Structural and Functional Analyses of Cone Snail Toxins


Abstract
:
1. Introduction
2. Conotoxin Families
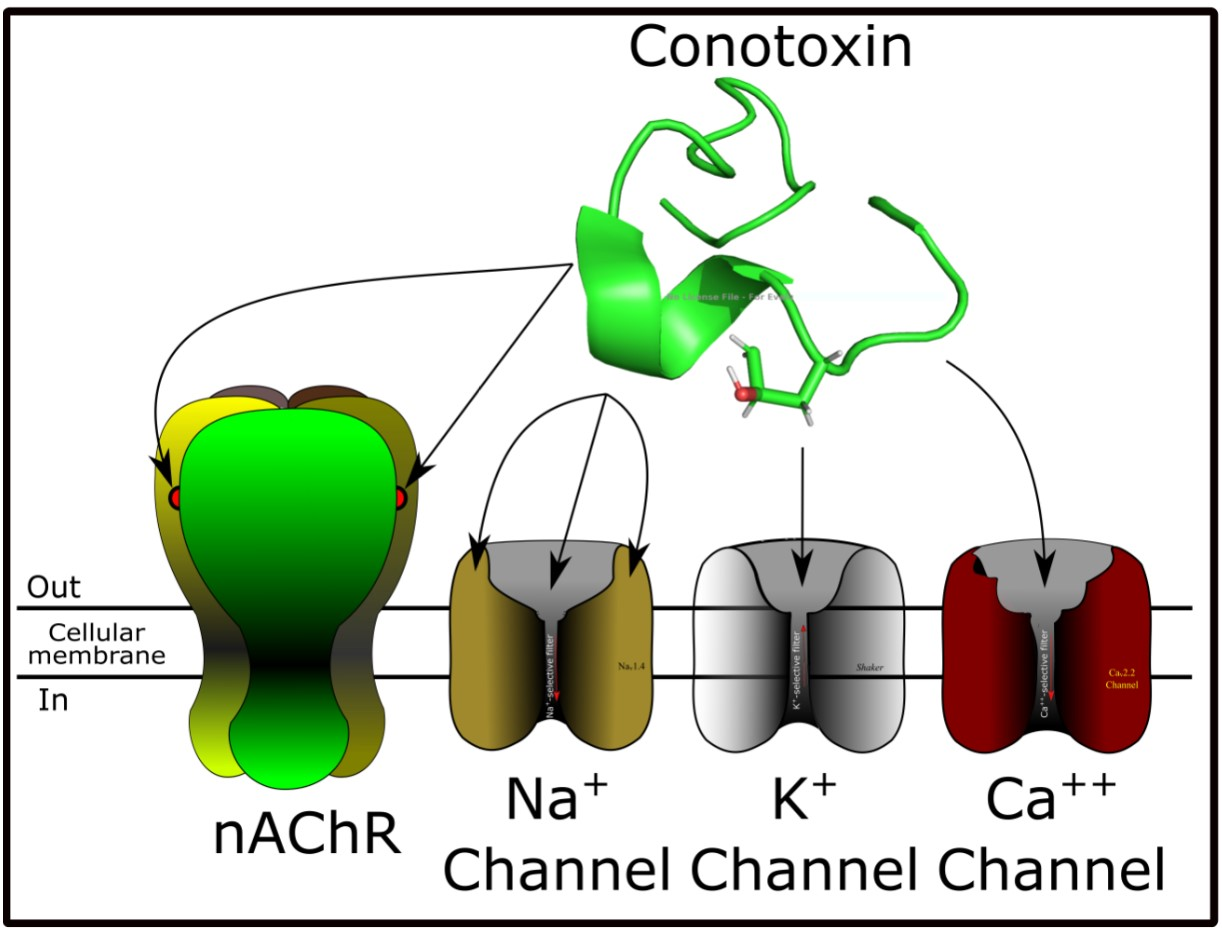
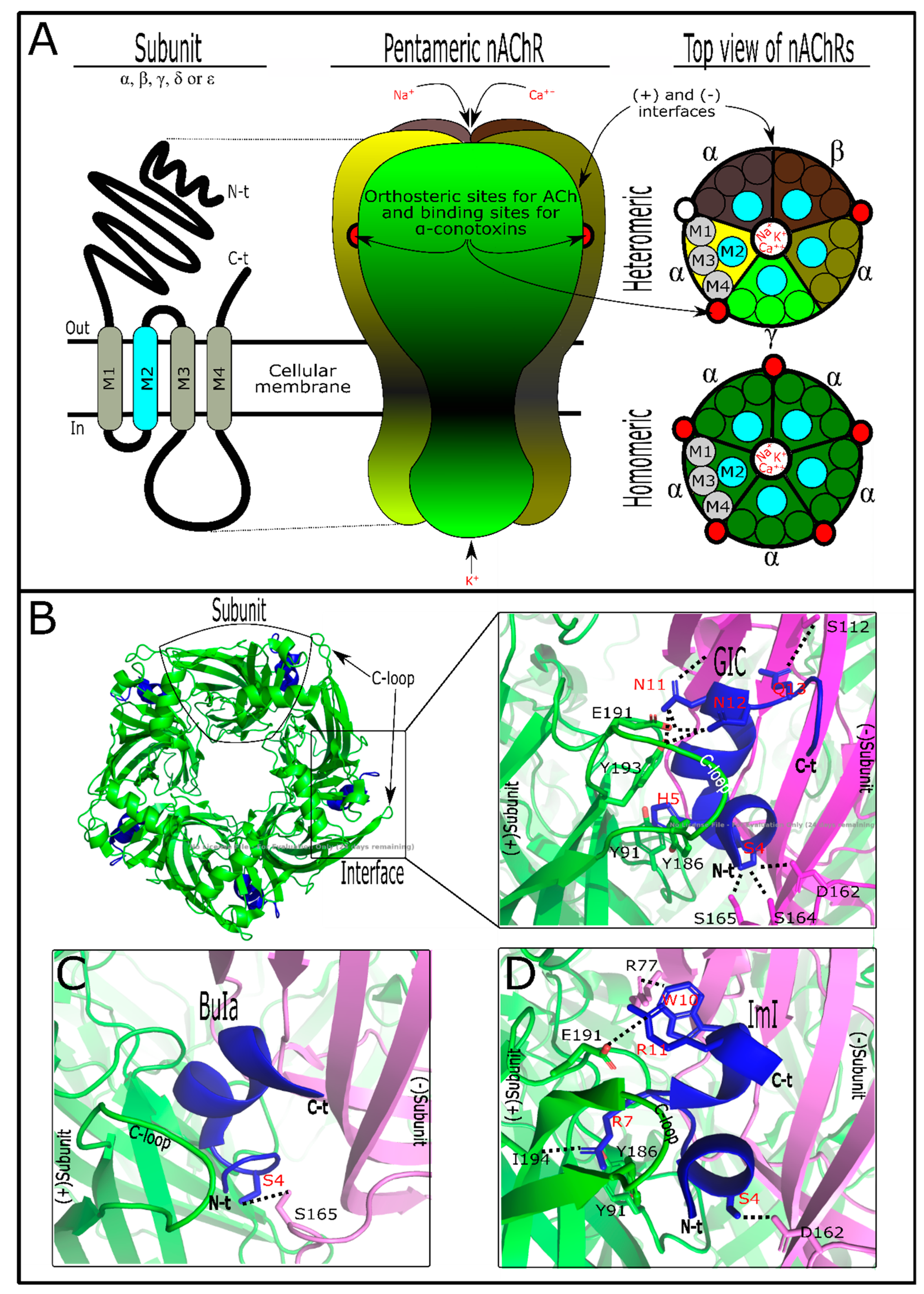
3. Conotoxins Interacting on Nicotinic Acetylcholine Receptors (nAChRs)
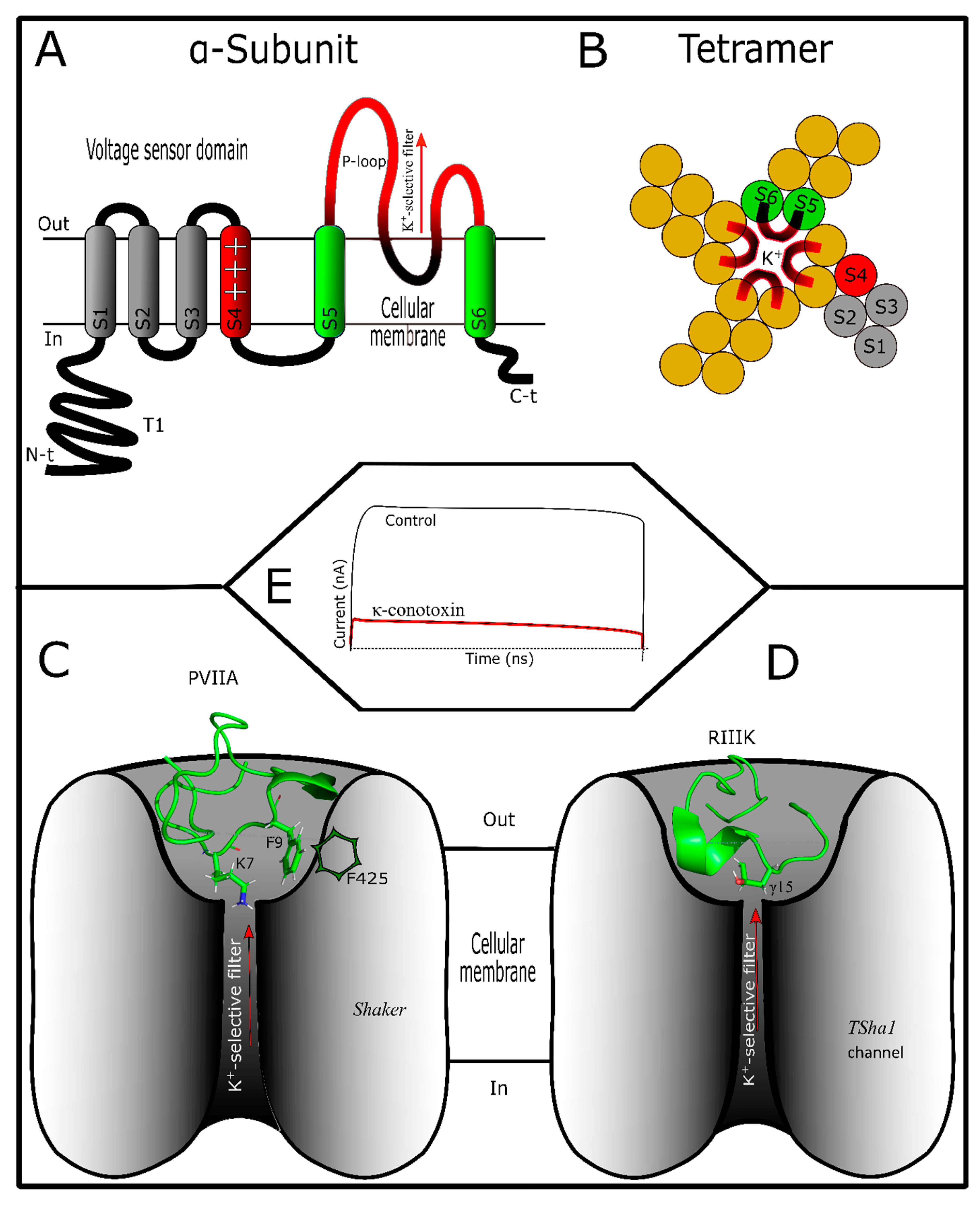
4. Conotoxins Interacting in Potassium Channels
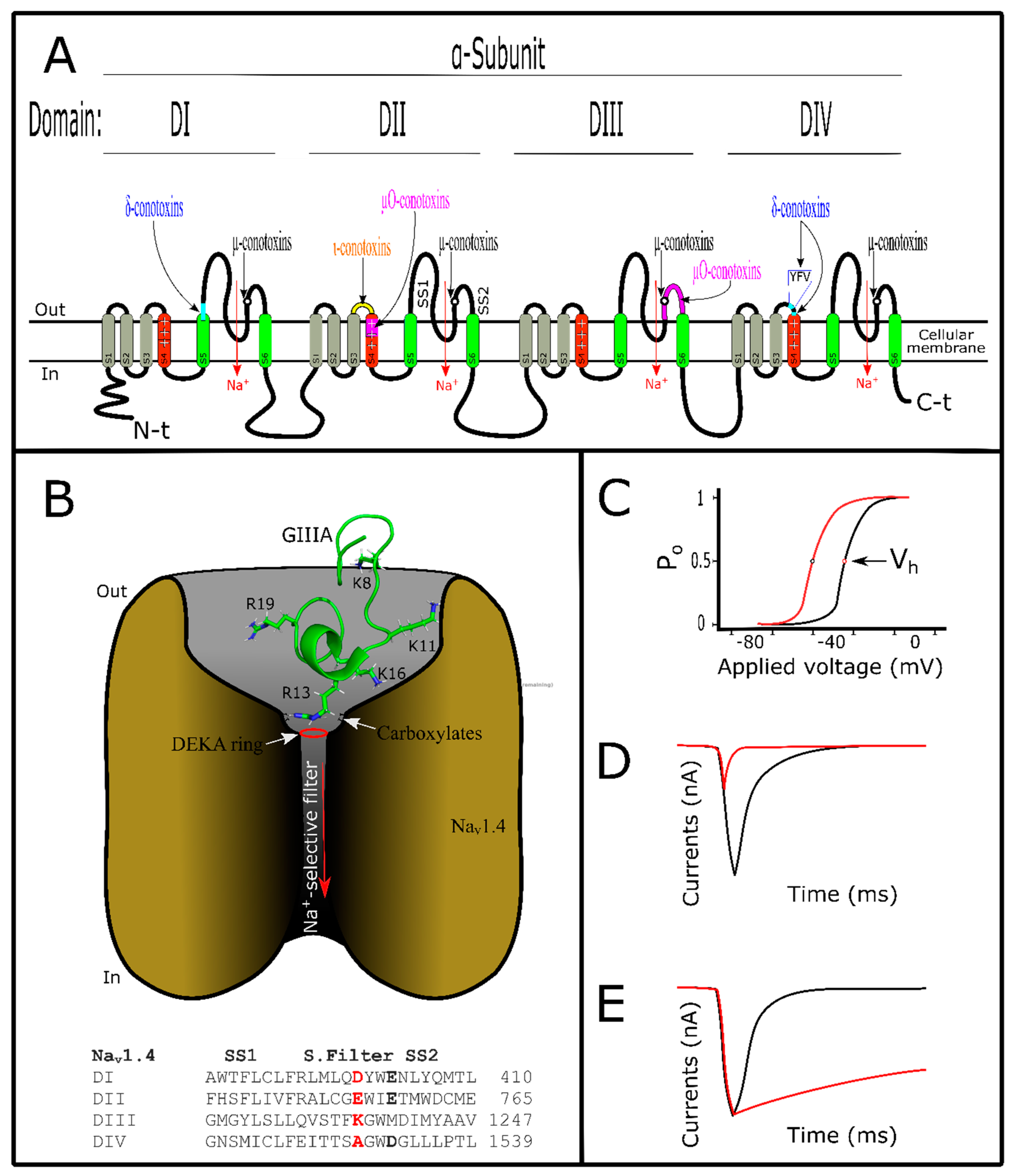
5. Conotoxins Interacting with Voltage-Gated Sodium Channels
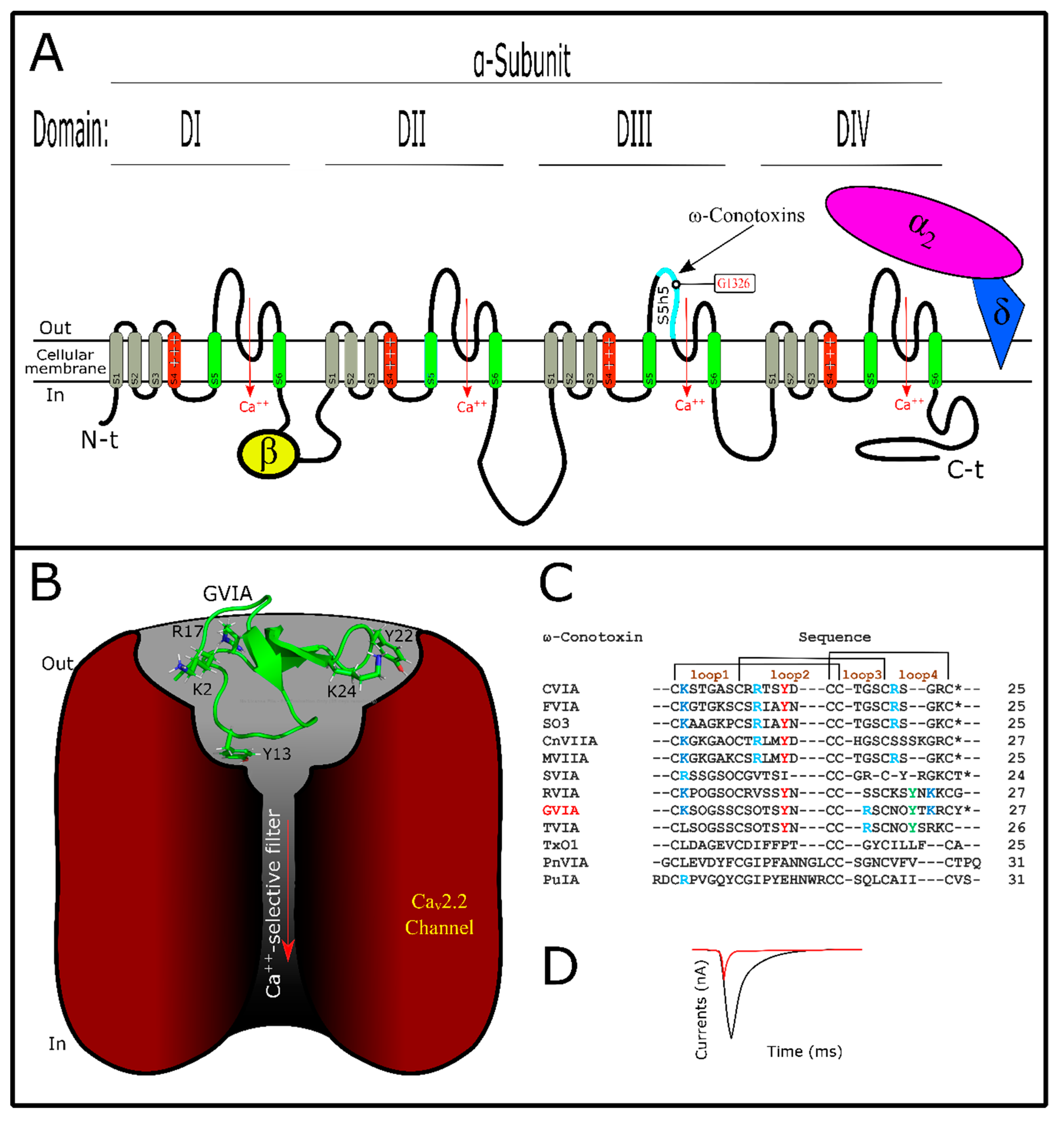
6. Conotoxins Interacting with Voltage-Gated Calcium Channels
7. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Board, W.E. Word Register of Marine Species. Available online: http://www.marinespecies.org (accessed on 14 May 2019).
- Olivera, B.M.; Seger, J.; Horvath, M.P.; Fedosov, A.E. Prey-capture strategies of fish-hunting cone snails: Behavior, neurobiology and evolution. Brain Behav. Evolut. 2015, 86, 58–74. [Google Scholar] [CrossRef] [PubMed]
- Cruz, L.; De Santos, V.; Zafaralla, G.; Ramilo, C.; Zeikus, R.; Gray, W.; Olivera, B. Invertebrate vasopressin/oxytocin homologs. Characterization of peptides from conus geographus and conus straitus venoms. J. Biol. Chem. 1987, 262, 15821–15824. [Google Scholar] [PubMed]
- Bayrhuber, M.; Vijayan, V.; Ferber, M.; Graf, R.; Korukottu, J.; Imperial, J.; Garrett, J.E.; Olivera, B.M.; Terlau, H.; Zweckstetter, M. Conkunitzin-s1 is the first member of a new kunitz-type neurotoxin family structural and functional characterization. J. Biol. Chem. 2005, 280, 23766–23770. [Google Scholar] [CrossRef] [PubMed]
- Lewis, R.J.; Dutertre, S.; Vetter, I.; Christie, M.J. Conus venom peptide pharmacology. Pharmacol. Rev. 2012, 64, 259–298. [Google Scholar] [CrossRef] [PubMed]
- Duda, T.F., Jr.; Kohn, A.J.; Palumbi, S.R. Origins of diverse feeding ecologies within conus, a genus of venomous marine gastropods. Biol. J. Linn. Soc. 2001, 73, 391–409. [Google Scholar] [CrossRef]
- Bergeron, Z.L.; Chun, J.B.; Baker, M.R.; Sandall, D.W.; Peigneur, S.; Peter, Y.; Thapa, P.; Milisen, J.W.; Tytgat, J.; Livett, B.G. A ‘conovenomic’analysis of the milked venom from the mollusk-hunting cone snail conus textile—The pharmacological importance of post-translational modifications. Peptides 2013, 49, 145–158. [Google Scholar] [CrossRef] [PubMed]
- Kohn, A.J. Human injuries and fatalities due to venomous marine snails of the family conidae. Int. J. Clin. Pharmacol. Ther. 2016, 54, 524. [Google Scholar] [CrossRef]
- Dajas-Bailador, F.; Wonnacott, S. Nicotinic acetylcholine receptors and the regulation of neuronal signalling. Trends Pharmacol. Sci. 2004, 25, 317–324. [Google Scholar] [CrossRef]
- Brown, D.A. Regulation of neural ion channels by muscarinic receptors. Neuropharmacology 2018, 136, 383–400. [Google Scholar] [CrossRef]
- Kutlu, M.G.; Gould, T.J. Nicotine modulation of fear memories and anxiety: Implications for learning and anxiety disorders. Biochem. Pharmacol. 2015, 97, 498–511. [Google Scholar] [CrossRef] [Green Version]
- Pérez-Verdaguer, M.; Capera, J.; Serrano-Novillo, C.; Estadella, I.; Sastre, D.; Felipe, A. The voltage-gated potassium channel kv1. 3 is a promising multitherapeutic target against human pathologies. Expert Opin. Ther. Targets 2016, 20, 577–591. [Google Scholar] [CrossRef] [PubMed]
- Zamponi, G.W. A Crash Course in Calcium Channels. ACS chem neurosci. 2017, 8, 2583–2585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Lera Ruiz, M.; Kraus, R.L. Voltage-gated sodium channels: Structure, function, pharmacology, and clinical indications. J. Med. Chem. 2015, 58, 7093–7118. [Google Scholar] [CrossRef] [PubMed]
- Gao, B.; Peng, C.; Yang, J.; Yi, Y.; Zhang, J.; Shi, Q. Cone snails: A big store of conotoxins for novel drug discovery. Toxins 2017, 9, 397. [Google Scholar] [CrossRef] [PubMed]
- Prashanth, J.R.; Brust, A.; Jin, A.-H.; Alewood, P.F.; Dutertre, S.; Lewis, R.J. Cone snail venomics: From novel biology to novel therapeutics. Future Med. Chem. 2014, 6, 1659–1675. [Google Scholar] [CrossRef] [PubMed]
- Bajaj, S.; Han, J. Venom-derived peptide modulators of cation-selective channels: Friend, foe or frenemy. Front. Pharmacol. 2019, 10, 58. [Google Scholar] [CrossRef] [PubMed]
- Akondi, K.B.; Muttenthaler, M.; Dutertre, S.; Kaas, Q.; Craik, D.J.; Lewis, R.J.; Alewood, P.F. Discovery, synthesis, and structure–activity relationships of conotoxins. Chem. Rev. 2014, 114, 5815–5847. [Google Scholar] [CrossRef]
- Mansbach, R.A.; Travers, T.; McMahon, B.H.; Fair, J.M.; Gnanakaran, S. Snails in silico: A review of computational studies on the conopeptides. Mar. Drugs 2019, 17, 145. [Google Scholar] [CrossRef]
- Kaas, Q.; Westermann, J.-C.; Craik, D.J. Conopeptide characterization and classifications: An analysis using conoserver. Toxicon 2010, 55, 1491–1509. [Google Scholar] [CrossRef]
- Robinson, S.; Norton, R. Conotoxin gene superfamilies. Mar. Drugs 2014, 12, 6058–6101. [Google Scholar] [CrossRef]
- Armishaw, C.J.; Alewood, P.F. Conotoxins as research tools and drug leads. Curr. Protein Pept. Sci. 2005, 6, 221–240. [Google Scholar] [CrossRef] [PubMed]
- Lebbe, E.K.; Tytgat, J. In the picture: Disulfide-poor conopeptides, a class of pharmacologically interesting compounds. J. Venom. Anim. Toxins Incl. Trop. Dis. 2016, 22, 30. [Google Scholar] [CrossRef] [PubMed]
- Tsetlin, V.; Utkin, Y.; Kasheverov, I. Polypeptide and peptide toxins, magnifying lenses for binding sites in nicotinic acetylcholine receptors. Biochem. Pharmacol. 2009, 78, 720–731. [Google Scholar] [CrossRef] [PubMed]
- Heimer, P.; Schmitz, T.; Bäuml, C.A.; Imhof, D. Synthesis and structure determination of µ-conotoxin piiia isomers with different disulfide connectivities. JoVE (J. Vis. Exp.) 2018, e58368. [Google Scholar] [CrossRef] [PubMed]
- Espiritu, M.J.; Cabalteja, C.C.; Sugai, C.K.; Bingham, J.-P. Incorporation of post-translational modified amino acids as an approach to increase both chemical and biological diversity of conotoxins and conopeptides. Amino Acids 2014, 46, 125–151. [Google Scholar] [CrossRef]
- Craik, Q.K.D. Conoserver, a database for conopeptide sequences and structures. Bioinformatics 2007, 24, 445–446. [Google Scholar]
- Dao, F.-Y.; Yang, H.; Su, Z.-D.; Yang, W.; Wu, Y.; Hui, D.; Chen, W.; Tang, H.; Lin, H. Recent advances in conotoxin classification by using machine learning methods. Molecules 2017, 22, 1057. [Google Scholar] [CrossRef] [PubMed]
- Himaya, S.; Lewis, R. Venomics-accelerated cone snail venom peptide discovery. Int. J. Mol. Sci. 2018, 19, 788. [Google Scholar] [CrossRef]
- Dutertre, S.; Nicke, A.; Tsetlin, V.I. Nicotinic acetylcholine receptor inhibitors derived from snake and snail venoms. Neuropharmacology 2017, 127, 196–223. [Google Scholar] [CrossRef]
- Ramírez, D.; Gonzalez, W.; Fissore, R.; Carvacho, I. Conotoxins as tools to understand the physiological function of voltage-gated calcium (cav) channels. Mar. Drugs 2017, 15, 313. [Google Scholar] [CrossRef]
- Terlau, H.; Olivera, B.M. Conus venoms: A rich source of novel ion channel-targeted peptides. Physiol. Rev. 2004, 84, 41–68. [Google Scholar] [CrossRef] [PubMed]
- Prashanth, J.R.; Dutertre, S.; Lewis, R.J. Pharmacology of predatory and defensive venom peptides in cone snails. Mol. Biosyst. 2017, 13, 2453–2465. [Google Scholar] [CrossRef] [PubMed]
- Gray, W.; Luque, A.; Olivera, B.; Barrett, J.; Cruz, L. Peptide toxins from conus geographus venom. J. Biol. Chem. 1981, 256, 4734–4740. [Google Scholar] [PubMed]
- Fainzilber, M.; Nakamura, T.; Lodder, J.C.; Zlotkin, E.; Kits, K.S.; Burlingame, A.L. Γ-conotoxin-pnviia, a γ-carboxyglutamate-containing peptide agonist of neuronal pacemaker cation currents. Biochemistry 1998, 37, 1470–1477. [Google Scholar] [CrossRef] [PubMed]
- Fainzilber, M.; Gordon, D.; Hasson, A.; Spira, M.E.; Zlotkin, E. Mollusc-specific toxins from the venom of conus textile neovicarius. Eur. J. Biochem. 1991, 202, 589–595. [Google Scholar] [CrossRef] [PubMed]
- Rigby, A.C.; Lucas-Meunier, E.; Kalume, D.E.; Czerwiec, E.; Hambe, B.; Dahlqvist, I.; Fossier, P.; Baux, G.; Roepstorff, P.; Baleja, J.D. A conotoxin from conus textile with unusual posttranslational modifications reduces presynaptic Ca2+ influx. Proc. Natl. Acad. Sci. 1999, 96, 5758–5763. [Google Scholar] [CrossRef] [PubMed]
- Buczek, O.; Wei, D.; Babon, J.J.; Yang, X.; Fiedler, B.; Chen, P.; Yoshikami, D.; Olivera, B.M.; Bulaj, G.; Norton, R.S. Structure and sodium channel activity of an excitatory i1-superfamily conotoxin. Biochemistry 2007, 46, 9929–9940. [Google Scholar] [CrossRef] [PubMed]
- Terlau, H.; Shon, K.-J.; Grilley, M.; Stocker, M.; Stühmer, W.; Olivera, B.M. Strategy for rapid immobilization of prey by a fish-hunting marine snail. Nature 1996, 381, 148. [Google Scholar] [CrossRef] [PubMed]
- Cruz, L.; Gray, W.; Olivera, B.; Zeikus, R.; Kerr, L.; Yoshikami, D.; Moczydlowski, E. Conus geographus toxins that discriminate between neuronal and muscle sodium channels. J. Biol. Chem. 1985, 260, 9280–9288. [Google Scholar]
- Sharpe, I.A.; Gehrmann, J.; Loughnan, M.L.; Thomas, L.; Adams, D.A.; Atkins, A.; Palant, E.; Craik, D.J.; Adams, D.J.; Alewood, P.F. Two new classes of conopeptides inhibit the α1-adrenoceptor and noradrenaline transporter. Nat. Neurosci. 2001, 4, 902. [Google Scholar] [CrossRef]
- England, L.J.; Imperial, J.; Jacobsen, R.; Craig, A.G.; Gulyas, J.; Akhtar, M.; Rivier, J.; Julius, D.; Olivera, B.M. Inactivation of a serotonin-gated ion channel by a polypeptide toxin from marine snails. Science 1998, 281, 575–578. [Google Scholar] [CrossRef] [PubMed]
- Petrel, C.; Hocking, H.; Reynaud, M.; Upert, G.; Favreau, P.; Biass, D.; Paolini-Bertrand, M.; Peigneur, S.; Tytgat, J.; Gilles, N. Identification, structural and pharmacological characterization of τ-cnva, a conopeptide that selectively interacts with somatostatin sst3 receptor. Biochem. Pharmacol. 2013, 85, 1663–1671. [Google Scholar] [CrossRef] [PubMed]
- Kerr, L.M.; Yoshikami, D. A venom peptide with a novel presynaptic blocking action. Nature 1984, 308, 282. [Google Scholar] [CrossRef] [PubMed]
- Zoli, M.; Pucci, S.; Vilella, A.; Gotti, C. Neuronal and extraneuronal nicotinic acetylcholine receptors. Curr. Neuropharmacol. 2018, 16, 338–349. [Google Scholar] [CrossRef] [PubMed]
- Millar, N.S.; Gotti, C. Diversity of vertebrate nicotinic acetylcholine receptors. Neuropharmacology 2009, 56, 237–246. [Google Scholar] [CrossRef] [Green Version]
- Gotti, C.; Clementi, F. Neuronal nicotinic receptors: From structure to pathology. Prog. Neurobiol. 2004, 74, 363–396. [Google Scholar] [CrossRef]
- Wessler, I.; Kirkpatrick, C. Acetylcholine beyond neurons: The non-neuronal cholinergic system in humans. Br. J. Pharmacol. 2008, 154, 1558–1571. [Google Scholar] [CrossRef]
- Gotti, C.; Clementi, F.; Fornari, A.; Gaimarri, A.; Guiducci, S.; Manfredi, I.; Moretti, M.; Pedrazzi, P.; Pucci, L.; Zoli, M. Structural and functional diversity of native brain neuronal nicotinic receptors. Biochem. Pharmacol. 2009, 78, 703–711. [Google Scholar] [CrossRef] [Green Version]
- Nicke, A.; Samochocki, M.; Loughnan, M.L.; Bansal, P.S.; Maelicke, A.; Lewis, R.J. A-conotoxins epi and auib switch subtype selectivity and activity in native versus recombinant nicotinic acetylcholine receptors. FEBS Lett. 2003, 554, 219–223. [Google Scholar] [CrossRef]
- Azam, L.; McIntosh, J.M. Alpha-conotoxins as pharmacological probes of nicotinic acetylcholine receptors. Acta Pharmacol. Sin. 2009, 30, 771. [Google Scholar] [CrossRef]
- Carstens, B.B.; Berecki, G.; Daniel, J.T.; Lee, H.S.; Jackson, K.A.; Tae, H.S.; Sadeghi, M.; Castro, J.; O’Donnell, T.; Deiteren, A. Structure–activity studies of cysteine-rich α-conotoxins that inhibit high-voltage-activated calcium channels via gabab receptor activation reveal a minimal functional motif. Angew. Chem. Int. Ed. 2016, 55, 4692–4696. [Google Scholar] [CrossRef] [PubMed]
- Janes, R.W. A-conotoxins as selective probes for nicotinic acetylcholine receptor subclasses. Curr. Opin. Pharmacol. 2005, 5, 280–292. [Google Scholar] [CrossRef] [PubMed]
- Turner, M.W.; Marquart, L.A.; Phillips, P.D.; McDougal, O.M. Mutagenesis of α-conotoxins for enhancing activity and selectivity for nicotinic acetylcholine receptors. Toxins 2019, 11, 113. [Google Scholar] [CrossRef] [PubMed]
- Cruz, L.J.; Gray, W.R.; Olivera, B.M. Purification and properties of a myotoxin from conus geographus venom. Arch. Biochem. Biophys. 1978, 190, 539–548. [Google Scholar] [CrossRef]
- McIntosh, J.M.; Yoshikami, D.; Mahe, E.; Nielsen, D.B.; Rivier, J.E.; Gray, W.R.; Olivera, B.M. A nicotinic acetylcholine receptor ligand of unique specificity, alpha-conotoxin imi. J. Biol. Chem. 1994, 269, 16733–16739. [Google Scholar] [PubMed]
- Azam, L.; Dowell, C.; Watkins, M.; Stitzel, J.A.; Olivera, B.M.; McIntosh, J.M. A-conotoxin buia, a novel peptide from conus bullatus, distinguishes among neuronal nicotinic acetylcholine receptors. J. Biol. Chem. 2005, 280, 80–87. [Google Scholar] [CrossRef] [PubMed]
- Luo, S.; Kulak, J.M.; Cartier, G.E.; Jacobsen, R.B.; Yoshikami, D.; Olivera, B.M.; McIntosh, J.M. A-conotoxin auib selectively blocks α3β4 nicotinic acetylcholine receptors and nicotine-evoked norepinephrine release. J. Neurosci. 1998, 18, 8571–8579. [Google Scholar] [CrossRef] [PubMed]
- Clark, R.J.; Fischer, H.; Nevin, S.T.; Adams, D.J.; Craik, D.J. The synthesis, structural characterization, and receptor specificity of the α-conotoxin vc1. 1. J. Biol. Chem. 2006, 281, 23254–23263. [Google Scholar] [CrossRef]
- Imperial, J.S.; Bansal, P.S.; Alewood, P.F.; Daly, N.L.; Craik, D.J.; Sporning, A.; Terlau, H.; López-Vera, E.; Bandyopadhyay, P.K.; Olivera, B.M. A novel conotoxin inhibitor of kv1. 6 channel and nachr subtypes defines a new superfamily of conotoxins. Biochemistry 2006, 45, 8331–8340. [Google Scholar] [CrossRef]
- Teichert, R.W.; Jimenez, E.C.; Olivera, B.M. As-conotoxin rviiia: A structurally unique conotoxin that broadly targets nicotinic acetylcholine receptors. Biochemistry 2005, 44, 7897–7902. [Google Scholar] [CrossRef]
- Loughnan, M.L.; Nicke, A.; Jones, A.; Adams, D.J.; Alewood, P.F.; Lewis, R.J. Chemical and functional identification and characterization of novel sulfated α-conotoxins from the cone snail conus a nemone. J. Med. Chem. 2004, 47, 1234–1241. [Google Scholar] [CrossRef] [PubMed]
- Armishaw, C.J. Synthetic α-conotoxin mutants as probes for studying nicotinic acetylcholine receptors and in the development of novel drug leads. Toxins 2010, 2, 1471–1499. [Google Scholar] [CrossRef] [PubMed]
- Weltzin, M.M.; George, A.A.; Lukas, R.J.; Whiteaker, P. Distinctive single-channel properties of α4β2-nicotinic acetylcholine receptor isoforms. PLoS ONE 2019, 14, e0213143. [Google Scholar] [CrossRef] [PubMed]
- Lebbe, E.; Peigneur, S.; Wijesekara, I.; Tytgat, J. Conotoxins targeting nicotinic acetylcholine receptors: An overview. Mar. Drugs 2014, 12, 2970–3004. [Google Scholar] [CrossRef] [PubMed]
- Martinez, J.S.; Olivera, B.M.; Gray, W.R.; Craig, A.G.; Groebe, D.R.; Abramson, S.N.; McIntosh, J.M. Alpha.-conotoxin ei, a new nicotinic acetylcholine receptor antagonist with novel selectivity. Biochemistry 1995, 34, 14519–14526. [Google Scholar] [CrossRef] [PubMed]
- Groebe, D.R.; Dumm, J.M.; Levitan, E.S.; Abramson, S.N. Alpha-conotoxins selectively inhibit one of the two acetylcholine binding sites of nicotinic receptors. Mol. Pharmacol. 1995, 48, 105–111. [Google Scholar] [PubMed]
- Gerwig, G.; Hocking, H.; Stöcklin, R.; Kamerling, J.; Boelens, R. Glycosylation of conotoxins. Mar. Drugs 2013, 11, 623–642. [Google Scholar] [CrossRef] [PubMed]
- Kang, T.S.; Radić, Z.; Talley, T.T.; Jois, S.D.; Taylor, P.; Kini, R.M. Protein folding determinants: Structural features determining alternative disulfide pairing in α-and χ/λ-conotoxins. Biochemistry 2007, 46, 3338–3355. [Google Scholar] [CrossRef]
- Lin, B.; Xiang, S.; Li, M. Residues responsible for the selectivity of α-conotoxins for ac-achbp or nachrs. Mar. Drugs 2016, 14, 173. [Google Scholar] [CrossRef]
- Whiteaker, P.; Christensen, S.; Yoshikami, D.; Dowell, C.; Watkins, M.; Gulyas, J.; Rivier, J.; Olivera, B.M.; McIntosh, J.M. Discovery, synthesis, and structure activity of a highly selective α7 nicotinic acetylcholine receptor antagonist. Biochemistry 2007, 46, 6628–6638. [Google Scholar] [CrossRef]
- McIntosh, J.M.; Dowell, C.; Watkins, M.; Garrett, J.E.; Yoshikami, D.; Olivera, B.M. A-conotoxin gic from conus geographus, a novel peptide antagonist of nicotinic acetylcholine receptors. J. Biol. Chem. 2002, 277, 33610–33615. [Google Scholar] [CrossRef] [PubMed]
- Nicke, A.; Loughnan, M.L.; Millard, E.L.; Alewood, P.F.; Adams, D.J.; Daly, N.L.; Craik, D.J.; Lewis, R.J. Isolation, structure, and activity of gid, a novel α4/7-conotoxin with an extended n-terminal sequence. J. Biol. Chem. 2003, 278, 3137–3144. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.S.; Martinez, J.; Elgoyhen, A.B.; Heinemann, S.F.; McIntosh, J.M. Alpha-conotoxin imi exhibits subtype-specific nicotinic acetylcholine receptor blockade: Preferential inhibition of homomeric alpha 7 and alpha 9 receptors. Mol. Pharmacol. 1995, 48, 194–199. [Google Scholar] [PubMed]
- Chen, J.; Liang, L.; Ning, H.; Cai, F.; Liu, Z.; Zhang, L.; Zhou, L.; Dai, Q. Cloning, synthesis and functional characterization of a novel α-conotoxin lt1. 3. Mar. Drugs 2018, 16, 112. [Google Scholar] [CrossRef] [PubMed]
- McIntosh, J.M.; Azam, L.; Staheli, S.; Dowell, C.; Lindstrom, J.M.; Kuryatov, A.; Garrett, J.E.; Marks, M.J.; Whiteaker, P. Analogs of α-conotoxin mii are selective for α6-containing nicotinic acetylcholine receptors. Mol. Pharmacol. 2004, 65, 944–952. [Google Scholar] [CrossRef] [PubMed]
- Hone, A.J.; Gajewiak, J.; Christensen, S.; Lindstrom, J.; McIntosh, J.M. A-conotoxin peia [s9h, v10a, e14n] potently and selectively blocks α6β2β3 versus α6β4 nicotinic acetylcholine receptors. Mol. Pharmacol. 2012, 82, 972–982. [Google Scholar] [CrossRef] [PubMed]
- Luo, S.; Nguyen, T.; Cartier, G.; Olivera, B.; Yoshikami, D.; McIntosh, J. Single-residue alteration in α-conotoxin pnia switches its nachr subtype selectivity. Biochemistry 1999, 38, 14542–14548. [Google Scholar] [CrossRef]
- Luo, S.; Zhangsun, D.; Wu, Y.; Zhu, X.; Hu, Y.; McIntyre, M.; Christensen, S.; Akcan, M.; Craik, D.J.; McIntosh, J.M. Characterization of a novel α-conotoxin from conus textile that selectively targets α6/α3β2β3 nicotinic acetylcholine receptors. J. Biol. Chem. 2013, 288, 894–902. [Google Scholar] [CrossRef]
- Luo, S.; Zhangsun, D.; Zhu, X.; Wu, Y.; Hu, Y.; Christensen, S.; Harvey, P.J.; Akcan, M.; Craik, D.J.; McIntosh, J.M. Characterization of a novel α-conotoxin txid from conus textile that potently blocks rat α3β4 nicotinic acetylcholine receptors. J. Med. Chem. 2013, 56, 9655–9663. [Google Scholar] [CrossRef]
- Yu, R.; Tabassum, N.; Jiang, T. Investigation of α-conotoxin unbinding using umbrella sampling. Bioorganic Med. Chem. Lett. 2016, 26, 1296–1300. [Google Scholar] [CrossRef]
- Changeux, J.-P. The nicotinic acetylcholine receptor: A typical ‘allosteric machine’. Philos. Trans. R. Soc. B Biol. Sci. 2018, 373, 20170174. [Google Scholar] [CrossRef] [PubMed]
- Delbart, F.; Brams, M.; Gruss, F.; Noppen, S.; Peigneur, S.; Boland, S.; Chaltin, P.; Brandao-Neto, J.; von Delft, F.; Touw, W.G. An allosteric binding site of the α7 nicotinic acetylcholine receptor revealed in a humanized acetylcholine-binding protein. J. Biol. Chem. 2018, 293, 2534–2545. [Google Scholar] [CrossRef] [PubMed]
- Lin, B.; Xu, M.; Zhu, X.; Wu, Y.; Liu, X.; Zhangsun, D.; Hu, Y.; Xiang, S.-H.; Kasheverov, I.E.; Tsetlin, V.I. From crystal structure of α-conotoxin gic in complex with ac-achbp to molecular determinants of its high selectivity for α3β2 nachr. Sci. Rep. 2016, 6, 22349. [Google Scholar] [CrossRef] [PubMed]
- Kasheverov, I.E.; Utkin, Y.N.; Tsetlin, V.I. Naturally occurring and synthetic peptides acting on nicotinic acetylcholine receptors. Curr. Pharm. Des. 2009, 15, 2430–2452. [Google Scholar] [CrossRef] [PubMed]
- Seung-Wook, C.; Do-Hyoung, K.; Olivera, B.M.; Mcintosh, J.M.; Kyou-Hoon, H. Solution conformation of alpha-conotoxin gic, a novel potent antagonist of alpha3beta2 nicotinic acetylcholine receptors. Biochem. J. 2004, 380, 347–352. [Google Scholar]
- Kim, H.-W.; McIntosh, J.M. A6 nachr subunit residues that confer α-conotoxin buia selectivity. FASEB J. 2012, 26, 4102–4110. [Google Scholar] [CrossRef]
- Ellison, M.; Gao, F.; Wang, H.-L.; Sine, S.M.; McIntosh, J.M.; Olivera, B.M. A-conotoxins imi and imii target distinct regions of the human α7 nicotinic acetylcholine receptor and distinguish human nicotinic receptor subtypes. Biochemistry 2004, 43, 16019–16026. [Google Scholar] [CrossRef]
- Ulens, C.; Hogg, R.C.; Celie, P.H.; Bertrand, D.; Tsetlin, V.; Smit, A.B.; Sixma, T.K. Structural determinants of selective α-conotoxin binding to a nicotinic acetylcholine receptor homolog achbp. Proc. Natl. Acad. Sci. 2006, 103, 3615–3620. [Google Scholar] [CrossRef]
- Kuo, M.M.-C.; Haynes, W.J.; Loukin, S.H.; Kung, C.; Saimi, Y. Prokaryotic k+ channels: From crystal structures to diversity. FEMS Microbiol. Rev. 2005, 29, 961–985. [Google Scholar] [CrossRef]
- Grider MH, G.C. Physiology, Action Potential; StatPearls Publishing LLC: Treasure Island, FL, USA, 2019. [Google Scholar]
- Capera, J.; Serrano-Novillo, C.; Navarro-Pérez, M.; Cassinelli, S.; Felipe, A. The potassium channel odyssey: Mechanisms of traffic and membrane arrangement. Int. J. Mol. Sci. 2019, 20, 734. [Google Scholar] [CrossRef]
- Choe, S. Ion channel structure: Potassium channel structures. Nat. Rev. Neurosci. 2002, 3, 115. [Google Scholar] [CrossRef] [PubMed]
- Kuang, Q.; Purhonen, P.; Hebert, H. Structure of potassium channels. Cell. Mol. Life Sci. 2015, 72, 3677–3693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gutman, G.A.; Chandy, K.G.; Grissmer, S.; Lazdunski, M.; Mckinnon, D.; Pardo, L.A.; Robertson, G.A.; Rudy, B.; Sanguinetti, M.C.; Stühmer, W. International union of pharmacology. Liii. Nomenclature and molecular relationships of voltage-gated potassium channels. Pharmacol. Rev. 2005, 57, 473–508. [Google Scholar] [CrossRef] [PubMed]
- Wei, A.D.; Gutman, G.A.; Aldrich, R.; Chandy, K.G.; Grissmer, S.; Wulff, H. International union of pharmacology. Lii. Nomenclature and molecular relationships of calcium-activated potassium channels. Pharmacol. Rev. 2005, 57, 463–472. [Google Scholar] [CrossRef] [PubMed]
- Kubo, Y.; Adelman, J.P.; Clapham, D.E.; Jan, L.Y.; Karschin, A.; Kurachi, Y.; Lazdunski, M.; Nichols, C.G.; Seino, S.; Vandenberg, C.A. International union of pharmacology. Liv. Nomenclature and molecular relationships of inwardly rectifying potassium channels. Pharmacol. Rev. 2005, 57, 509–526. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, S.A.; Bayliss, D.A.; Kim, D.; Lesage, F.; Plant, L.D.; Rajan, S. International union of pharmacology. Lv. Nomenclature and molecular relationships of two-p potassium channels. Pharmacol. Rev. 2005, 57, 527–540. [Google Scholar] [CrossRef] [PubMed]
- Kuzmenkov, A.; Grishin, E.; Vassilevski, A. Diversity of potassium channel ligands: Focus on scorpion toxins. Biochemistry 2015, 80, 1764–1799. [Google Scholar] [CrossRef] [PubMed]
- Miller, C. An overview of the potassium channel family. Genome Biol. 2000, 1, reviews0004.0001. [Google Scholar] [CrossRef]
- Panyi, G.; Deutsch, C. Cross talk between activation and slow inactivation gates of shaker potassium channels. J. Gen. Physiol. 2006, 128, 547–559. [Google Scholar] [CrossRef]
- Massilia, G.R.; Eliseo, T.; Grolleau, F.; Lapied, B.; Barbier, J.; Bournaud, R.; Molgó, J.; Cicero, D.O.; Paci, M.; Schinina, M.E. Contryphan-vn: A modulator of Ca2+-dependent k+ channels. Biochem. Biophys. Res. Commun. 2003, 303, 238–246. [Google Scholar] [CrossRef]
- Hocking, H.G.; Gerwig, G.J.; Dutertre, S.; Violette, A.; Favreau, P.; Stöcklin, R.; Kamerling, J.P.; Boelens, R. Structure of the o-glycosylated conopeptide cctx from conus consors venom. Chem. A Eur. J. 2013, 19, 870–879. [Google Scholar] [CrossRef] [PubMed]
- Fan, C.-X.; Chen, X.-K.; Zhang, C.; Wang, L.-X.; Duan, K.-L.; He, L.-L.; Cao, Y.; Liu, S.-Y.; Zhong, M.-N.; Ulens, C. A novel conotoxin from conus betulinus, κ-btx, unique in cysteine pattern and in function as a specific bk channel modulator. J. Biol. Chem. 2003, 278, 12624–12633. [Google Scholar] [CrossRef] [PubMed]
- Finol-Urdaneta, R.K.; Remedi, M.S.; Raasch, W.; Becker, S.; Clark, R.B.; Strüver, N.; Pavlov, E.; Nichols, C.G.; French, R.J.; Terlau, H. Block of kv1. 7 potassium currents increases glucose-stimulated insulin secretion. EMBO Mol. Med. 2012, 4, 424–434. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Dendorfer, A.; Finol-Urdaneta, R.K.; Terlau, H.; Olivera, B.M. Biochemical characterization of κm-riiij, a kv1. 2 channel blocker evaluation of cardioprotective effects of κm-conotoxins. J. Biol. Chem. 2010, 285, 14882–14889. [Google Scholar] [CrossRef]
- Craig, A.G.; Zafaralla, G.; Cruz, L.J.; Santos, A.D.; Hillyard, D.R.; Dykert, J.; Rivier, J.E.; Gray, W.R.; Imperial, J.; DelaCruz, R.G. An o-glycosylated neuroexcitatory conus peptide. Biochemistry 1998, 37, 16019–16025. [Google Scholar] [CrossRef]
- Kauferstein, S.; Huys, I.; Lamthanh, H.; Stöcklin, R.; Sotto, F.; Menez, A.; Tytgat, J.; Mebs, D. A novel conotoxin inhibiting vertebrate voltage-sensitive potassium channels. Toxicon 2003, 42, 43–52. [Google Scholar] [CrossRef]
- Aguilar, M.B.; Pérez-Reyes, L.I.; López, Z.; de la Cotera, E.P.H.; Falcón, A.; Ayala, C.; Galván, M.; Salvador, C.; Escobar, L.I. Peptide sr11a from conus spurius is a novel peptide blocker for kv1 potassium channels. Peptides 2010, 31, 1287–1291. [Google Scholar] [CrossRef]
- Naranjo, D. Inhibition of single shaker k channels by κ− conotoxin-pviia. Biophys. J. 2002, 82, 3003–3011. [Google Scholar] [CrossRef]
- De la Vega, R.C.R.; Possani, L.D. Current views on scorpion toxins specific for k+-channels. Toxicon 2004, 43, 865–875. [Google Scholar] [CrossRef]
- Jacobsen, R.B.; Koch, E.D.; Lange-Malecki, B.; Stocker, M.; Verhey, J.; Van Wagoner, R.M.; Vyazovkina, A.; Olivera, B.M.; Terlau, H. Single amino acid substitutions in κ-conotoxin pviia disrupt interaction with the shaker k+ channel. J. Biol. Chem. 2000, 275, 24639–24644. [Google Scholar] [CrossRef]
- Huang, X.; Dong, F.; Zhou, H.-X. Electrostatic recognition and induced fit in the κ-pviia toxin binding to shaker potassium channel. J. Am. Chem. Soc. 2005, 127, 6836–6849. [Google Scholar] [CrossRef] [PubMed]
- Kwon, S.; Bosmans, F.; Kaas, Q.; Cheneval, O.; Conibear, A.C.; Rosengren, K.J.; Wang, C.K.; Schroeder, C.I.; Craik, D.J. Efficient enzymatic cyclization of an inhibitory cystine knot-containing peptide. Biotechnol. Bioeng. 2016, 113, 2202–2212. [Google Scholar] [CrossRef] [PubMed]
- Cordeiro, S.; Finol-Urdaneta, R.K.; Köpfer, D.; Markushina, A.; Song, J.; French, R.J.; Kopec, W.; de Groot, B.L.; Giacobassi, M.J.; Leavitt, L.S. Conotoxin κm-riiij, a tool targeting asymmetric heteromeric kv1 channels. Proc. Natl. Acad. Sci. 2019, 116, 1059–1064. [Google Scholar] [CrossRef] [PubMed]
- Al-Sabi, A.; Lennartz, D.; Ferber, M.; Gulyas, J.; Rivier, J.E.; Olivera, B.M.; Carlomagno, T.; Terlau, H. Κm-conotoxin riiik, structural and functional novelty in a k+ channel antagonist. Biochemistry 2004, 43, 8625–8635. [Google Scholar] [CrossRef] [PubMed]
- Verdier, L.; Al-Sabi, A.; Rivier, J.E.; Olivera, B.M.; Terlau, H.; Carlomagno, T. Identification of a novel pharmacophore for peptide toxins interacting with k+ channels. J. Biol. Chem. 2005, 280, 21246–21255. [Google Scholar] [CrossRef] [PubMed]
- Rashid, M.; Mahdavi, S.; Kuyucak, S. Computational studies of marine toxins targeting ion channels. Mar. Drugs 2013, 11, 848–869. [Google Scholar] [CrossRef] [PubMed]
- Dauplais, M.; Lecoq, A.; Song, J.; Cotton, J.; Jamin, N.; Gilquin, B.; Roumestand, C.; Vita, C.; de Medeiros, C.L.; Rowan, E.G. On the convergent evolution of animal toxins conservation of a diad of functional residues in potassium channel-blocking toxins with unrelated structures. J. Biol. Chem. 1997, 272, 4302–4309. [Google Scholar] [CrossRef] [PubMed]
- Shon, K.-J.; Stocker, M.; Terlau, H.; Stühmer, W.; Jacobsen, R.; Walker, C.; Grilley, M.; Watkins, M.; Hillyard, D.R.; Gray, W.R. Κ-conotoxin pviia is a peptide inhibiting theshaker k+ channel. J. Biol. Chem. 1998, 273, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Catterall, W.A. Voltage-gated sodium channels at 60: Structure, function and pathophysiology. J. Physiol. 2012, 590, 2577–2589. [Google Scholar] [CrossRef] [PubMed]
- Catterall, W.A.; Goldin, A.L.; Waxman, S.G. International union of pharmacology. Xlvii. Nomenclature and structure-function relationships of voltage-gated sodium channels. Pharmacol. Rev. 2005, 57, 397–409. [Google Scholar] [CrossRef] [PubMed]
- Kwong, K.; Carr, M.J. Voltage-gated sodium channels. Curr. Opin. Pharmacol. 2015, 22, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Cestèle, S.; Catterall, W.A. Molecular mechanisms of neurotoxin action on voltage-gated sodium channels. Biochimie 2000, 82, 883–892. [Google Scholar] [CrossRef]
- Payandeh, J.; El-Din, T.M.G.; Scheuer, T.; Zheng, N.; Catterall, W.A. Crystal structure of a voltage-gated sodium channel in two potentially inactivated states. Nature 2012, 486, 135. [Google Scholar] [CrossRef] [PubMed]
- Catterall, W.A. Structure and function of voltage-gated sodium channels at atomic resolution. Exp. Physiol. 2014, 99, 35–51. [Google Scholar] [CrossRef] [PubMed]
- Catterall, W.A. From ionic currents to molecular mechanisms: The structure and function of voltage-gated sodium channels. Neuron 2000, 26, 13–25. [Google Scholar] [CrossRef]
- Zhorov, B.S. Structural models of ligand-bound sodium channels. In Voltage-Gated Sodium Channels: Structure, Function and Channelopathies; Springer: New York, NY, USA, 2017; Volume 246, pp. 251–269. [Google Scholar]
- Jover, E.; Martin-Moutot, N.; Couraud, F.; Rochat, H. Binding of scorpion toxins to rat brain synaptosomal fraction. Effects of membrane potential, ions, and other neurotoxins. Biochemistry 1980, 19, 463–467. [Google Scholar] [CrossRef] [PubMed]
- Tikhonov, D.B.; Zhorov, B.S. Predicting structural details of the sodium channel pore basing on animal toxin studies. Front. Pharmacol. 2018, 9, 880. [Google Scholar] [CrossRef]
- Ekberg, J.; Craik, D.J.; Adams, D.J. Conotoxin modulation of voltage-gated sodium channels. Int. J. Biochem. Cell Biol. 2008, 40, 2363–2368. [Google Scholar] [CrossRef]
- Buczek, O.; Yoshikami, D.; Bulaj, G.; Jimenez, E.C.; Olivera, B.M. Post-translational amino acid isomerization a functionally important d-amino acid in an excitatory peptide. J. Biol. Chem. 2005, 280, 4247–4253. [Google Scholar] [CrossRef]
- Fiedler, B.; Zhang, M.-M.; Buczek, O.; Azam, L.; Bulaj, G.; Norton, R.S.; Olivera, B.M.; Yoshikami, D. Specificity, affinity and efficacy of iota-conotoxin rxia, an agonist of voltage-gated sodium channels nav1. 2, 1.6 and 1.7. Biochem. Pharmacol. 2008, 75, 2334–2344. [Google Scholar] [CrossRef]
- Tietze, A.A.; Tietze, D.; Ohlenschläger, O.; Leipold, E.; Ullrich, F.; Kühl, T.; Mischo, A.; Buntkowsky, G.; Görlach, M.; Heinemann, S.H. Structurally diverse μ-conotoxin piiia isomers block sodium channel nav1. 4. Angew. Chem. Int. Ed. 2012, 51, 4058–4061. [Google Scholar] [CrossRef] [PubMed]
- Khoo, K.K.; Gupta, K.; Green, B.R.; Zhang, M.-M.; Watkins, M.; Olivera, B.M.; Balaram, P.; Yoshikami, D.; Bulaj, G.; Norton, R.S. Distinct disulfide isomers of μ-conotoxins kiiia and kiiib block voltage-gated sodium channels. Biochemistry 2012, 51, 9826–9835. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Liu, J.; Pi, C.; Zeng, X.; Zhou, M.; Jiang, X.; Chen, S.; Ren, Z.; Xu, A. Identification of a novel m-superfamily conotoxin with the ability to enhance tetrodotoxin sensitive sodium currents. Arch. Toxicol. 2009, 83, 925–932. [Google Scholar] [CrossRef] [PubMed]
- Jimenez, E.C.; Shetty, R.P.; Lirazan, M.; Rivier, J.; Walker, C.; Abogadie, F.C.; Yoshikami, D.; Cruz, L.J.; Olivera, B.M. Novel excitatory conus peptides define a new conotoxin superfamily. J. Neurochem. 2003, 85, 610–621. [Google Scholar] [CrossRef] [PubMed]
- Green, B.; Olivera, B. Venom peptides from cone snails: Pharmacological probes for voltage-gated sodium channels. In Current Topics in Membranes; Elsevier: Amsterdam, The Netherlands, 2016; Volume 78, pp. 65–86. [Google Scholar]
- Daly, N.L.; Ekberg, J.A.; Thomas, L.; Adams, D.J.; Lewis, R.J.; Craik, D.J. Structures of μo-conotoxins from conus marmoreus inhibitors of tetrodotoxin (ttx)-sensitive and ttx-resistant sodium channels in mammalian sensory neurons. J. Biol. Chem. 2004, 279, 25774–25782. [Google Scholar] [CrossRef] [PubMed]
- Fainzilber, M.; van der Schors, R.; Lodder, J.C.; Li, K.W.; Geraerts, W.P.; Kits, K.S. New sodium channel-blocking conotoxins also affect calcium currents in lymnaea neurons. Biochemistry 1995, 34, 5364–5371. [Google Scholar] [CrossRef] [PubMed]
- McIntosh, J.M.; Hasson, A.; Spira, M.E.; Gray, W.R.; Li, W.; Marsh, M.; Hillyard, D.R.; Olivera, B.M. A new family of conotoxins that blocks voltage-gated sodium channels. J. Biol. Chem. 1995, 270, 16796–16802. [Google Scholar] [CrossRef] [PubMed]
- Vetter, I.; Dekan, Z.; Knapp, O.; Adams, D.J.; Alewood, P.F.; Lewis, R.J. Isolation, characterization and total regioselective synthesis of the novel μo-conotoxin mfvia from conus magnificus that targets voltage-gated sodium channels. Biochem. Pharmacol. 2012, 84, 540–548. [Google Scholar] [CrossRef]
- Zorn, S.; Leipold, E.; Hansel, A.; Bulaj, G.; Olivera, B.M.; Terlau, H.; Heinemann, S.H. The μo-conotoxin mrvia inhibits voltage-gated sodium channels by associating with domain-3. FEBS Lett. 2006, 580, 1360–1364. [Google Scholar] [CrossRef]
- Leipold, E.; DeBie, H.; Zorn, S.; Adolfo, B.; Olivera, B.M.; Terlau, H.; Heinemann, S.H. µo-conotoxins inhibit nav channels by interfering with their voltage sensors in domain-2. Channels 2007, 1, 253–262. [Google Scholar] [CrossRef]
- Leipold, E.; Hansel, A.; Borges, A.; Heinemann, S.H. Subtype specificity of scorpion β-toxin tz1 interaction with voltage-gated sodium channels is determined by the pore loop of domain 3. Mol. Pharmacol. 2006, 70, 340–347. [Google Scholar] [CrossRef]
- Cohen, L.; Ilan, N.; Gur, M.; Stühmer, W.; Gordon, D.; Gurevitz, M. Design of a specific activator for skeletal muscle sodium channels uncovers channel architecture. J. Biol. Chem. 2007, 282, 29424–29430. [Google Scholar] [CrossRef] [PubMed]
- Leipold, E.; Borges, A.; Heinemann, S.H. Scorpion β-toxin interference with nav channel voltage sensor gives rise to excitatory and depressant modes. J. Gen. Physiol. 2012, 139, 305–319. [Google Scholar] [CrossRef] [PubMed]
- Deuis, J.R.; Dekan, Z.; Inserra, M.C.; Lee, T.-H.; Aguilar, M.-I.; Craik, D.J.; Lewis, R.J.; Alewood, P.F.; Mobli, M.; Schroeder, C.I. Development of a μo-conotoxin analogue with improved lipid membrane interactions and potency for the analgesic sodium channel nav1. 8. J. Biol. Chem. 2016, 291, 11829–11842. [Google Scholar] [CrossRef] [PubMed]
- Heinemann, S.; Leipold, E. Conotoxins of the o-superfamily affecting voltage-gated sodium channels. Cell. Mol. Life Sci. 2007, 64, 1329–1340. [Google Scholar] [CrossRef] [PubMed]
- Fainzilber, M.; Kofman, O.; Zlotkin, E.; Gordon, D. A new neurotoxin receptor site on sodium channels is identified by a conotoxin that affects sodium channel inactivation in molluscs and acts as an antagonist in rat brain. J. Biol. Chem. 1994, 269, 2574–2580. [Google Scholar] [PubMed]
- Shon, K.-J.; Hasson, A.; Spira, M.E.; Cruz, L.J.; Gray, W.R.; Olivera, B.M. Delta.-conotoxin gmvia, a novel peptide from the venom of conus gloriamaris. Biochemistry 1994, 33, 11420–11425. [Google Scholar] [CrossRef] [PubMed]
- Peigneur, S.; Paolini-Bertrand, M.; Gaertner, H.; Biass, D.; Violette, A.; Stöcklin, R.; Favreau, P.; Tytgat, J.; Hartley, O. Δ-conotoxins synthesized using an acid-cleavable solubility tag approach reveal key structural determinants for nav subtype selectivity. J. Biol. Chem. 2014, 289, 35341–35350. [Google Scholar] [CrossRef] [PubMed]
- Fainzilber, M.; Lodder, J.C.; Kits, K.S.; Kofman, O.; Vinnitsky, I.; Van Rietschoten, J.; Zlotkin, E.; Gordon, D. A new conotoxin affecting sodium current inactivation interacts with the-conotoxin receptor site. J. Biol. Chem. 1995, 270, 1123–1129. [Google Scholar] [CrossRef]
- Leipold, E.; Hansel, A.; Olivera, B.M.; Terlau, H.; Heinemann, S.H. Molecular interaction of δ-conotoxins with voltage-gated sodium channels. FEBS Lett. 2005, 579, 3881–3884. [Google Scholar] [CrossRef]
- Tietze, D.; Leipold, E.; Heimer, P.; Böhm, M.; Winschel, W.; Imhof, D.; Heinemann, S.H.; Tietze, A.A. Molecular interaction of δ-conopeptide evia with voltage-gated na+ channels. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2016, 1860, 2053–2063. [Google Scholar] [CrossRef] [PubMed]
- Green, B.R.; Bulaj, G.; Norton, R.S. Structure and function of μ-conotoxins, peptide-based sodium channel blockers with analgesic activity. Future Med. Chem. 2014, 6, 1677–1698. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wu, Q.; Pi, C.; Zhao, Y.; Zhou, M.; Wang, L.; Chen, S.; Xu, A. Isolation and characterization of a t-superfamily conotoxin from conus litteratus with targeting tetrodotoxin-sensitive sodium channels. Peptides 2007, 28, 2313–2319. [Google Scholar] [CrossRef]
- Norton, R.S. µ-conotoxins as leads in the development of new analgesics. Molecules 2010, 15, 2825–2844. [Google Scholar] [CrossRef]
- Zhang, M.M.; McArthur, J.R.; Azam, L.; Bulaj, G.; Olivera, B.M.; French, R.J.; Yoshikami, D. Unexpected synergism between tetrodotoxin and μ-conotoxin in blocking voltage-gated sodium channels. Channels 2009, 3, 32–38. [Google Scholar] [CrossRef]
- Stephan, M.; Potts, J.; Agnew, W. The μi skeletal muscle sodium channel: Mutation e403q eliminates sensitivity to tetrodotoxin but not to μ-conotoxins giiia and giiib. J. Membr. Biol. 1994, 137, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Leipold, E.; Ullrich, F.; Thiele, M.; Tietze, A.A.; Terlau, H.; Imhof, D.; Heinemann, S.H. Subtype-specific block of voltage-gated k+ channels by μ-conopeptides. Biochem. Biophys. Res. Commun. 2017, 482, 1135–1140. [Google Scholar] [CrossRef]
- Kaufmann, D.; Tietze, A.A.; Tietze, D. In silico analysis of the subtype selective blockage of kcna ion channels through the µ-conotoxins piiia, siiia, and giiia. Mar. Drugs 2019, 17, 180. [Google Scholar] [CrossRef]
- Xue, T.; Ennis, I.L.; Sato, K.; French, R.J.; Li, R.A. Novel interactions identified between μ-conotoxin and the na+ channel domain i p-loop: Implications for toxin-pore binding geometry. Biophys. J. 2003, 85, 2299–2310. [Google Scholar] [CrossRef]
- French, R.J.; Yoshikami, D.; Sheets, M.F.; Olivera, B.M. The tetrodotoxin receptor of voltage-gated sodium channels—Perspectives from interactions with μ-conotoxins. Mar. Drugs 2010, 8, 2153–2161. [Google Scholar] [CrossRef]
- Choudhary, G.; Aliste, M.P.; Tieleman, D.P.; French, R.J.; Dudley, J.; Samuel, C. Docking of μ-conotoxin giiia in the sodium channel outer vestibule. Channels 2007, 1, 344–352. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.; Romantseva, L.; Lam, A.; Lipkind, G.; Fozzard, H. Role of outer ring carboxylates of the rat skeletal muscle sodium channel pore in proton block. J. Physiol. 2002, 543, 71–84. [Google Scholar] [CrossRef] [PubMed]
- Mahdavi, S.; Kuyucak, S. Systematic study of binding of μ-conotoxins to the sodium channel nav1. 4. Toxins 2014, 6, 3454–3470. [Google Scholar] [CrossRef] [PubMed]
- Korkosh, V.S.; Zhorov, B.S.; Tikhonov, D.B. Folding similarity of the outer pore region in prokaryotic and eukaryotic sodium channels revealed by docking of conotoxins giiia, piiia, and kiiia in a navab-based model of nav1. 4. J. Gen. Physiol. 2014, 144, 231–244. [Google Scholar] [CrossRef] [PubMed]
- Patel, D.; Mahdavi, S.; Kuyucak, S. Computational study of binding of μ-conotoxin giiia to bacterial sodium channels navab and navrh. Biochemistry 2016, 55, 1929–1938. [Google Scholar] [CrossRef]
- Mahdavi, S.; Kuyucak, S. Molecular dynamics study of binding of µ-conotoxin giiia to the voltage-gated sodium channel nav1. 4. PLoS ONE 2014, 9, e105300. [Google Scholar] [CrossRef]
- Cummins, T.R.; Aglieco, F.; Dib-Hajj, S.D. Critical molecular determinants of voltage-gated sodium channel sensitivity to μ-conotoxins giiia/b. Mol. Pharmacol. 2002, 61, 1192–1201. [Google Scholar] [CrossRef]
- Catterall, W.A. Structure and regulation of voltage-gated Ca2+ channels. Annu. Rev. Cell Develop. Biol. 2000, 16, 521–555. [Google Scholar] [CrossRef]
- Catterall, W.A.; Swanson, T.M. Structural basis for pharmacology of voltage-gated sodium and calcium channels. Mol. Pharmacol. 2015, 88, 141–150. [Google Scholar] [CrossRef]
- Wu, J.; Yan, Z.; Li, Z.; Qian, X.; Lu, S.; Dong, M.; Zhou, Q.; Yan, N. Structure of the voltage-gated calcium channel ca v 1.1 at 3.6 å resolution. Nature 2016, 537, 191. [Google Scholar] [CrossRef]
- Hering, S.; Zangerl-Plessl, E.-M.; Beyl, S.; Hohaus, A.; Andranovits, S.; Timin, E. Calcium channel gating. Pflügers Archiv-Eur. J. Physiol. 2018, 470, 1291–1309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dolphin, A.C. Voltage-gated calcium channels: Their discovery, function and importance as drug targets. Brain Neurosci. Adv. 2018, 2, 2398212818794805. [Google Scholar] [CrossRef] [PubMed]
- Zamponi, G.W.; Striessnig, J.; Koschak, A.; Dolphin, A.C. The physiology, pathology, and pharmacology of voltage-gated calcium channels and their future therapeutic potential. Pharmacol. Rev. 2015, 67, 821–870. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, K.J.; Schroeder, T.; Lewis, R. Structure–activity relationships of ω-conotoxins at n-type voltage-sensitive calcium channels. J. Mol. Recognit. 2000, 13, 55–70. [Google Scholar] [CrossRef]
- Neumaier, F.; Dibue-Adjei, M.; Hescheler, J.; Schneider, T. Voltage-gated calcium channels: Determinants of channel function and modulation by inorganic cations. Prog. Neurobiol. 2015, 129, 1–36. [Google Scholar] [CrossRef]
- Jurkovicova-Tarabova, B.; Lacinova, L. Structure, function and regulation of cav 2.2 n-type calcium channels. Gen. Physiol. Biophys. 2019, 38, 101–110. [Google Scholar] [CrossRef] [PubMed]
- Miljanich, G. Ziconotide: Neuronal calcium channel blocker for treating severe chronic pain. Curr. Med. Chem. 2004, 11, 3029–3040. [Google Scholar] [CrossRef]
- Pallaghy, P.K.; Norton, R.S.; Nielsen, K.J.; Craik, D.J. A common structural motif incorporating a cystine knot and a triple-stranded β-sheet in toxic and inhibitory polypeptides. Protein Sci. 1994, 3, 1833–1839. [Google Scholar] [CrossRef]
- Kohno, T.; Kim, J.I.; Kobayashi, K.; Kodera, Y.; Maeda, T.; Sato, K. Three-dimensional structure in solution of the calcium channel blocker. Omega.-conotoxin mviia. Biochemistry 1995, 34, 10256–10265. [Google Scholar] [CrossRef]
- Adams, D.J.; Berecki, G. Mechanisms of conotoxin inhibition of n-type (cav2. 2) calcium channels. Biochim. Biophys. Acta (BBA)-Biomembr. 2013, 1828, 1619–1628. [Google Scholar] [CrossRef]
- Hansson, K.; Ma, X.; Eliasson, L.; Czerwiec, E.; Furie, B.; Furie, B.C.; Rorsman, P.; Stenflo, J. The first γ-carboxyglutamic acid-containing contryphan a selective l-type calcium ion channel blocker isolated from the venom of conus marmoreus. J. Biol. Chem. 2004, 279, 32453–32463. [Google Scholar] [CrossRef] [PubMed]
- Bernáldez, J.; Román-González, S.; Martínez, O.; Jiménez, S.; Vivas, O.; Arenas, I.; Corzo, G.; Arreguín, R.; García, D.; Possani, L. A conus regularis conotoxin with a novel eight-cysteine framework inhibits cav2. 2 channels and displays an anti-nociceptive activity. Mar. Drugs 2013, 11, 1188–1202. [Google Scholar] [CrossRef] [PubMed]
- Flinn, J.P.; Pallaghy, P.K.; Lew, M.J.; Murphy, R.; Angus, J.A.; Norton, R.S. Roles of key functional groups in ω-conotoxin gvia: Synthesis, structure and functional assay of selected peptide analogues. Eur. J. Biochem. 1999, 262, 447–455. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, C.; Doering, C.; Zamponi, G.; Lewis, R. N-type calcium channel blockers: Novel therapeutics for the treatment of pain. Med. Chem. 2006, 2, 535–543. [Google Scholar] [CrossRef] [PubMed]
- Sato, K.; Park, N.G.; Kohno, T.; Maeda, T.; Kim, J.I.; Kato, R.; Takahashi, M. Role of basic residues for the binding of ω-conotoxin gvia to n-type calcium channels. Biochem. Biophys. Res. Commun. 1993, 194, 1292–1296. [Google Scholar] [CrossRef] [PubMed]
- Lew, M.J.; Flinn, J.P.; Pallaghy, P.K.; Murphy, R.; Whorlow, S.L.; Wright, C.E.; Norton, R.S.; Angus, J.A. Structure-function relationships of ω-conotoxin gvia synthesis, structure, calcium channel binding, and functional assay of alanine-substituted analogues. J. Biol. Chem. 1997, 272, 12014–12023. [Google Scholar] [CrossRef] [PubMed]
- Ellinor, P.T.; Zhang, J.-F.; Horne, W.A.; Tsien, R.W. Structural determinants of the blockade of n-type calcium channels by a peptide neurotoxin. Nature 1994, 372, 272. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.-P.; Hamid, J.; Doering, C.; Bosey, G.M.; Snutch, T.P.; Zamponi, G.W. Residue gly1326 of the n-type calcium channel α1b subunit controls reversibility of ω-conotoxin gvia and mviia block. J. Biol. Chem. 2001, 276, 15728–15735. [Google Scholar] [CrossRef]
- Chen, R.; Chung, S.-H. Complex structures between the n-type calcium channel (cav2. 2) and ω-conotoxin gvia predicted via molecular dynamics. Biochemistry 2013, 52, 3765–3772. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Family | Target and Mode of Action | Reference |
|---|---|---|
| α-conotoxins | Inhibitory competitors of nicotinic acetylcholine receptors (nAChR) | [34] |
| γ-conotoxins | Acting on neuronal pacemaker currents affecting inward cation currents | [35] |
| δ-conotoxins | Acting on voltage-gated sodium (Na+) channel VGSCs, activating and inactivating them | [36] |
| ε-conotoxins | Acting on G-protein-coupled presynaptic receptors or calcium channels | [37] |
| ι-conotoxins | Activating VGSCs | [38] |
| κ-conotoxins | Blocking voltage-gated potassium (K+) channel VGKCs | [39] |
| µ-conotoxins | Blocking VGSCs | [40] |
| ρ-conotoxins | Inhibitors of alpha1-adrenoreceptors (GPCR) | [41] |
| σ-conotoxins | Acting on serotonin gated ion channels 5-HT3 | [42] |
| τ-conotoxins | Acting on somatostatin receptors | [43] |
| χ-conotoxins | Inhibitors of neuronal noradrenaline transporters | [41] |
| ω-conotoxins | Acting on voltage-gated calcium (Ca++) channel VGCCs | [44] |
| α-CTx | Primary Sequence | Loop Class | Reference |
|---|---|---|---|
| Framework | and Cys pair connectivity  | m/n | |
| GI | ECCNPACGRHYSCGK * | 3/5 | [55] |
| ImI | GCCSDPRCAWRC * | 4/3 | [56] |
| BuIA | GCCSTPPCAVLYC* | 4/4 | [57] |
| AuIB | GCCSYPPCFATNPDC * | 4/6 | [58] |
| Vc1.1 | GCCSDPRCNYDHPEIC * | 4/7 | [59] |
| Other frameworks | |||
| αJ-pl14a | FPRPRICNLACRAGIGHKYPFCHCR * | X | [60] |
| αS-RVIIIA | KCNFDKCKGTGVYNCG(Gla)SCSC(Gla)GLHSCRCTYNIGSMKSGCACICTYY | X | [61] |
| αD-VxXXB | DD(Gla)S(Gla)CIINTRDSPWGRCCRTRMCGSMCCPRNGCTCVYHWRRGHGCSCPG (dimer) | X | [62] |
| α-Conotoxin | nAChR Type Target (IC50) | Reference |
|---|---|---|
| ArIB | rα7 (1.81 nM) > rα6/α3β2β3 (6.45 nM) > rα3β2 (60.1 nM) | [71] |
| BuIA | rα6/α3β2 (0.258 nM) > rα6/α3β4 (1.54 nM) > rα3β2 (5.72 nM) > rα3β4 (27.7 nM) | [57] |
| GIC | hα3β2 (1.1 nM) > hα4β2 (309 nM) > hα3β4 (755 nM) | [72] |
| GID | rα3β2 (3.1 nM) > rα7 (4.5 nM) > rα4β2 (152 nM) | [73] |
| ImI | rα7 (220 nM) > rα7 (1.8 μM) > mα1β1γδ (51 μM)hα3β2 (40.8 nM) > hα7 (595 nM) | [74] |
| Lt1.3 | rα3β2 (44.8 nM) | [75] |
| MII | rα6/α3β2β3 (0.39 nM) > rα3β2 (2.18 nM) | [76] |
| PeIA | rα9α10 (6.9 nM) > rα6/α3β2β3 (17.2 nM) > rα3β2 (19.2 nM) > rα3β4 (480 nM) | [77] |
| PnIA | rα3β2 (9.56 nM) > rα7 (252 nM) | [78] |
| TxIB | rα6/α3β2β3 (28 nM) | [79] |
| TxID | rα3β4 (12.5 nM) > rα6/α3β4 (94 nM) > rα3β4 (4.5μM) rα3β4 (3.6 nM) > rα6/α3β4 (34 nM) | [80] |
| Vc1.1 | rα3β4 (4.2 μM) > rα3α5β2 (7.2 μM) > rα3β2 (7.3 μM) | [59] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morales Duque, H.; Campos Dias, S.; Franco, O.L. Structural and Functional Analyses of Cone Snail Toxins. Mar. Drugs 2019, 17, 370. https://doi.org/10.3390/md17060370
Morales Duque H, Campos Dias S, Franco OL. Structural and Functional Analyses of Cone Snail Toxins. Marine Drugs. 2019; 17(6):370. https://doi.org/10.3390/md17060370
Chicago/Turabian StyleMorales Duque, Harry, Simoni Campos Dias, and Octávio Luiz Franco. 2019. "Structural and Functional Analyses of Cone Snail Toxins" Marine Drugs 17, no. 6: 370. https://doi.org/10.3390/md17060370
APA StyleMorales Duque, H., Campos Dias, S., & Franco, O. L. (2019). Structural and Functional Analyses of Cone Snail Toxins. Marine Drugs, 17(6), 370. https://doi.org/10.3390/md17060370