A Marine Alkaloid, Ascomylactam A, Suppresses Lung Tumorigenesis via Inducing Cell Cycle G1/S Arrest through ROS/Akt/Rb Pathway

 and
and
Abstract
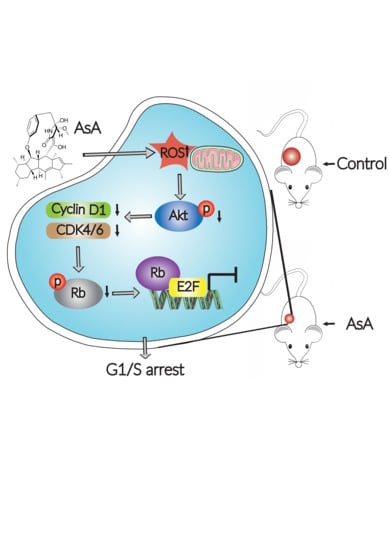
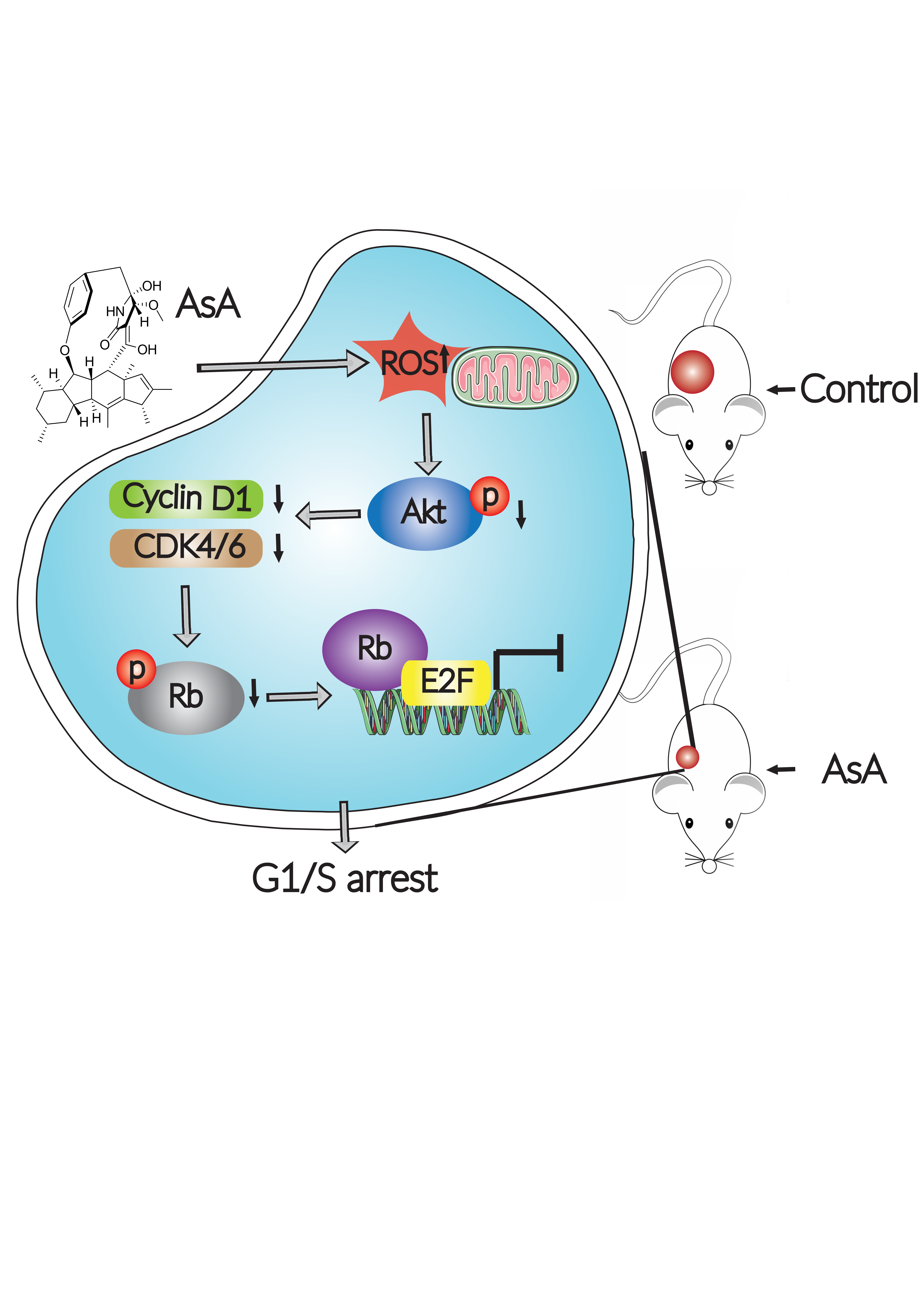
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
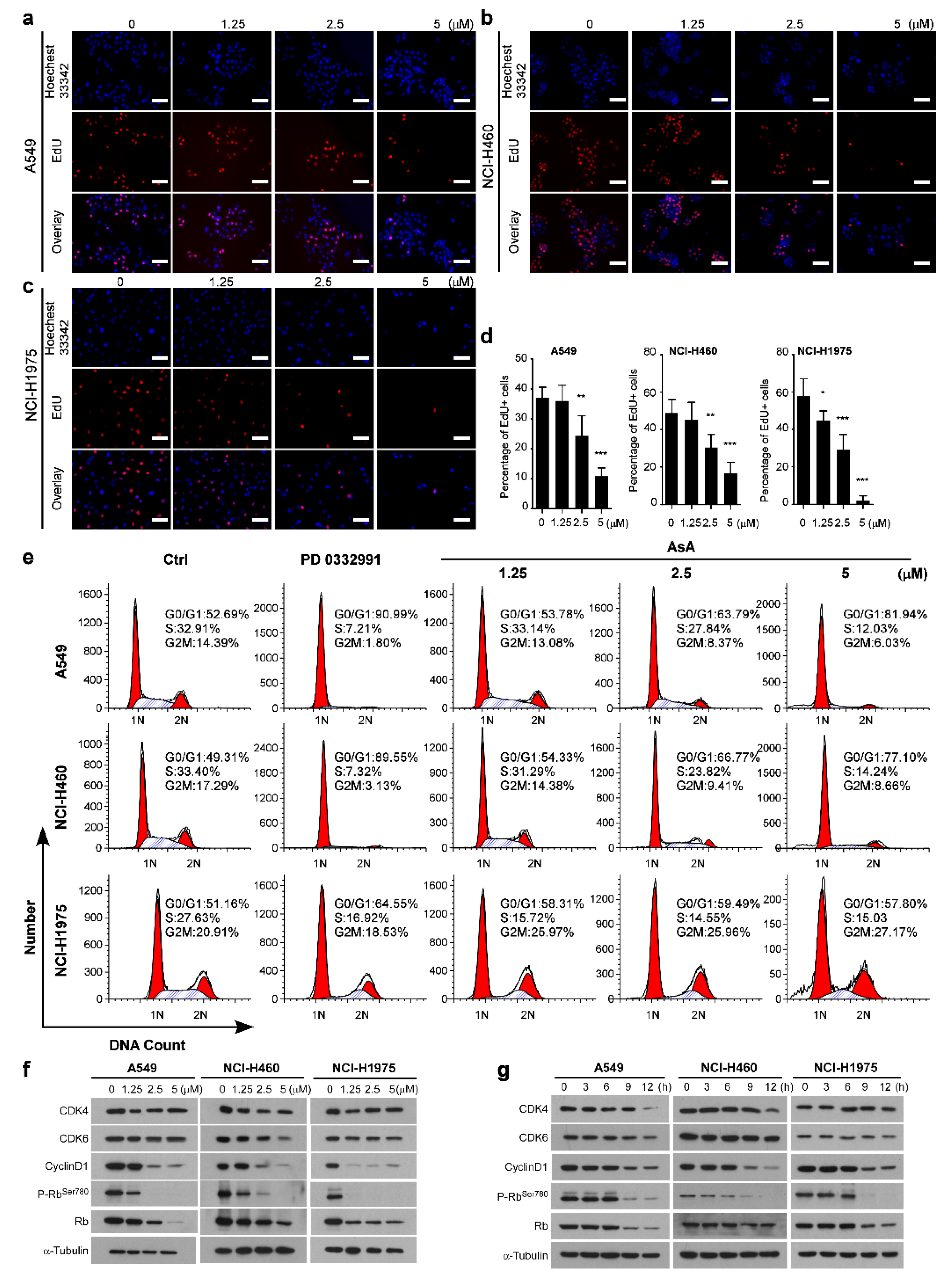
2.1. AsA Inhibits the Proliferation of Lung Cancer Cells In Vitro
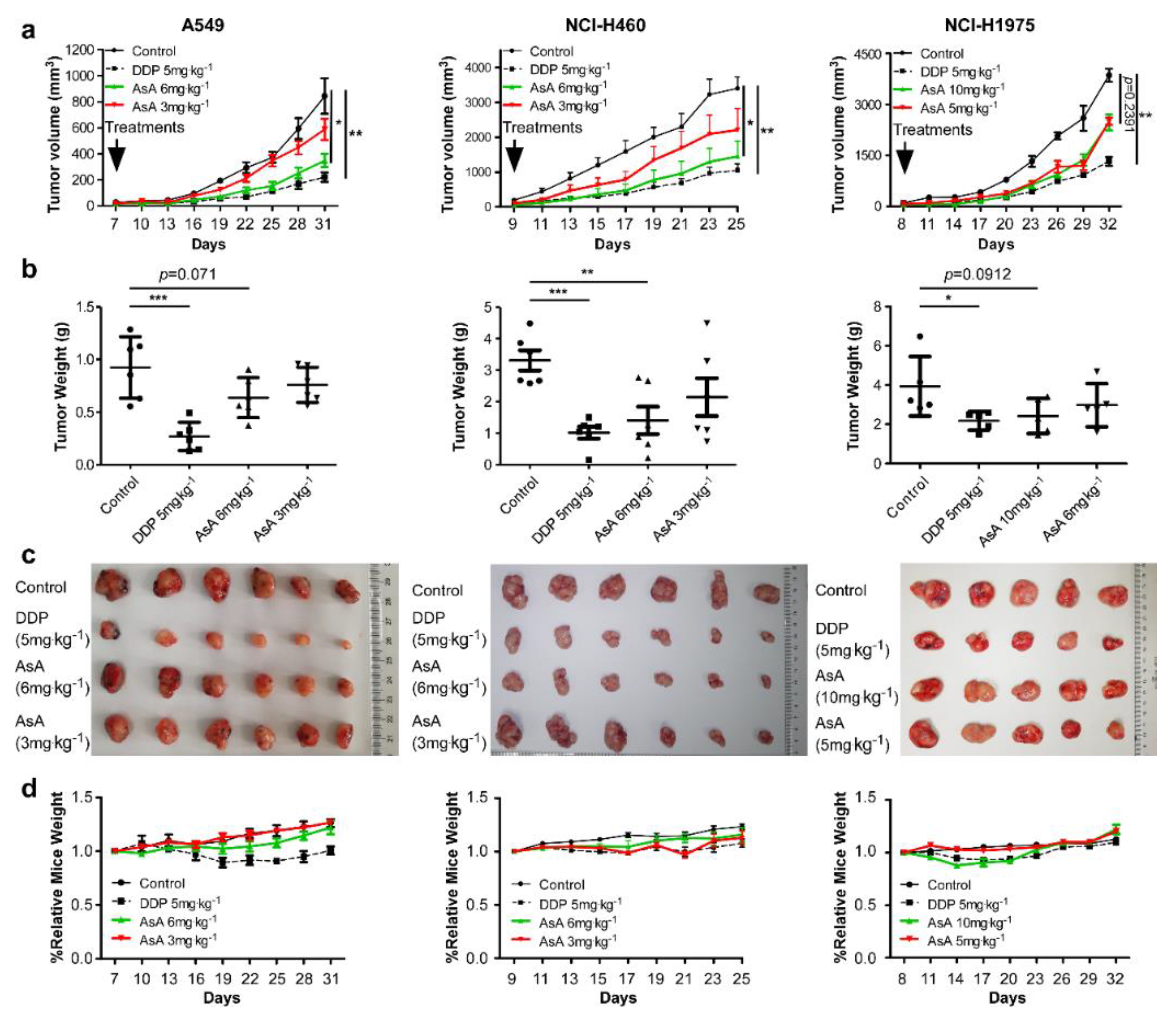
2.2. AsA Suppresses NSCLC Cells Growth In Vivo
2.3. AsA Induces G1/S Cell Cycle Arrest in Lung Cancer Cells
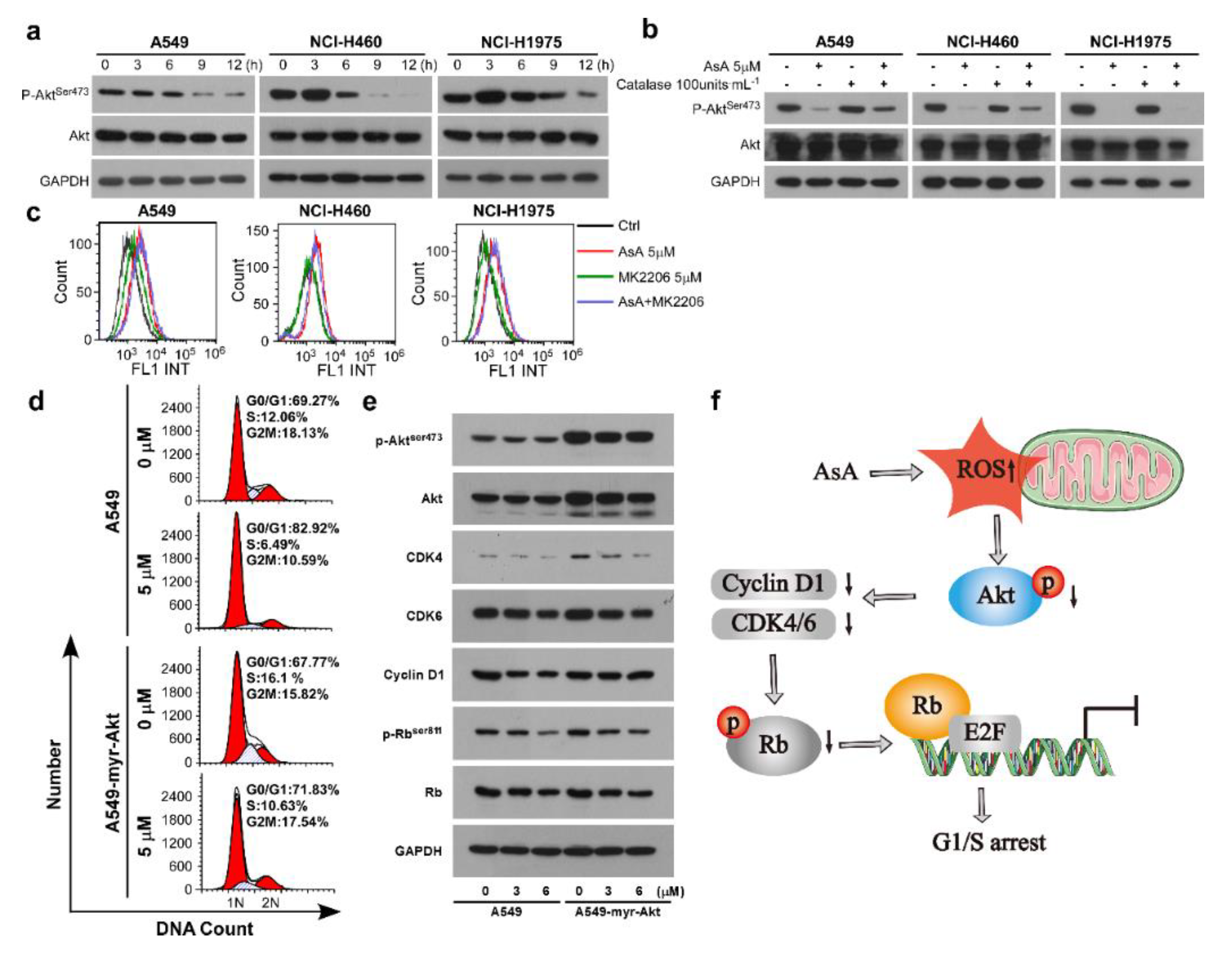
2.4. AsA Blocks the Cell Cycle via Upregulating ROS
2.5. AsA Induces the ROS-Dependent Inactivation of PI3K/Akt Pathway in Lung Cancer Cells
3. Discussion
4. Materials and Methods
4.1. Preparation of AsA
4.2. Cell Culture
4.3. Cell Viability Assay
4.4. Clonogenic Assay
4.5. Anchorage-Independent Soft Agar Assay
4.6. 5-Ethynyl-20-Deoxyuridine Assay
4.7. Cell Cycle Analysis
4.8. Western Blotting Analysis
4.9. Detection of Intracellular ROS
4.10. Chemicals and Fluorescent Probes for Studying ROS Generation
4.11. In Vivo Assay
4.12. Statistical Analysis
4.13. Materials
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Global Burden of Disease Cancer Collaboration; Fitzmaurice, C.; Abate, D.; Abbasi, N.; Abbastabar, H.; Abd-Allah, F.; Abdel-Rahman, O.; Abdelalim, A.; Abdoli, A.; Abdollahpour, I.; et al. Global, Regional, and National Cancer Incidence, Mortality, Years of Life Lost, Years Lived With Disability, and Disability-Adjusted Life-Years for 29 Cancer Groups, 1990 to 2017. JAMA Oncol. 2019, 5, 1749. [Google Scholar] [CrossRef] [Green Version]
- Siegel, R.L.; Mph, K.D.M.; Jemal, A. Cancer statistics, 2020. CA A Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Molina, J.R.; Yang, P.; Cassivi, S.D.; Schild, S.E.; Adjei, A.A. Non-Small Cell Lung Cancer: Epidemiology, Risk Factors, Treatment, and Survivorship. Mayo Clin. Proc. 2008, 83, 584–594. [Google Scholar] [CrossRef]
- Herbst, R.S.; Morgensztern, D.; Boshoff, C. The biology and management of non-small cell lung cancer. Nature 2018, 553, 446–454. [Google Scholar] [CrossRef] [PubMed]
- Newman, D.J.; Cragg, G.M. Natural Products as Sources of New Drugs from 1981 to 2014. J. Nat. Prod. 2016, 79, 629–661. [Google Scholar] [CrossRef] [Green Version]
- Olano, C.; Méndez, C.; Salas, J.A. Antitumor Compounds from Marine Actinomycetes. Mar. Drugs 2009, 7, 210–248. [Google Scholar] [CrossRef] [Green Version]
- Shin, S.-W.; Seo, C.-Y.; Han, H.; Han, J.-Y.; Jeong, J.-S.; Kwak, J.-Y.; I Park, J. 15d-PGJ2 Induces Apoptosis by Reactive Oxygen Species-mediated Inactivation of Akt in Leukemia and Colorectal Cancer Cells and Shows In vivo Antitumor Activity. Clin. Cancer Res. 2009, 15, 5414–5425. [Google Scholar] [CrossRef] [Green Version]
- Zhou, M.; Shen, S.; Zhao, X.; Gong, X. Luteoloside induces G0/G1 arrest and pro-death autophagy through the ROS-mediated AKT/mTOR/p70S6K signalling pathway in human non-small cell lung cancer cell lines. Biochem. Biophys. Res. Commun. 2017, 494, 263–269. [Google Scholar] [CrossRef]
- Yang, X.; Guo, F.; Peng, Q.; Liu, Y.; Yang, B. Suppression of in vitro and in vivo human ovarian cancer growth by isoacteoside is mediated via sub-G1 cell cycle arrest, ROS generation, and modulation of AKT/PI3K/m-TOR signalling pathway. J. BUON Off. J. Balk. Union Oncol. 2019, 24, 285–290. [Google Scholar]
- Schumacker, P.T. Reactive oxygen species in cancer cells: Live by the sword, die by the sword. Cancer Cell 2006, 10, 175–176. [Google Scholar] [CrossRef] [Green Version]
- Sabharwal, S.S.; Schumacker, P.T. Mitochondrial ROS in cancer: Initiators, amplifiers or an Achilles’ heel? Nat. Rev. Cancer 2014, 14, 709–721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Song, Y.C.; Tan, R.-X. Biology and chemistry of endophytes. Nat. Prod. Rep. 2006, 23, 753–771. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Liu, Z.; Huang, Y.; Liu, L.; He, J.; Wang, L.; Yuan, J.; She, Z. Ascomylactams A–C, Cytotoxic 12- or 13-Membered-Ring Macrocyclic Alkaloids Isolated from the Mangrove Endophytic Fungus Didymella sp. CYSK-4, and Structure Revisions of Phomapyrrolidones A and C. J. Nat. Prod. 2019, 82, 1752–1758. [Google Scholar] [CrossRef] [PubMed]
- Dias, R.B.; De Araújo, T.B.S.; Freitas, R.; Rodrigues, A.C.B.D.C.; Sousa, L.P.; Sales, C.B.S.; Valverde, L.D.F.; Soares, M.B.P.; Dos Reis, M.G.; Della Coletta, R.; et al. β-Lapachone and its iodine derivatives cause cell cycle arrest at G2/M phase and reactive oxygen species-mediated apoptosis in human oral squamous cell carcinoma cells. Free Radic. Boil. Med. 2018, 126, 87–100. [Google Scholar] [CrossRef]
- Al-Oqail, M.M.; Siddiqui, M.; Al-Sheddi, E.S.; Saquib, Q.; Musarrat, J.; Alkhedhairy, A.A.; Farshori, N.N. Verbesina encelioides: Cytotoxicity, cell cycle arrest, and oxidative DNA damage in human liver cancer (HepG2) cell line. BMC Complement. Altern. Med. 2016, 16, 126. [Google Scholar] [CrossRef] [Green Version]
- Shahrestanaki, M.K.; Bagheri, M.; Ghanadian, M.; Aghaei, M.; Jafari, S.M. Centaurea cyanus extracted 13-O-acetylsolstitialin A decrease Bax/Bcl-2 ratio and expression of cyclin D1/Cdk-4 to induce apoptosis and cell cycle arrest in MCF-7 and MDA-MB-231 breast cancer cell lines. J. Cell. Biochem. 2019, 120, 18309–18319. [Google Scholar] [CrossRef]
- Luo, J.; Manning, B.D.; Cantley, L.C. Targeting the PI3K-Akt pathway in human cancer. Cancer Cell 2003, 4, 257–262. [Google Scholar] [CrossRef] [Green Version]
- Hu, Y.; Chen, J.; Hu, G.-P.; Yu, J.; Zhu, X.; Lin, Y.-C.; Chen, S.-P.; Yuan, J. Statistical Research on the Bioactivity of New Marine Natural Products Discovered during the 28 Years from 1985 to 2012. Mar. Drugs 2015, 13, 202–221. [Google Scholar] [CrossRef]
- Dias, D.A.; Urban, S.; Roessner, U. A Historical Overview of Natural Products in Drug Discovery. Metabolites 2012, 2, 303–336. [Google Scholar] [CrossRef] [Green Version]
- Shiono, Y.; Shimanuki, K.; Hiramatsu, F.; Koseki, T.; Tetsuya, M.; Fujisawa, N.; Kimura, K.-I. Pyrrospirones A and B, apoptosis inducers in HL-60 cells, from an endophytic fungus, Neonectria ramulariae Wollenw KS-246. Bioorganic Med. Chem. Lett. 2008, 18, 6050–6053. [Google Scholar] [CrossRef]
- Uesugi, S.; Fujisawa, N.; Yoshida, J.; Watanabe, M.; Dan, S.; Yamori, T.; Shiono, Y.; Kimura, K.-I. Pyrrocidine A, a metabolite of endophytic fungi, has a potent apoptosis-inducing activity against HL60 cells through caspase activation via the Michael addition. J. Antibiot. 2015, 69, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Ebrahim, W.; Aly, A.H.; Wray, V.; Mándi, A.; Teiten, M.-H.; Gaascht, F.; Orlikova, B.; Kassack, M.U.; Lin, W.-H.; Diederich, M.; et al. Embellicines A and B: Absolute Configuration and NF-κB Transcriptional Inhibitory Activity. J. Med. Chem. 2013, 56, 2991–2999. [Google Scholar] [CrossRef] [PubMed]
- Kang, M.J.; Wu, T.; Wijeratne, E.M.K.; Lau, E.C.; Mason, D.J.; Mesa, C.; Tillotson, J.; Zhang, N.D.; Gunatilaka, A.A.L.; La Clair, J.J.; et al. Functional chromatography reveals three natural products that target the same protein with distinct mechanisms of action. ChemBioChem 2014, 15, 2125–2131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, M.; Liu, H.; Chen, J. Mechanisms of the CDK4/6 inhibitor palbociclib (PD 0332991) and its future application in cancer treatment (Review). Oncol. Rep. 2018. [Google Scholar] [CrossRef] [Green Version]
- Hei, T.K.; Liu, S.X.; Waldren, C. Mutagenicity of arsenic in mammalian cells: Role of reactive oxygen species. Proc. Natl. Acad. Sci. USA 1998, 95, 8103–8107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adler, V.; Yin, Z.; Tew, K.D.; Ronai, Z.A. Role of redox potential and reactive oxygen species in stress signaling. Oncogene 1999, 18, 6104–6111. [Google Scholar] [CrossRef] [Green Version]
- Gao, N.; Ding, M.; Zheng, J.Z.; Zhang, Z.; Leonard, S.S.; Liu, K.J.; Shi, X.; Jiang, B. Vanadate-induced Expression of Hypoxia-inducible Factor 1α and Vascular Endothelial Growth Factor through Phosphatidylinositol 3-Kinase/Akt Pathway and Reactive Oxygen Species. J. Boil. Chem. 2002, 277, 31963–31971. [Google Scholar] [CrossRef] [Green Version]
- Reuter, S.; Gupta, S.C.; Chaturvedi, M.M.; Aggarwal, B.B. Oxidative stress, inflammation, and cancer: How are they linked? Free Radic. Boil. Med. 2010, 49, 1603–1616. [Google Scholar] [CrossRef] [Green Version]
- Tafani, M.; Sansone, L.; Limana, F.; Arcangeli, T.; De Santis, E.; Polese, M.; Fini, M.; Russo, M.A. The Interplay of Reactive Oxygen Species, Hypoxia, Inflammation, and Sirtuins in Cancer Initiation and Progression. Oxidative Med. Cell. Longev. 2015, 2016, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Galadari, S.; Rahman, A.; Pallichankandy, S.; Thayyullathil, F. Reactive oxygen species and cancer paradox: To promote or to suppress? Free Radic. Boil. Med. 2017, 104, 144–164. [Google Scholar] [CrossRef]
- Weng, M.; Chang, J.-H.; Hung, W.-Y.; Yang, Y.-C.; Chien, M.-H. The interplay of reactive oxygen species and the epidermal growth factor receptor in tumor progression and drug resistance. J. Exp. Clin. Cancer Res. 2018, 37, 61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, Y.; Wang, X.; Hu, D. Furanodienone induces G0/G1 arrest and causes apoptosis via the ROS/MAPKs-mediated caspase-dependent pathway in human colorectal cancer cells: A study in vitro and in vivo. Cell Death Dis. 2017, 8, e2815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Qin, Y.; Yang, C.; Zhang, H.; Li, Y.; Wu, B.; Huang, J.; Zhou, X.; Huang, B.; Yang, K.; et al. Cardamonin induces ROS-mediated G2/M phase arrest and apoptosis through inhibition of NF-κB pathway in nasopharyngeal carcinoma. Cell Death Dis. 2017, 8, e3024. [Google Scholar] [CrossRef]
- Brazil, D.P.; Hemmings, B.A. Ten years of protein kinase B signalling: A hard Akt to follow. Trends Biochem. Sci. 2001, 26, 657–664. [Google Scholar] [CrossRef]
- Xie, G.; Zhu, X.; Li, Q.; Gu, M.; He, Z.; Wu, J.; Li, J.; Lin, Y.; Li, M.; She, Z.; et al. SZ-685C, a marine anthraquinone, is a potent inducer of apoptosis with anticancer activity by suppression of the Akt/FOXO pathway. Br. J. Pharmacol. 2010, 159, 689–697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, J.; Li, R.; Zhu, X.; Zhang, H.; Wu, J.; Zeng, M.; Song, E.; He, Y.; Yin, Y.; Li, J.; et al. CK1α suppresses lung tumour growth by stabilizing PTEN and inducing autophagy. Nature 2018, 20, 465–478. [Google Scholar] [CrossRef]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, L.; Huang, Y.; Huang, C.-h.; Yu, J.-c.; Zheng, Y.-c.; Chen, Y.; She, Z.-g.; Yuan, J. A Marine Alkaloid, Ascomylactam A, Suppresses Lung Tumorigenesis via Inducing Cell Cycle G1/S Arrest through ROS/Akt/Rb Pathway. Mar. Drugs 2020, 18, 494. https://doi.org/10.3390/md18100494
Wang L, Huang Y, Huang C-h, Yu J-c, Zheng Y-c, Chen Y, She Z-g, Yuan J. A Marine Alkaloid, Ascomylactam A, Suppresses Lung Tumorigenesis via Inducing Cell Cycle G1/S Arrest through ROS/Akt/Rb Pathway. Marine Drugs. 2020; 18(10):494. https://doi.org/10.3390/md18100494
Chicago/Turabian StyleWang, Lan, Yun Huang, Cui-hong Huang, Jian-chen Yu, Ying-chun Zheng, Yan Chen, Zhi-gang She, and Jie Yuan. 2020. "A Marine Alkaloid, Ascomylactam A, Suppresses Lung Tumorigenesis via Inducing Cell Cycle G1/S Arrest through ROS/Akt/Rb Pathway" Marine Drugs 18, no. 10: 494. https://doi.org/10.3390/md18100494
APA StyleWang, L., Huang, Y., Huang, C.-h., Yu, J.-c., Zheng, Y.-c., Chen, Y., She, Z.-g., & Yuan, J. (2020). A Marine Alkaloid, Ascomylactam A, Suppresses Lung Tumorigenesis via Inducing Cell Cycle G1/S Arrest through ROS/Akt/Rb Pathway. Marine Drugs, 18(10), 494. https://doi.org/10.3390/md18100494