Neuronal Nicotinic Acetylcholine Receptor Modulators from Cone Snails


Abstract
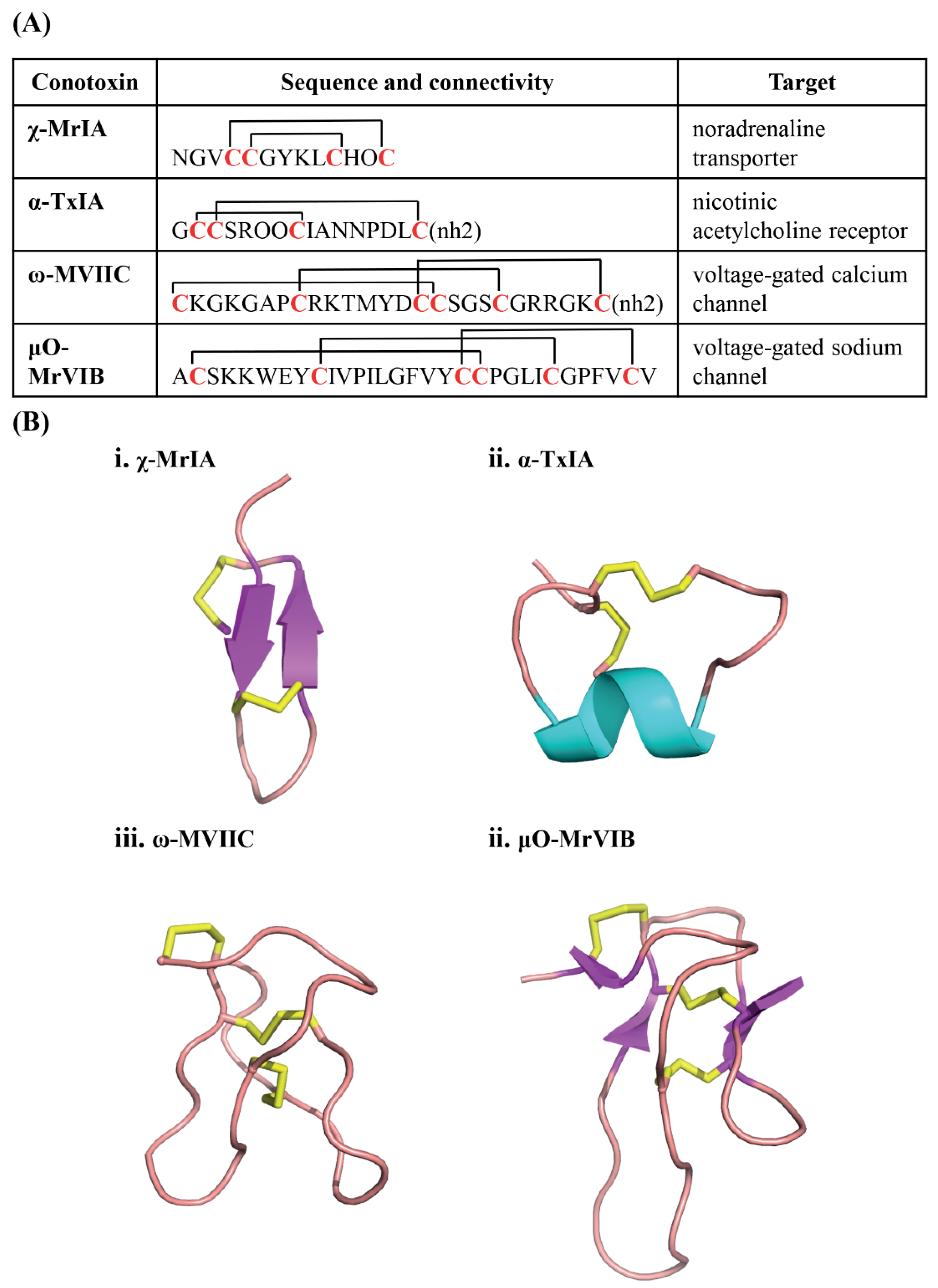
:1. Conotoxins—Venom Peptides from Marine Cone Snails
2. α-Conotoxins
3. α-Conotoxin Contributions to Determining nAChR Ligand Recognition Properties
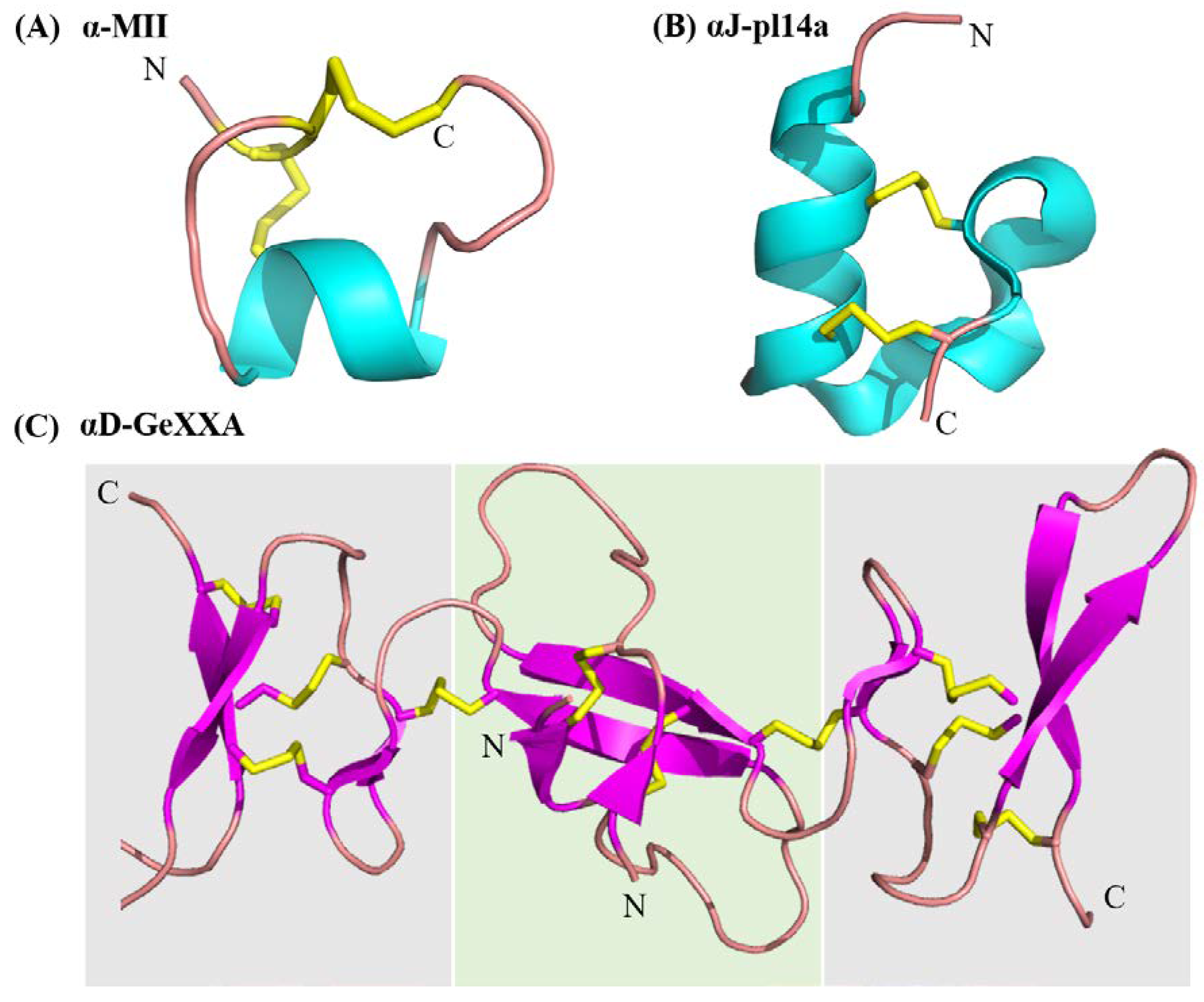
4. The Non-Classical α-Conotoxins
4.1. α-Conotoxins from the A Superfamily Exhibiting Unusual Characteristics
4.2. α-Conotoxins from Other Superfamilies Targeting nAChRs
4.2.1. S Superfamily
4.2.2. D Superfamily
4.2.3. B3 Superfamily
4.2.4. O1 Superfamily
4.2.5. T Superfamily
4.2.6. J Superfamily
4.2.7. M Superfamily
5. Applications
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Dutertre, S.; Jin, A.-H.; Kaas, Q.; Jones, A.; Alewood, P.F.; Lewis, R.J. Deep venomics reveals the mechanism for expanded peptide diversity in cone snail venom. Mol. Cell. Proteom. 2013, 12, 312–329. [Google Scholar] [CrossRef] [PubMed]
- Lewis, R.J.; Dutertre, S.; Vetter, I.; Christie, M.J. Conus venom peptide pharmacology. Pharmacol. Rev. 2012, 64, 259–298. [Google Scholar] [CrossRef] [PubMed]
- Dutertre, S.; Jin, A.-H.; Vetter, I.; Hamilton, B.; Sunagar, K.; Lavergne, V.; Dutertre, V.; Fry, B.G.; Antunes, A.; Venter, D.J.; et al. Evolution of separate predation-and defence-evoked venoms in carnivorous cone snails. Nat. Commun. 2014, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewis, R.J.; Garcia, M.L. Therapeutic potential of venom peptides. Nat. Rev. Drug. Discov. 2003, 2, 790–802. [Google Scholar] [CrossRef] [PubMed]
- Dutertre, S.; Jin, A.-H.; Alewood, P.F.; Lewis, R.J. Intraspecific variations in Conus geographus defence-evoked venom and estimation of the human lethal dose. Toxicon 2014, 91, 135–144. [Google Scholar] [CrossRef] [PubMed]
- Davis, J.; Jones, A.; Lewis, R.J. Remarkable inter-and intra-species complexity of conotoxins revealed by LC/MS. Peptides 2009, 30, 1222–1227. [Google Scholar] [CrossRef] [PubMed]
- Vetter, I.; Davis, J.L.; Rash, L.D.; Anangi, R.; Mobli, M.; Alewood, P.F.; Lewis, R.J.; King, G.F. Venomics: A new paradigm for natural products-based drug discovery. Amino Acids 2011, 40, 15–28. [Google Scholar] [CrossRef] [PubMed]
- Kaas, Q.; Craik, D.J. Conotoxins and other conopeptides. Outst. Mar. Mol. 2014. [Google Scholar] [CrossRef]
- Teichert, R.W.; Olivera, B.M.; McIntosh, J.M.; Bulaj, G.; Horvath, M.P. The molecular diversity of conoidean venom peptides and their targets: From basic research to therapeutic applications. Venom Drugs 2015, 2015, 163–203. [Google Scholar]
- Norton, R.S.; Olivera, B.M. Conotoxins down under. Toxicon 2006, 48, 780–798. [Google Scholar] [CrossRef] [PubMed]
- Azam, L.; McIntosh, J.M. Alpha-conotoxins as pharmacological probes of nicotinic acetylcholine receptors. Acta Pharmacol. Sin. 2009, 30, 771–783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prashanth, J.R.; Lewis, R.J.; Dutertre, S. Towards an integrated venomics approach for accelerated conopeptide discovery. Toxicon 2012, 60, 470–477. [Google Scholar] [CrossRef] [PubMed]
- Prashanth, J.R.; Brust, A.; Jin, A.; Alewood, P.F.; Dutertre, S.; Lewis, R.J. Cone snail venomics: From novel biology to novel therapeutics. Future Med. Chem. 2014, 6, 1659–1675. [Google Scholar] [CrossRef] [PubMed]
- Jin, A.-H.; Dutertre, S.; Kaas, Q.; Lavergne, V.; Kubala, P.; Lewis, R.J.; Alewood, P.F. Transcriptomic messiness in the venom duct of Conus miles contributes to conotoxin diversity. Mol. Cell. Proteom. 2013, 12, 3824–3833. [Google Scholar] [CrossRef] [PubMed]
- Lavergne, V.; Harliwong, I.; Jones, A.; Miller, D.; Taft, R.J.; Alewood, P.F. Optimized deep-targeted proteotranscriptomic profiling reveals unexplored Conus toxin diversity and novel cysteine frameworks. Proc. Natl. Acad. Sci. USA 2015, 112, E3782–E3791. [Google Scholar] [CrossRef] [PubMed]
- Halai, R.; Craik, D.J. Conotoxins: Natural product drug leads. Nat. Prod. Rep. 2009, 26, 526–536. [Google Scholar] [CrossRef] [PubMed]
- Brust, A.; Palant, E.; Croker, D.E.; Colless, B.; Drinkwater, R.; Patterson, B.; Schroeder, C.I.; Wilson, D.; Nielsen, C.K.; Smith, M.T.; et al. χ-Conopeptide pharmacophore development: Toward a novel class of norepinephrine transporter inhibitor (Xen2174) for pain. J. Med. Chem. 2009, 52, 6991–7002. [Google Scholar] [CrossRef] [PubMed]
- Kang, T.S.; Vivekanandan, S.; Jois, S.D.S.; Kini, R.M. Effect of C-terminal amidation on folding and disulfide-pairing of α-conotoxin ImI. Angew. Chem. Int. Ed. 2005, 44, 6333–6337. [Google Scholar] [CrossRef] [PubMed]
- Lovelace, E.S.; Gunasekera, S.; Alvarmo, C.; Clark, R.J.; Nevin, S.T.; Grishin, A.A.; Adams, D.J.; Craik, D.J.; Daly, N.L. Stabilization of α-conotoxin AuIB: Influences of disulfide connectivity and backbone cyclization. Antioxid. Redox Signal. 2011, 14, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Lewis, R.J. Discovery and development of the χ-conopeptide class of analgesic peptides. Toxicon 2012, 59, 524–528. [Google Scholar] [CrossRef] [PubMed]
- Dutertre, S.; Ulens, C.; Büttner, R.; Fish, A.; van Elk, R.; Kendel, Y.; Hopping, G.; Alewood, P.F.; Schroeder, C.; Nicke, A.; et al. AChBP-targeted α-conotoxin correlates distinct binding orientations with nAChR subtype selectivity. EMBO J. 2007, 26, 3858–3867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hillyard, D.R.; Monje, V.D.; Mintz, I.M.; Bean, B.P.; Nadasdi, L.; Ramachandran, J.; Miljanich, G.; Azimi-Zoonooz, A.; McIntosh, J.M.; Cruz, L.J.; et al. A new Conus peptide ligand for mammalian presynaptic Ca2+ channels. Neuron 1992, 9, 69–77. [Google Scholar] [CrossRef]
- Bulaj, G.; Zhang, M.-M.; Green, B.R.; Fiedler, B.; Layer, R.T.; Wei, S.; Nielsen, J.S.; Low, S.J.; Klein, B.D.; et al. Synthetic μO-conotoxin MrVIB blocks TTX-resistant sodium channel NaV1.8 and has a long-lasting analgesic activity. Biochemistry 2006, 45, 7404–7414. [Google Scholar]
- Dutertre, S.; Nicke, A.; Tsetlin, V.I. Nicotinic acetylcholine receptor inhibitors derived from snake and snail venoms. Neuropharmacology 2017, 127, 196–223. [Google Scholar] [CrossRef] [PubMed]
- McIntosh, J.M.; Santos, A.D.; Olivera, B.M. Conus peptides targeted to specific nicotinic acetylcholine receptor subtypes. Annu. Rev. Biochem. 1999, 68, 59–88. [Google Scholar] [CrossRef] [PubMed]
- Luo, S.; Christensen, S.; Zhangsun, D.; Wu, Y.; Hu, Y.; Zhu, X.; Chhabra, S.; Norton, R.S.; McIntosh, J.M. A novel inhibitor of α9α10 nicotinic acetylcholine receptors from Conus vexillum delineates a new conotoxin superfamily. PLoS ONE 2013, 8. [Google Scholar] [CrossRef] [PubMed]
- Loughnan, M.; Nicke, A.; Jones, A.; Schroeder, C.I.; Nevin, S.T.; Adams, D.J.; Alewood, P.F.; Lewis, R.J. Identification of a novel class of nicotinic receptor antagonists dimeric conotoxins VxXIIA, VxXIIB, and VxXIIC from conus vexillum. J. Biol. Chem. 2006, 281, 24745–24755. [Google Scholar] [CrossRef] [PubMed]
- Imperial, J.S.; Bansal, P.S.; Alewood, P.F.; Daly, N.L.; Craik, D.J.; Sporning, A.; Terlau, H.; López-Vera, E.; Bandyopadhyay, P.K.; Olivera, B.M. A novel conotoxin inhibitor of Kv1. 6 channel and nAChR subtypes defines a new superfamily of conotoxins. Biochemistry 2006, 45, 8331–8340. [Google Scholar] [CrossRef] [PubMed]
- Luo, S.; Zhangsun, D.; Harvey, P.J.; Kaas, Q.; Wu, Y.; Zhu, X.; Hu, Y.; Li, X.; Tsetlin, V.I.; Christensen, S.; et al. Cloning, synthesis, and characterization of αO-conotoxin GeXIVA, a potent α9α10 nicotinic acetylcholine receptor antagonist. Proc. Natl. Acad. Sci. USA 2015, 112, E4026–E4035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christensen, S.B.; Bandyopadhyay, P.K.; Olivera, B.M.; McIntosh, J.M. αS-conotoxin GVIIIB potently and selectively blocks α9α10 nicotinic acetylcholine receptors. Biochem. Pharmacol. 2015, 96, 349–356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.; Du, T.; Liu, Z.; Wang, S.; Wu, Y.; Ding, J.; Jiang, L.; Dai, Q. Characterization of a T-superfamily conotoxin TxVC from conus textile that selectively targets neuronal nAChR subtypes. Biochem. Biophys. Res. Commun. 2014, 454, 151–156. [Google Scholar] [CrossRef] [PubMed]
- Loughnan, M.L.; Nicke, A.; Lawrence, N.; Lewis, R.J. Novel αD-conopeptides and their precursors identified by cDNA cloning define the D-conotoxin superfamily. Biochemistry 2009, 48, 3717–3729. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Zhang, T.; Kompella, S.N.; Yan, M.; Lu, A.; Wang, Y.; Shao, X.; Chi, C.; Adams, D.J.; Ding, J.; et al. Conotoxin αD-GeXXA utilizes a novel strategy to antagonize nicotinic acetylcholine receptors. Sci. Rep. 2015, 5. [Google Scholar] [CrossRef] [PubMed]
- Kaas, Q.; Yu, R.; Jin, A.; Dutertre, S.; Craik, D.J. ConoServer: Updated content, knowledge, and discovery tools in the conopeptide database. Nucl. Acids Res. 2011, 40, D325–D330. [Google Scholar] [CrossRef] [PubMed]
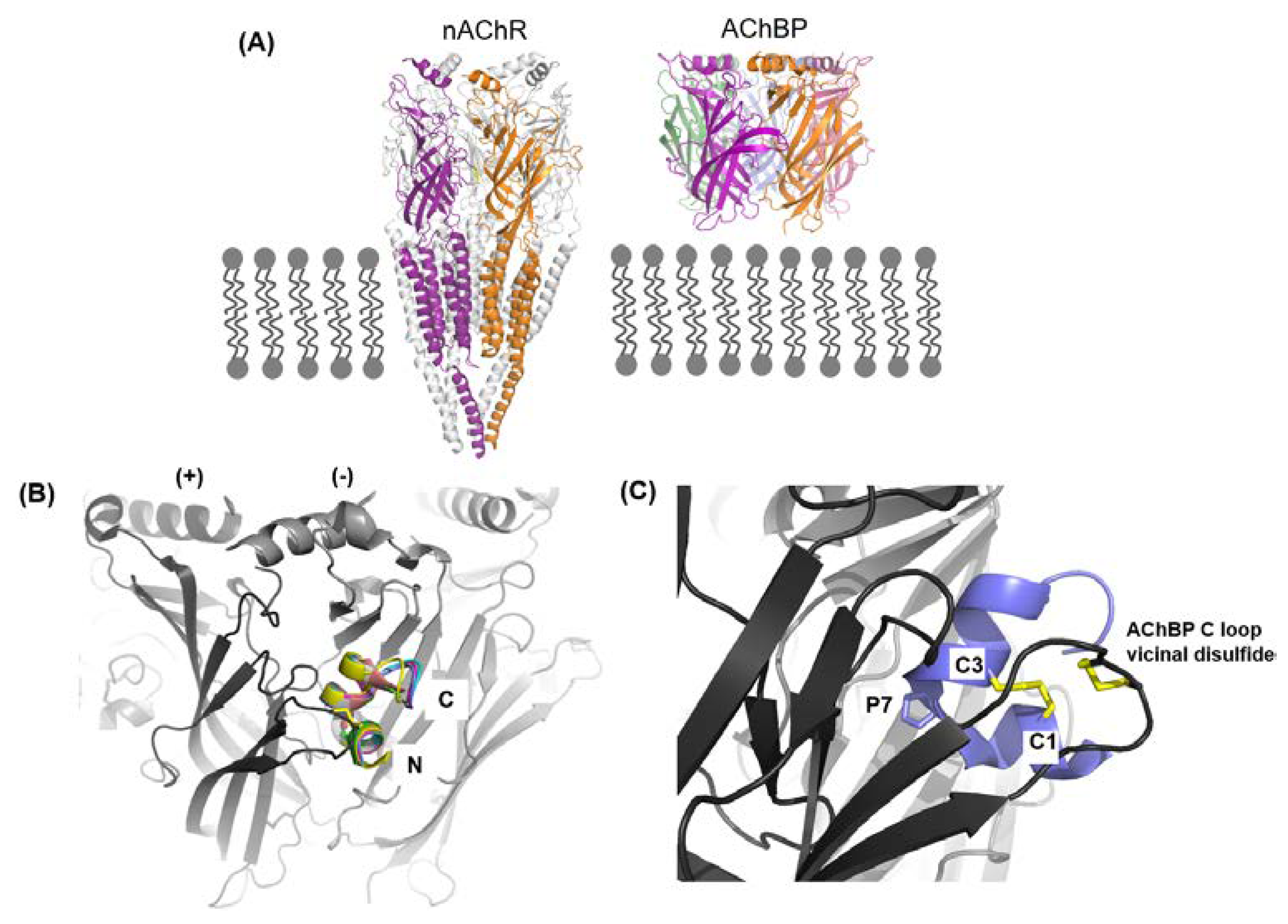
- Hurst, R.; Rollema, H.; Bertrand, D. Nicotinic acetylcholine receptors: From basic science to therapeutics. Pharmacol. Ther. 2013, 137, 22–54. [Google Scholar] [CrossRef] [PubMed]
- Changeux, J.-P. The nicotinic acetylcholine receptor: The founding father of the pentameric ligand-gated ion channel superfamily. J. Biol. Chem. 2012, 287, 40207–40215. [Google Scholar] [CrossRef] [PubMed]
- Sharma, G.; Vijayaraghavan, S. Nicotinic receptor signaling in nonexcitable cells. J. Neurobiol. 2002, 53, 524–534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Yu, M.; Ochani, M.; Amella, C.A.; Tanovic, M.; Susarla, S.; Li, J.H.; Wang, H.; Yang, H.; Ulloa, L.; et al. Nicotinic acetylcholine receptor alpha7 subunit is an essential regulator of inflammation. Nature 2003, 6921, 384–388. [Google Scholar]
- Stokes, C.; Treinin, M.; Papke, R.L. Looking below the surface of nicotinic acetylcholine receptors. Trends Pharmacol. Sci. 2015, 36, 514–523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dani, J.A. Overview of nicotinic receptors and their roles in the central nervous system. Biol. Psychiatr. 2001, 49, 166–174. [Google Scholar] [CrossRef]
- Papke, R.L. Merging old and new perspectives on nicotinic acetylcholine receptors. Biochem. Pharm. 2014, 89, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lebbe, E.K.; Peigneur, S.; Wijesekara, I.; Tytga, J. Conotoxins targeting nicotinic acetylcholine receptors: An overview. Mar. Drugs 2014, 12, 2970–3004. [Google Scholar] [CrossRef] [PubMed]
- Giribaldi, J.; Dutertre, S. α-Conotoxins to explore the molecular, physiological and pathophysiological functions of neuronal nicotinic acetylcholine receptors. Neurosci. Lett. 2017. [Google Scholar] [CrossRef] [PubMed]
- Dutton, J.L.; Craik, D.J. Alpha conotoxins nicotinic acetylcholine receptor antagonists as pharmacological tools and potential drug leads. Curr. Med. Chem. 2001, 8, 327–344. [Google Scholar] [CrossRef] [PubMed]
- Nicke, A.; Wonnacott, S.; Lewis, R.J. α-Conotoxins as tools for the elucidation of structure and function of neuronal nicotinic acetylcholine receptor subtypes. Eur. J. Biochem. 2004, 271, 2305–2319. [Google Scholar] [CrossRef] [PubMed]
- Corzo, G.; Escoubas, P.; Stankiewicz, M.; Pelhate, M.; Kristensen, C.P.; Nakajima, T. Isolation, synthesis and pharmacological characterization of δ-palutoxins IT, novel insecticidal toxins from the spider Paracoelotes luctuosus (Amaurobiidae). Eur. J. Biochem. 2000, 267, 5783–5795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dajas-Bailador, F.; Wonnacott, S. Nicotinic acetylcholine receptors and the regulation of neuronal signalling. Trends Pharmacol. Sci. 2004, 25, 317–324. [Google Scholar] [CrossRef] [PubMed]
- Dani, J.A.; Bertrand, D. Nicotinic acetylcholine receptors and nicotinic cholinergic mechanisms of the central nervous system. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 699–729. [Google Scholar] [CrossRef] [PubMed]
- Jensen, A.A.; Frølund, B.; Liljefors, T.; Krogsgaard-Larsen, P. Neuronal nicotinic acetylcholine receptors: Structural revelations, target identifications, and therapeutic inspirations. J. Med. Chem. 2005, 48, 4705–4745. [Google Scholar] [CrossRef] [PubMed]
- Kalamida, D.; Poulas, K.; Avramopoulou, V.; Fostieri, E.; Lagoumintzis, G.; Lazaridis, K.; Sideri, A.; Zouridakis, M.; Tzartos, S.J. Muscle and neuronal nicotinic acetylcholine receptors. FEBS J. 2007, 274, 3799–3845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lagoumintzis, G.; Poulas, K.; Avramopoulou, V.; Fostieri, E.; Lagoumintzis, G.; Lazaridis, K.; Sideri, A.; Zouridakis, M.; Tzartos, S.J. Muscle and neuronal nicotinic acetylcholine receptors Structure, function and pathogenicity. FEBS J. 2007, 274, 3799–3845. [Google Scholar]
- Lindstrom, J. Neuronal nicotinic acetylcholine receptors. In Ion Channels; Springer: New York, NY, USA, 1996. [Google Scholar]
- Lloyd, G.K.; Williams, M. Neuronal nicotinic acetylcholine receptors as novel drug targets. J. Pharmacol. Exp. Ther. 2000, 292, 461–467. [Google Scholar] [PubMed]
- Millar, N.S.; Gotti, C. Diversity of vertebrate nicotinic acetylcholine receptors. Neuropharmacology 2009, 56, 237–246. [Google Scholar] [CrossRef] [PubMed]
- Taly, A.; Charon, S. α7 nicotinic acetylcholine receptors: A therapeutic target in the structure era. Curr. Drug Targets 2012, 13, 695–706. [Google Scholar] [CrossRef] [PubMed]
- Unwin, N. Refined structure of the nicotinic acetylcholine receptor at 4 Å resolution. J. Mol. Biol. 2005, 346, 967–989. [Google Scholar] [CrossRef] [PubMed]
- Rucktooa, P.; Smit, A.B.; Sixma, T.K. Insight in nAChR subtype selectivity from AChBP crystal structures. Biochem. Pharmacol. 2009, 78, 777–787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, P.; Talley, T.T.; Hansen, S.B.; Hibbs, R.E.; Shi, J. Structure-guided drug design: Conferring selectivity among neuronal nicotinic receptor and acetylcholine-binding protein subtypes. Biochem. Pharmacol. 2007, 74, 1164–1171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shahsavar, A.; Gajhede, M.; Kastrup, J.S.; Balle, T. Structural studies of nicotinic acetylcholine receptors: Using acetylcholine-binding protein as a structural surrogate. Basic Clin. Pharmacol. Toxicol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Sixma, T.K.; Smit, A.B. Acetylcholine binding protein (AChBP): A secreted glial protein that provides a high-resolution model for the extracellular domain of pentameric ligand-gated ion channels. Annu. Rev. Biophys. Biomol. Struct. 2003, 32, 311–334. [Google Scholar] [CrossRef] [PubMed]
- Smit, A.B.; Celie, P.H.N.; Kasheverov, I.E.; Mordvintsev, D.Y.; van Nierop, P.; Bertrand, D.; Tsetlin, V.; Sixma, T.K. Acetylcholine-binding proteins. J. Mol. Neurosci. 2006, 30, 9–10. [Google Scholar] [CrossRef]
- Brejc, K.; van Dijk, W.J.; Klaassen, R.V.; Schuurmans, M.; van der Oost, J.; Smit, A.B.; Sixma, T.K. Crystal structure of an ACh-binding protein reveals the ligand-binding domain of nicotinic receptors. Nature 2001, 411, 269–276. [Google Scholar]
- Zouridakis, M.; Giastas, P.; Zarkadas, E.; Chroni-Tzartou, D.; Bregestovski, P.; Tzartos, S.J. Crystal structures of free and antagonist-bound states of human α9 nicotinic receptor extracellular domain. Nat. Struct. Mol. Biol. 2014, 21, 976–980. [Google Scholar] [CrossRef] [PubMed]
- Kouvatsos, N.; Giastas, P.; Chroni-Tzartou, D.; Poulopoulou, C.; Tzartos, S.J. Crystal structure of a human neuronal nAChR extracellular domain in pentameric assembly: Ligand-bound α2 homopentamer. Proc. Natl. Acad. Sci. USA 2016, 113, 9635–9640. [Google Scholar] [CrossRef] [PubMed]
- Morales-Perez, C.L.; Noviello, C.M.; Hibbs, R.E. X-ray structure of the human α4β2 nicotinic receptor. Nature 2016, 538, 411–415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
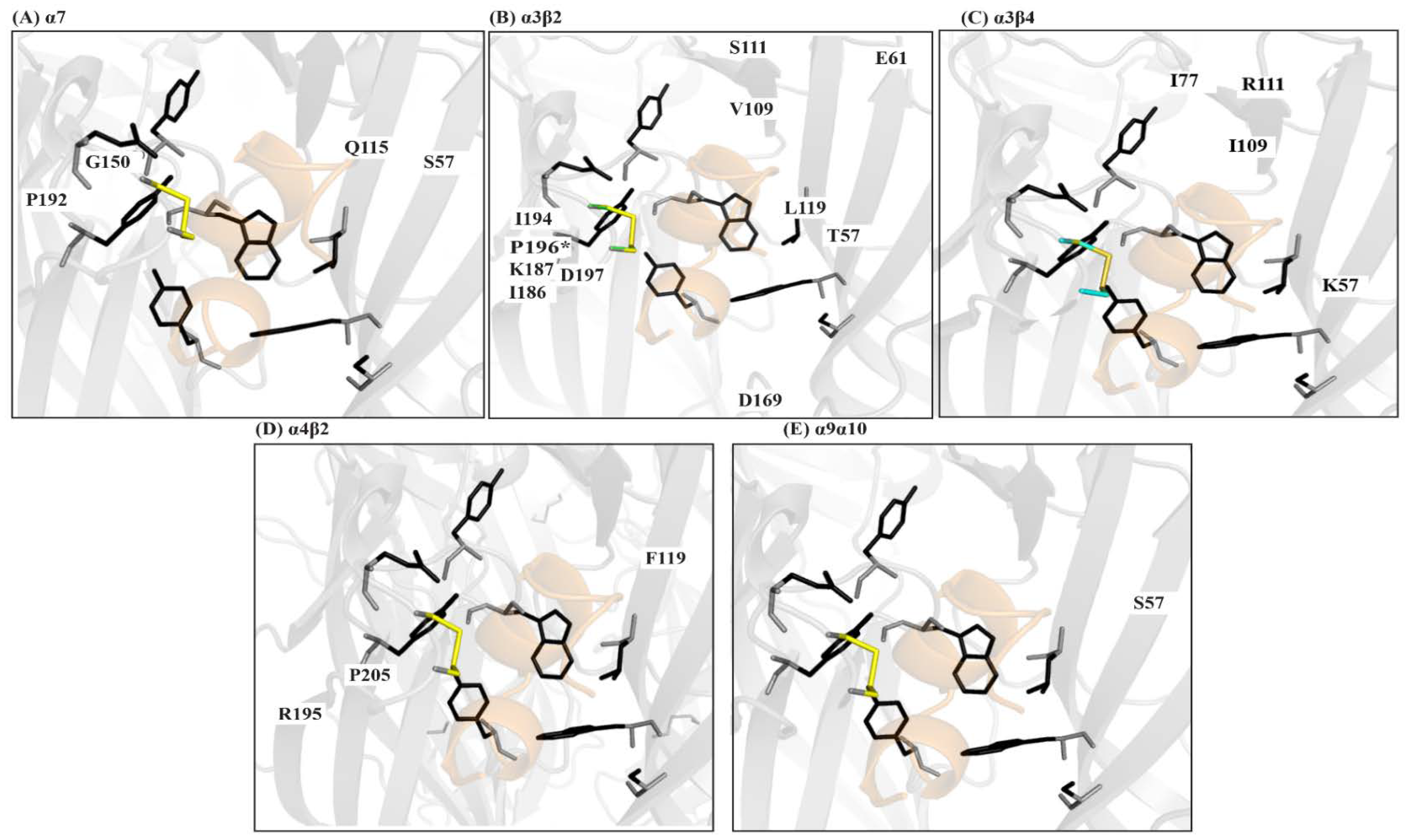
- Xu, M.; Zhu, X.; Yu, J.; Yu, J.; Luo, S.; Wang, X. The crystal structure of Ac-AChBP in complex with α-conotoxin LvIA reveals the mechanism of its selectivity towards different nAChR subtypes. Protein Cell 2017, 8, 675–685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hansen, S.B.; Sulzenbacher, G.; Huxford, T.; Marchot, P.; Taylor, P.; Bourne, Y. Structures of aplysia AChBP complexes with nicotinic agonists and antagonists reveal distinctive binding interfaces and conformations. EMBO J. 2005, 24, 3635–3646. [Google Scholar] [CrossRef] [PubMed]
- Celie, P.H.; Kasheverov, I.E.; Mordvintsev, D.Y.; Hogg, R.C.; van Nierop, P.; van Elk, R.; van Rossum-Fikkert, S.E.; Zhmak, M.N.; Bertrand, D.; Tsetlin, V.; et al. Crystal structure of nicotinic acetylcholine receptor homolog AChBP in complex with an α-conotoxin PnIA variant. Nat. Struct. Mol. Biol. 2005, 12, 582–588. [Google Scholar] [CrossRef] [PubMed]
- Abraham, N.; Healy, M.; Ragnarsson, L.; Brust, A.; Alewood, P.F.; Lewis, R.J. Structural mechanisms for α-conotoxin activity at the human α3β4 nicotinic acetylcholine receptor. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Lin, B.; Xu, M.; Zhu, X.; Wu, Y.; Liu, X.; Zhangsun, D.; Hu, Y.; Xiang, S.; Kasheverov, I.E.; Tsetlin, V.I.; et al. From crystal structure of α-conotoxin GIC in complex with Ac-AChBP to molecular determinants of its high selectivity for α3β2 nAChR. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Dutertre, S.; Lewis, R.J. Toxin insights into nicotinic acetylcholine receptors. Biochem. Pharmcol. 2006, 72, 661–670. [Google Scholar] [CrossRef] [PubMed]
- Fainzilber, M.; Hasson, A.; Oren, R.; Burlingame, A.L.; Gordon, D.; Spira, M.E.; Zlotkin, E. New mollusk-specific. alpha.-conotoxins block Aplysia neuronal acetylcholine receptors. Biochemistry 1994, 33, 9523–9529. [Google Scholar] [CrossRef] [PubMed]
- Hogg, R.C.; Miranda, L.P.; Craik, D.J.; Lewis, R.J.; Alewood, P.F.; Adams, D.J. Single amino acid substitutions in α-conotoxin PnIA shift selectivity for subtypes of the mammalian neuronal nicotinic acetylcholine receptor. J. Biol. Chem. 1999, 274, 36559–36564. [Google Scholar] [CrossRef] [PubMed]
- Luo, S.; Nguyen, T.A.; Cartier, G.E.; Olivera, B.M.; Yoshikami, D.; McIntosh, J.M. Single-residue alteration in α-conotoxin PnIA switches its nAChR subtype selectivity. Biochemistry 1999, 38, 14542–14548. [Google Scholar] [CrossRef] [PubMed]
- McIntosh, J.M.; Yoshikami, D.; Mahe, E.; Nielsen, D.B.; Rivier, J.E.; Gray, W.R.; Olivera, B.M. A nicotinic acetylcholine receptor ligand of unique specificity, alpha-conotoxin ImI. J. Biol. Chem. 1994, 269, 16733–16739. [Google Scholar] [PubMed]
- Inserra, M.C.; Kompella, S.N.; Vetter, I.; Brust, A.; Daly, N.L.; Cuny, H.; Craik, D.J.; Alewood, P.F.; Adams, D.J.; Lewis, R.J. Isolation and characterization of α-conotoxin LsIA with potent activity at nicotinic acetylcholine receptors. Biochem. Pharmacol. 2013, 86, 791–799. [Google Scholar] [CrossRef] [PubMed]
- Jin, A.-H.; Vetter, I.; Dutertre, S.; Abraham, N.; Emidio, N.B.; Inserra, M.; Murali, S.S.; Christie, M.J.; Alewood, P.F.; Lewis, R.J. MrIC, a novel α-conotoxin agonist in the presence of PNU at endogenous α7 nicotinic acetylcholine receptors. Biochemistry 2014. [Google Scholar] [CrossRef] [PubMed]
- Mueller, A.; Starobova, H.; Inserra, M.C.; Jin, A.H.; Deuis, J.R.; Dutertre, S.; Lewis, R.J.; Alewood, P.F.; Daly, N.L.; Vetter, I. α-conotoxin MrIC is a biased agonist at α 7 nicotinic acetylcholine receptors. Biochem. Pharmacol. 2015, 94, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Dutertre, S.; Nicke, A.; Lewis, R.J. β2 subunit contribution to 4/7 α-conotoxin binding to the nicotinic acetylcholine receptor. J. Biol. Chem. 2005, 280, 30460–30468. [Google Scholar] [CrossRef] [PubMed]
- Luo, S.; Zhangsun, D.; Schroeder, C.I.; Zhu, X.; Hu, Y.; Wu, Y.; Weltzin, M.M.; Eberhard, S.; Kaas, Q.; Craik, D.J.; et al. A novel α4/7-conotoxin LvIA from Conus lividus that selectively blocks α3β2 vs. α6/α3β2β3 nicotinic acetylcholine receptors. FASEB J. 2014, 28, 1842–1853. [Google Scholar] [PubMed] [Green Version]
- Zhangsun, D.; Zhu, X.; Wu, Y.; Hu, Y.; Kaas, Q.; Craik, D.J.; McIntosh, J.M.; Luo, S. Key residues in the nicotinic acetylcholine receptor β2 subunit contribute to α-conotoxin LvIA binding. J. Biol. Chem. 2015, 290, 9855–9862. [Google Scholar] [CrossRef] [PubMed]
- Kompella, S.N.; Cuny, H.; Hung, A.; Adams, D.J. Molecular basis for differential sensitivity of α-conotoxin RegIIA at rat and human neuronal nicotinic acetylcholine receptors. Mol. Pharmacol. 2015, 88, 993–1001. [Google Scholar] [CrossRef] [PubMed]
- Luo, S.; Zhangsun, D.; Zhu, X.; Wu, Y.; Hu, Y.; Christensen, S.; Harvey, P.J.; Akcan, M.; Craik, D.J.; McIntosh, J.M. Characterization of a novel α-conotoxin TxID from Conus textile that potently blocks rat α3β4 nicotinic acetylcholine receptors. J. Med. Chem. 2013, 56, 9655–9663. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Zhangsun, D.; Zhu, X.; Kaas, Q.; Zhangsun, M.; Harvey, P.J.; Craik, D.J.; McIntosh, J.M.; Luo, S. α-Conotoxin [S9A] TxID potently discriminates between α3β4 and α6/α3β4 nicotinic acetylcholine receptors. J. Med. Chem. 2017, 60, 5826–5833. [Google Scholar] [CrossRef] [PubMed]
- Kompella, S.N.; Hung, A.; Clark, R.J.; Marí, F.; Adams, D.J. Alanine scan of α-conotoxin RegIIA reveals a selective α3β4 nicotinic acetylcholine receptor antagonist. J. Biol. Chem. 2015, 290, 1039–1048. [Google Scholar] [CrossRef] [PubMed]
- Ellison, M.; Haberlandt, C.; Gomez-Casati, M.E.; Watkins, M.; Elgoyhen, A.B.; McIntosh, J.M.; Olivera, B.M. α-RgIA: A novel conotoxin that specifically and potently blocks the α9α10 nAChR. Biochemistry 2006, 45, 1511–1517. [Google Scholar] [CrossRef] [PubMed]
- Ellison, M.; Feng, Z.; Park, A.J.; Zhang, X.; Olivera, B.M.; McIntosh, J.M.; Norton, R.S. α-RgIA, a novel conotoxin that blocks the α9α10 nAChR: Structure and identification of key receptor-binding residues. J. Mol. Biol. 2008, 377, 1216–1227. [Google Scholar] [CrossRef] [PubMed]
- Millard, E.L.; Nevin, S.T.; Loughnan, M.L.; Nicke, A.; Clark, R.J.; Alewood, P.F.; Lewis, R.J.; Adams, D.J.; Craik, D.J.; Daly, N.L. Inhibition of neuronal nicotinic acetylcholine receptor subtypes by α-Conotoxin GID and analogues. J. Biol. Chem. 2009, 284, 4944–4951. [Google Scholar] [CrossRef] [PubMed]
- Nicke, A.; Loughnan, M.L.; Millard, E.L.; Alewood, P.F.; Adams, D.J.; Daly, N.L.; Craik, D.J.; Lewis, R.J. Isolation, structure, and activity of GID, a novel α4/7-conotoxin with an extended N-terminal sequence. J. Biol. Chem. 2003, 278, 3137–3144. [Google Scholar] [CrossRef] [PubMed]
- Suresh, A.; Hung, A. Molecular simulation study of the unbinding of α-conotoxin [Υ4E] GID at the α7 and α4β2 neuronal nicotinic acetylcholine receptors. J. Mol. Graph. Model. 2016, 70, 109–121. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, J.; Yongye, A.B.; Chang, Y.; Gyanda, R.; Medina-Franco, J.L.; Armishaw, C.J. Design and synthesis of α-conotoxin GID analogues as selective α4β2 nicotinic acetylcholine receptor antagonists. Pept. Sci. 2014, 102, 78–87. [Google Scholar] [CrossRef] [PubMed]
- Beissner, M.; Dutertre, S.; Schemm, R.; Danker, T.; Sporning, A.; Grubmüller, H.; Nicke, A. Efficient binding of 4/7 α-conotoxins to nicotinic α4β2 receptors is prevented by Arg185 and Pro195 in the α4 subunit. Mol. Pharmacol. 2012, 82, 711–718. [Google Scholar] [CrossRef] [PubMed]
- Dutertre, S.; Nicke, A.; Tyndall, J.D.A.; Lewis, R.J. Determination of α-conotoxin binding modes on neuronal nicotinic acetylcholine receptors. J. Mol. Recognit. 2004, 17, 339–347. [Google Scholar] [CrossRef] [PubMed]
- Ellison, M.; Gao, F.; Wang, H.; Sine, S.M.; McIntosh, J.M.; Olivera, B.M. α-Conotoxins ImI and ImII target distinct regions of the human α7 nicotinic acetylcholine receptor and distinguish human nicotinic receptor subtypes. Biochemistry 2004, 43, 16019–16026. [Google Scholar] [CrossRef] [PubMed]
- Quiram, P.A.; Jones, J.J.; Sine, S.M. Pairwise Interactions between Neuronal α7Acetylcholine Receptors and α-Conotoxin ImI. J. Biol. Chem. 1999, 274, 19517–19524. [Google Scholar] [CrossRef] [PubMed]
- Yu, R.; Craik, D.J.; Kaas, Q. Blockade of neuronal α7-nAChR by α-conotoxin ImI explained by computational scanning and energy calculations. PLoS Comput. Biol. 2011, 7. [Google Scholar] [CrossRef] [PubMed]
- Everhart, D.; Cartier, G.E.; Malhotra, A.; Gomes, A.V.; McIntosh, J.M.; Luetje, C.W. Determinants of potency on α-conotoxin MII, a peptide antagonist of neuronal nicotinic receptors. Biochemistry 2004, 43, 2732–2737. [Google Scholar] [CrossRef] [PubMed]
- Azam, L.; McIntosh, J.M. Molecular basis for the differential sensitivity of rat and human α9α10 nAChRs to α-conotoxin RgIA. J. Neurochem. 2012, 122, 1137–1144. [Google Scholar] [CrossRef] [PubMed]
- Dutton, J.L.; Bansal, P.S.; Hogg, R.C.; Adams, D.J.; Alewood, P.F.; Craik, D.J. A new level of conotoxin diversity, a non-native disulfide bond connectivity in α-conotoxin AuIB reduces structural definition but increases biological activity. J. Biol. Chem. 2002, 277, 48849–48857. [Google Scholar] [CrossRef] [PubMed]
- Grishin, A.A.; Wang, C.A.; Muttenthaler, M.; Alewood, P.F.; Lewis, R.J.; Adams, D.J. α-Conotoxin AuIB isomers exhibit distinct inhibitory mechanisms and differential sensitivity to stoichiometry of α3β4 nicotinic acetylcholine receptors. J. Biol. Chem. 2010, 285, 22254–22263. [Google Scholar] [CrossRef] [PubMed]
- Lebbe, E.K.; Peigneur, S.; Maiti, M.; Mille, B.G.; Devi, P.; Ravichandran, S.; Lescrinier, E.; Waelkens, E.; D’Souza, L.; Herdewijn, P.; et al. Discovery of a new subclass of α-conotoxins in the venom of Conus australis. Toxicon 2014, 91, 145–154. [Google Scholar] [CrossRef] [PubMed]
- Ellison, M.; McIntosh, J.M.; Olivera, B.M. α-Conotoxins ImI and ImII similar α7 nicotinic receptor antagonists act at different sites. J. Biol. Chem. 2003, 278, 757–764. [Google Scholar] [CrossRef] [PubMed]
- Luo, S.; Akondi, K.B.; Zhangsun, D.; Wu, Y.; Zhu, X.; Hu, Y.; Christensen, S.; Dowell, C.; Daly, N.L.; Craik, D.J.; et al. Atypical α-conotoxin LtIA from Conus litteratus targets a novel microsite of the α3β2 nicotinic receptor. J. Biol. Chem. 2010, 285, 12355–12366. [Google Scholar] [CrossRef] [PubMed]
- Peng, C.; Han, Y.; Sanders, T.; Chew, G.; Liu, J.; Hawrot, E.; Chi, C.; Wang, C. α4/7-conotoxin Lp1. 1 is a novel antagonist of neuronal nicotinic acetylcholine receptors. Peptides 2008, 29, 1700–1707. [Google Scholar] [PubMed]
- Franco, A.; Pisarewicz, K.; Moller, C.; Mora, D.; Fields, G.B.; Marí, F. Hyperhydroxylation: A new strategy for neuronal targeting by venomous marine molluscs. In Molluscs; Springer: Boca Raton, FL, USA, 2006. [Google Scholar]
- Yang, L.; Tae, H.; Fan, Z.; Shao, X.; Xu, S.; Zhao, S.; Adams, D.J.; Wang, C. A novel lid-covering peptide inhibitor of nicotinic acetylcholine receptors derived from αd-conotoxin GeXXA. Mar. Drugs 2017, 15, 164. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Bartels, P.; Sadeghi, M.; Du, T.; Dai, Q.; Zhu, C.; Yu, S.; Wang, S.; Dong, M.; Sun, T.; et al. A novel α-conopeptide Eu1. 6 inhibits N-type (Ca V 2.2) calcium channels and exhibits potent analgesic activity. Sci. Rep. 2018, 8. [Google Scholar] [CrossRef]
- Kudryavtsev, D.; Shelukhina, I.; Vulfius, C.; Makarieva, T.; Stonik, V.; Zhmak, M.; Ivanov, I.; Kasheverov, I.; Utkin, Y.; Tsetlin, V. Natural compounds interacting with nicotinic acetylcholine receptors: From low-molecular weight ones to peptides and proteins. Toxins 2015, 7, 1683–1701. [Google Scholar] [CrossRef] [PubMed]
- Favreau, P.; Benoit, E.; Hocking, H.G.; Carlier, L.; D’hoedt, D.; Leipold, E.; Markgraf, R.; Schlumberger, S.; Córdova, M.A.; Gaertner, H.; et al. A novel µ-conopeptide, CnIIIC, exerts potent and preferential inhibition of NaV1. 2/1.4 channels and blocks neuronal nicotinic acetylcholine receptors. Br. J. Pharmacol. 2012, 166, 1654–1668. [Google Scholar] [CrossRef] [PubMed]
- Sakai, R.; Swanson, G.T. Recent progress in neuroactive marine natural products. Nat. Prod. Rep. 2014, 31, 273–309. [Google Scholar] [CrossRef] [PubMed]
- Bourne, Y.; Sulzenbacher, G.; Radić, Z.; Aráoz, R.; Reynaud, M.; Benoit, E.; Zakarian, A.; Servent, D.; Molgó, J.; Taylor, P. Marine macrocyclic imines, pinnatoxins A and G: Structural determinants and functional properties to distinguish neuronal α7 from muscle α12βγδ nAChRs. Structure 2015, 23, 1106–1115. [Google Scholar] [CrossRef] [PubMed]
- Champtiaux, N.; Han, Z.; Bessis, A.; Rossi, F.M.; Zoli, M.; Marubio, L.; McIntosh, J.M.; Changeux, J. Distribution and pharmacology of α6-containing nicotinic acetylcholine receptors analyzed with mutant mice. J. Neurosci. 2002, 22, 1208–1217. [Google Scholar] [CrossRef] [PubMed]
- Grady, S.R.; Meinerz, N.M.; Cao, J.; Reynolds, A.M.; Picciotto, M.R.; Changeux, J.; McIntosh, J.M.; Marks, M.J.; Collins, A.C. Nicotinic agonists stimulate acetylcholine release from mouse interpeduncular nucleus: A function mediated by a different nAChR than dopamine release from striatum. J. Neurochem. 2001, 76, 258–268. [Google Scholar] [CrossRef] [PubMed]
- Quik, M.; Polonskaya, Y.; Kulak, J.M.; McIntosh, J.M. Vulnerability of 125I-α-conotoxin MII binding sites to nigrostriatal damage in monkey. J. Neurosci. 2001, 21, 5494–5500. [Google Scholar] [CrossRef] [PubMed]
- Improgo, M.R.; Soll, L.G.; Tapper, A.R.; Gardner, P.D. Nicotinic acetylcholine receptors mediate lung cancer growth. Front. Physiol. 2013, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blanchfield, J.T.; Dutton, J.L.; Hogg, R.C.; Gallagher, O.P.; Craik, D.J.; Jones, A.; Adams, D.J.; Lewis, R.J.; Alewood, P.F.; Toth, I. Synthesis, structure elucidation, in vitro biological activity, toxicity, and Caco-2 cell permeability of lipophilic analogues of α-conotoxin MII. J. Med. Chem. 2003, 46, 1266–1272. [Google Scholar] [CrossRef] [PubMed]
- Clark, R.J.; Fischer, H.; Dempster, L.; Daly, N.L.; Rosengren, K.J.; Nevin, S.T.; Meunier, F.A.; Adams, D.J.; Craik, D.J. Engineering stable peptide toxins by means of backbone cyclization: Stabilization of the α-conotoxin MII. Proc. Natl. Acad. Sci. USA 2005, 102, 13767–13772. [Google Scholar] [CrossRef] [PubMed]
- Armishaw, C.J.; Daly, N.L.; Nevin, S.T.; Adams, D.J.; Craik, D.J.; Alewood, P.F. α-Selenoconotoxins, a new class of potent α7 neuronal nicotinic receptor antagonists. J. Biol. Chem. 2006, 281, 14136–14143. [Google Scholar] [CrossRef] [PubMed]
- Clark, R.J.; Craik, D.J. Engineering cyclic peptide toxins. Methods Enzymol. 2012, 503, 57–74. [Google Scholar] [PubMed]
- Clark, R.J.; Akcan, M.; Kaas, Q.; Daly, N.L.; Craik, D.J. Cyclization of conotoxins to improve their biopharmaceutical properties. Toxicon 2012, 59, 446–455. [Google Scholar] [CrossRef] [PubMed]
- MacRaild, C.A.; Illesinghe, J.; van Lierop, B.J.; Townsend, A.L.; Chebib, M.; Livett, B.G.; Robinson, A.J.; Norton, R.S. Structure and Activity of (2,8)-Dicarba-(3,12)-cystino α-ImI, an α-Conotoxin containing a nonreducible cystine analogue. J. Med. Chem. 2009, 52, 755–762. [Google Scholar] [CrossRef] [PubMed]
- Kessler, H. Conformation and biological activity of cyclic peptides. Angew. Chem. Int. Ed. Engl. 1982, 21, 512–523. [Google Scholar] [CrossRef]
- Swedberg, J.E.; Schroeder, C.I.; Mitchell, J.M.; Durek, T.; Fairlie, D.P.; Edmonds, D.J.; Griffith, D.A.; Ruggeri, R.B.; Derksen, D.R.; Loria, P.M.; et al. Cyclic alpha-conotoxin peptidomimetic chimeras as potent GLP-1R agonists. Eur. J. Med. Chem. 2015, 103, 175–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wan, J.; Huang, J.X.; Vetter, I.; Mobli, M.; Lawson, J.; Tae, H.-S.; Abraham, N.; Paul, B.; Cooper, M.A.; Adams, D.J.; et al. α-Conotoxin dendrimers have enhanced potency and selectivity for homomeric nicotinic acetylcholine receptors. J. Am. Chem. Soc. 2015, 137, 3209–3212. [Google Scholar] [CrossRef] [PubMed]
- Mei, D.; Lin, Z.; Fu, J.; He, B.; Gao, W.; Ma, L.; Dai, W.; Zhang, H.; Wang, X.; Wang, J.; et al. The use of α-conotoxin ImI to actualize the targeted delivery of paclitaxel micelles to α7 nAChR-overexpressing breast cancer. Biomaterials 2015, 42, 52–65. [Google Scholar] [CrossRef] [PubMed]
- Akondi, K.B.; Muttenthaler, M.; Dutertre, S.; Kaas, Q.; Craik, D.J.; Lewis, R.J.; Alewood, P.F. Discovery, synthesis, and structure-activity relationships of conotoxins. Chem. Rev. 2014, 114, 5815–5847. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.; Banerjee, J.; Dowell, C.; Wu, J.; Gyanda, R.; Houghten, R.A.; Toll, L.; McIntosh, J.M.; Armishaw, C.J. Discovery of a potent and selective α3β4 nicotinic acetylcholine receptor antagonist from an α-conotoxin synthetic combinatorial library. J. Med. Chem. 2014, 57, 3511–3521. [Google Scholar] [CrossRef] [PubMed]
- Satkunanathan, N.; Livett, B.; Gayler, K.; Sandall, D.; Down, J.; Khalil, Z. Alpha-conotoxin Vc1.1 alleviates neuropathic pain and accelerates functional recovery of injured neurons. Brain Res. 2005, 1059, 149–158. [Google Scholar] [CrossRef] [PubMed]
- Vincler, M.; Wittenauer, S.; Parker, R.; Ellison, M.; Olivera, B.M.; McIntosh, J.M. Molecular mechanism for analgesia involving specific antagonism of alpha9alpha10 nicotinic acetylcholine receptors. Proc. Natl. Acad. Sci. USA 2006, 103, 17880–17884. [Google Scholar] [CrossRef] [PubMed]
- Nevin, S.T.; Clark, R.J.; Klimis, H.; Christie, M.J.; Craik, D.J.; Adams, D.J. Are alpha9alpha10 nicotinic acetylcholine receptors a pain target for alpha-conotoxins? Mol. Pharmacol. 2007, 72, 1406–1410. [Google Scholar] [CrossRef] [PubMed]
- Tsetlin, V.; Utkin, Y.; Kasheverov, I. Polypeptide and peptide toxins, magnifying lenses for binding sites in nicotinic acetylcholine receptors. Biochem. Pharmacol. 2009, 78, 720–731. [Google Scholar] [CrossRef] [PubMed]
- Lin, B.; Xiang, S.; Li, M. Residues responsible for the selectivity of α-conotoxins for Ac-AChBP or nAChRs. Mar. Drugs 2016, 14, 173. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Superfamily | Sequence, Cysteine Framework and Connectivity | Pharmacology |
|---|---|---|
| A superfamily | ||
| AuIB |  | non-competitive inhibitor of the α3β4 [99] |
| AuIB |  | 10-fold more potent in rat parasympathetic ganglions than the globular isomer |
| AusIA |  | Defines a new 5/5 subclass. Both globular and ribbon isomers are equipotent at α7 [101] |
| ImII |  | Lacks conserved proline in loop 1. Allosteric inhibitor of the α7 |
| LtIA |  | Ala-Xaa-Ala motif substitutes the conserved Ser-Xaa-Ser motif. Competitive blocker with a shallow binding pocket. |
| Lp1.1 |  | Ala-Xaa-Ala motif substitutes the conserved Ser-Xaa-Ser motif |
| MrIC |  | State dependent activator of the α7 |
| Eu1.6 |  | α-conotoxin inhibiting Cav 2.2 [107] |
| S superfamily | ||
| RVIIIA | KCNFDKCKGTGVYNCG(Gla)SCSC(Gla)GLHSCRCTYNIGSMKSGCACICTYY | Atypical cysteine framework. Cysteine connectivity unknown. No C-terminal amidation. Two γ-carboxyglutamates. Broad selectivity: α7, α6/α3β2β3, α3β2 and to a smaller extent the α3β4 and α4β2 [9] |
| GVIIIA | GCTRTCGGOKCTGTCTCTNSSKCGCRYNVHPSG(BTr)GCGCACS * | Cysteine connectivity unknown. C-terminal amidation and bromo-tyrosine present as post-translational modification. Selective α9α10 inhibitor |
| D superfamily | ||
| VxXXA | DVQDCQVSTOGSKWGRCCLNRVCGPMCCPASHCYCVYHRGRGHGCSC | Dimeric peptides. Allosteric inhibitors of the α7, α3β2 and α4β2. |
| VxXXB | DD(Gla)S(Gla)CIINTRDSPWGRCCRTRMCGSMCCPRNGCTCVYHWRRGHGCSCPG | |
| VxXXC | DLRQCTRNAPGSTWGRCCLNPMCGNFCCPRSGCTCAYNWRRGIYCSC | |
| GeXXA |  | Dimeric peptide. Allosteric inhibitors of the α9α10. ‘Lid covering’ binding mode. |
| B3 superfamily | ||
| VxXXIVA |  | Potency dependent on disulfide connectivity—[1,2] 1.2 µM > [1,3] 3.9 µM > [1,4] > 30 µM, suggesting that the novel cysteine framework is important for its mode of action [26] |
| O1 superfamily | ||
| GeXIVA |  |
|
| T superfamily | ||
| TxVC |  | α4β2 inhibitor. Members of this superfamily typically target Cav channels. |
| J superfamily | ||
| pl14a |  | Targets Kv1.6 and α3β4 |
| M superfamily | ||
| CnIIIC |  | Targets Nav 1.2, 1.4 together with α3β2 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abraham, N.; Lewis, R.J. Neuronal Nicotinic Acetylcholine Receptor Modulators from Cone Snails. Mar. Drugs 2018, 16, 208. https://doi.org/10.3390/md16060208
Abraham N, Lewis RJ. Neuronal Nicotinic Acetylcholine Receptor Modulators from Cone Snails. Marine Drugs. 2018; 16(6):208. https://doi.org/10.3390/md16060208
Chicago/Turabian StyleAbraham, Nikita, and Richard J. Lewis. 2018. "Neuronal Nicotinic Acetylcholine Receptor Modulators from Cone Snails" Marine Drugs 16, no. 6: 208. https://doi.org/10.3390/md16060208
APA StyleAbraham, N., & Lewis, R. J. (2018). Neuronal Nicotinic Acetylcholine Receptor Modulators from Cone Snails. Marine Drugs, 16(6), 208. https://doi.org/10.3390/md16060208