8) | 0.84, d (6.8) |
| 17 | 1.12, d (6.8) | 0.92, d (6.8) | 0.98, d (6.7) | 1.00, d (6.8) | 0.89, d (6.8) |
| 18 | 1.96, d (1.1) | 1.29, s | 1.26, s | 1.27, s | 1.31, s |
| 19 | 1.43, s | 1.43, s | 1.23, s | 1.31, s | 1.32 s |
| 21 | 3.66, s |
| No. | 3 a | 4 a | 5 a | 6 a | 7 b |
|---|---|---|---|---|---|
| 1 | 164.1, C | 78.2, C | 88.1, C | 89.6, C | 76.1, C |
| 2 | 119.3, CH | 131.8, CH | 128.7, CH | 129.8, CH | 134.6, CH |
| 3 | 136.6, CH | 134.9, CH | 139.8, CH | 139.9, CH | 132.4, CH |
| 4 | 121.7, C | 73.6, C | 73.5, C | 74.0, C | 75.2, CH |
| 5 | 187.7, C | 34.3, CH2 | 38.1, CH2 | 36.9, CH2 | 34.3, CH2 |
| 6 | 100.4, CH | 34.3, CH2 | 36.1, CH2 | 34.5, CH2 | 25.7, CH2 |
| 7 | 209.6, C | 212.0, C | 217.8, C | 215.1, C | 66.1, CH |
| 8 | 91.3, C | 88.1, C | 79.8, C | 90.0, C | 83.6, C |
| 9 | 40.5, CH2 | 37.4, CH2 | 42.1, CH2 | 36.7, CH2 | 34.8, CH2 |
| 10 | 25.8, CH2 | 24.8, CH2 | 25.9, CH2 | 27.5, CH2 | 27.4, CH2 |
| 11 | 152.1, CH | 133.8, CH | 150.9, CH | 80.8, CH | 141.0, CH |
| 12 | 130.1, C | 137.5, C | 125.2, C | 44.1, CH | 134.1, C |
| 13 | 36.5, CH2 | 26.5, CH2 | 25.1, CH2 | 17.9, CH2 | 31.0, CH2 |
| 14 | 31.6, CH2 | 33.0, CH2 | 29.1, CH2 | 28.2, CH2 | 39.7, CH2 |
| 15 | 38.6, CH | 38.8, CH | 38.3, CH | 38.0, CH | 36.3, CH |
| 16 | 21.8, CH3 | 18.1, CH3 | 17.5, CH3 | 17.6, CH3 | 17.2, CH3 |
| 17 | 21.4, CH3 | 17.0, CH3 | 16.9, CH3 | 17.0, CH3 | 16.0, CH3 |
| 18 | 12.2, CH3 | 31.4, CH3 | 26.9, CH3 | 24.4, CH3 | 33.5, CH3 |
| 19 | 20.9, CH3 | 28.0, CH3 | 28.3, CH3 | 24.0, CH3 | 21.6, CH3 |
| 20 | 169.2, C | 172.1, C | 168.9, C | 175.3, C | 168.4, C |
| 21 | 51.5, CH3 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, W.; Zou, Y.-H.; Ge, M.-X.; Lou, L.-L.; Xu, Y.-S.; Ahmed, A.; Chen, Y.-Y.; Zhang, J.-S.; Tang, G.-H.; Yin, S. Biscembranoids and Cembranoids from the Soft Coral Sarcophyton elegans. Mar. Drugs 2017, 15, 85. https://doi.org/10.3390/md15040085
Li W, Zou Y-H, Ge M-X, Lou L-L, Xu Y-S, Ahmed A, Chen Y-Y, Zhang J-S, Tang G-H, Yin S. Biscembranoids and Cembranoids from the Soft Coral Sarcophyton elegans. Marine Drugs. 2017; 15(4):85. https://doi.org/10.3390/md15040085
Chicago/Turabian StyleLi, Wei, Yi-Hong Zou, Man-Xi Ge, Lan-Lan Lou, Yun-Shao Xu, Abrar Ahmed, Yun-Yun Chen, Jun-Sheng Zhang, Gui-Hua Tang, and Sheng Yin. 2017. "Biscembranoids and Cembranoids from the Soft Coral Sarcophyton elegans" Marine Drugs 15, no. 4: 85. https://doi.org/10.3390/md15040085
APA StyleLi, W., Zou, Y. -H., Ge, M. -X., Lou, L. -L., Xu, Y. -S., Ahmed, A., Chen, Y. -Y., Zhang, J. -S., Tang, G. -H., & Yin, S. (2017). Biscembranoids and Cembranoids from the Soft Coral Sarcophyton elegans. Marine Drugs, 15(4), 85. https://doi.org/10.3390/md15040085