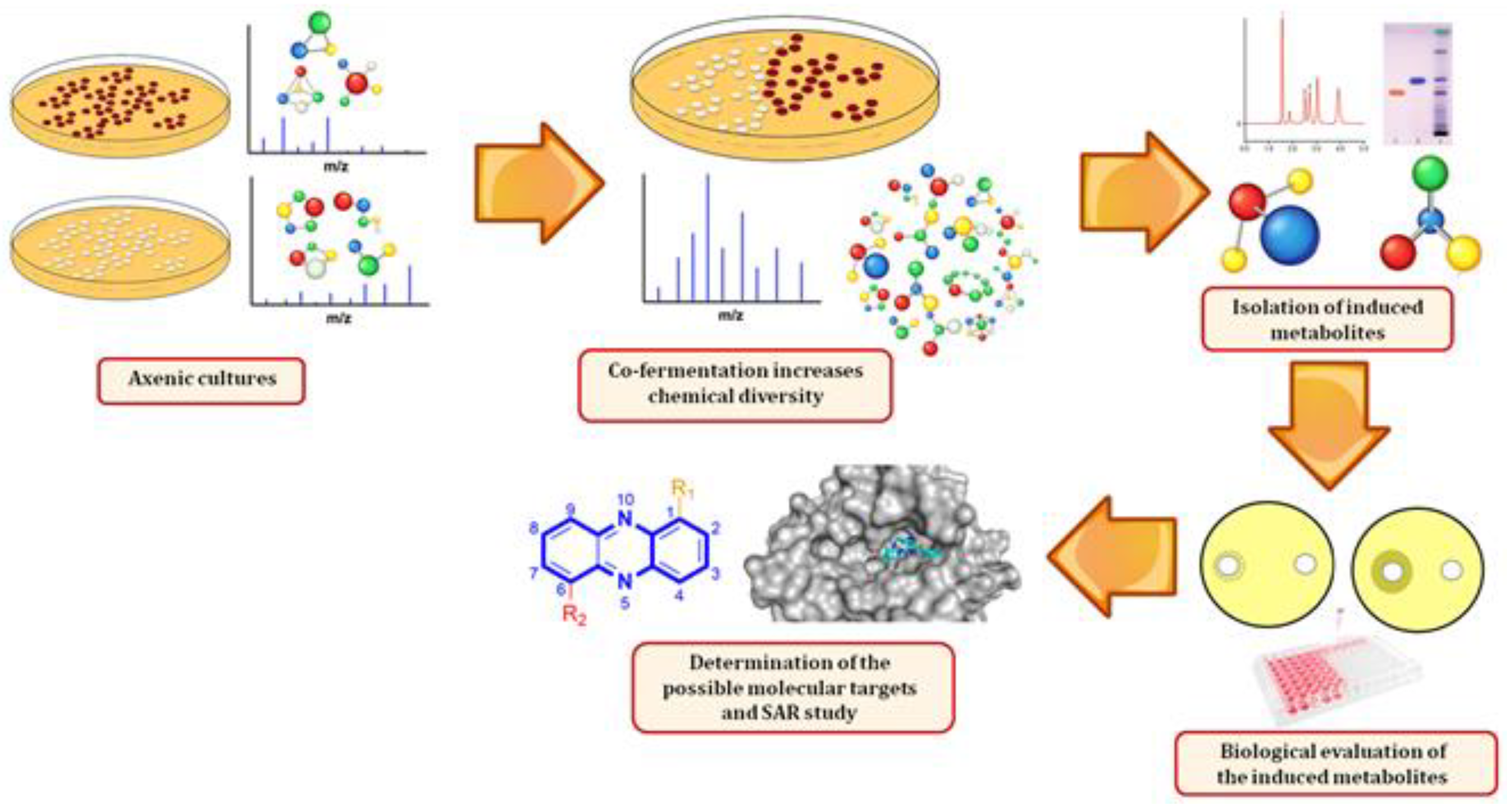
Induction of Antibacterial Metabolites by Co-Cultivation of Two Red-Sea-Sponge-Associated Actinomycetes Micromonospora sp. UR56 and Actinokinespora sp. EG49

 ,
, 
 ,
,  , ,
, ,  and
and 
Abstract
:
1. Introduction
2. Results and Discussion
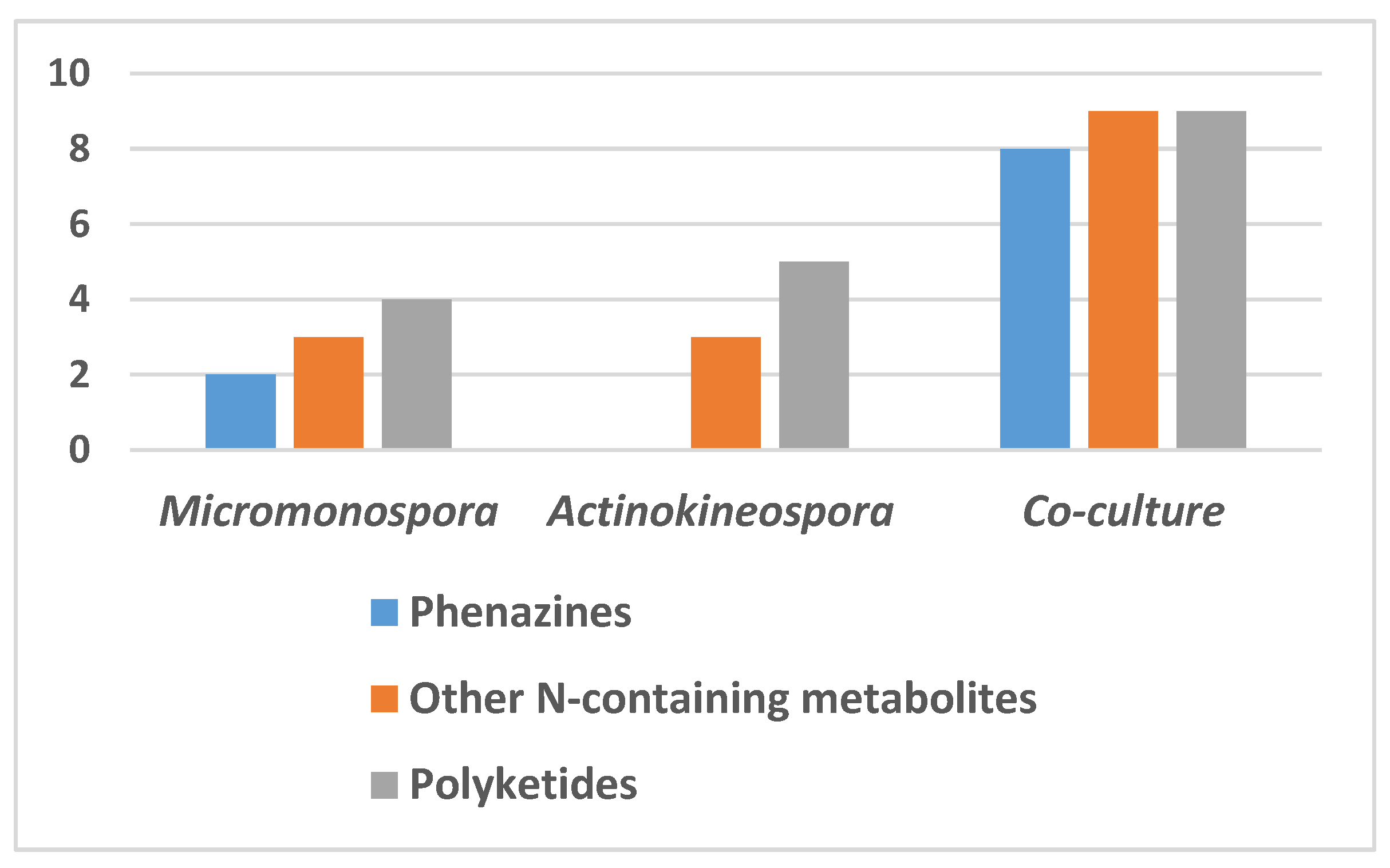
2.1. Metabolomic Profiles of the Axenic and Co-Culture Extracts
2.2. Bioactivity Testing
2.2.1. Antibacterial Activity
2.2.2. Antibiofilm Activity
2.2.3. Cytotoxic Activity
2.2.4. In Vitro Enzyme Assay
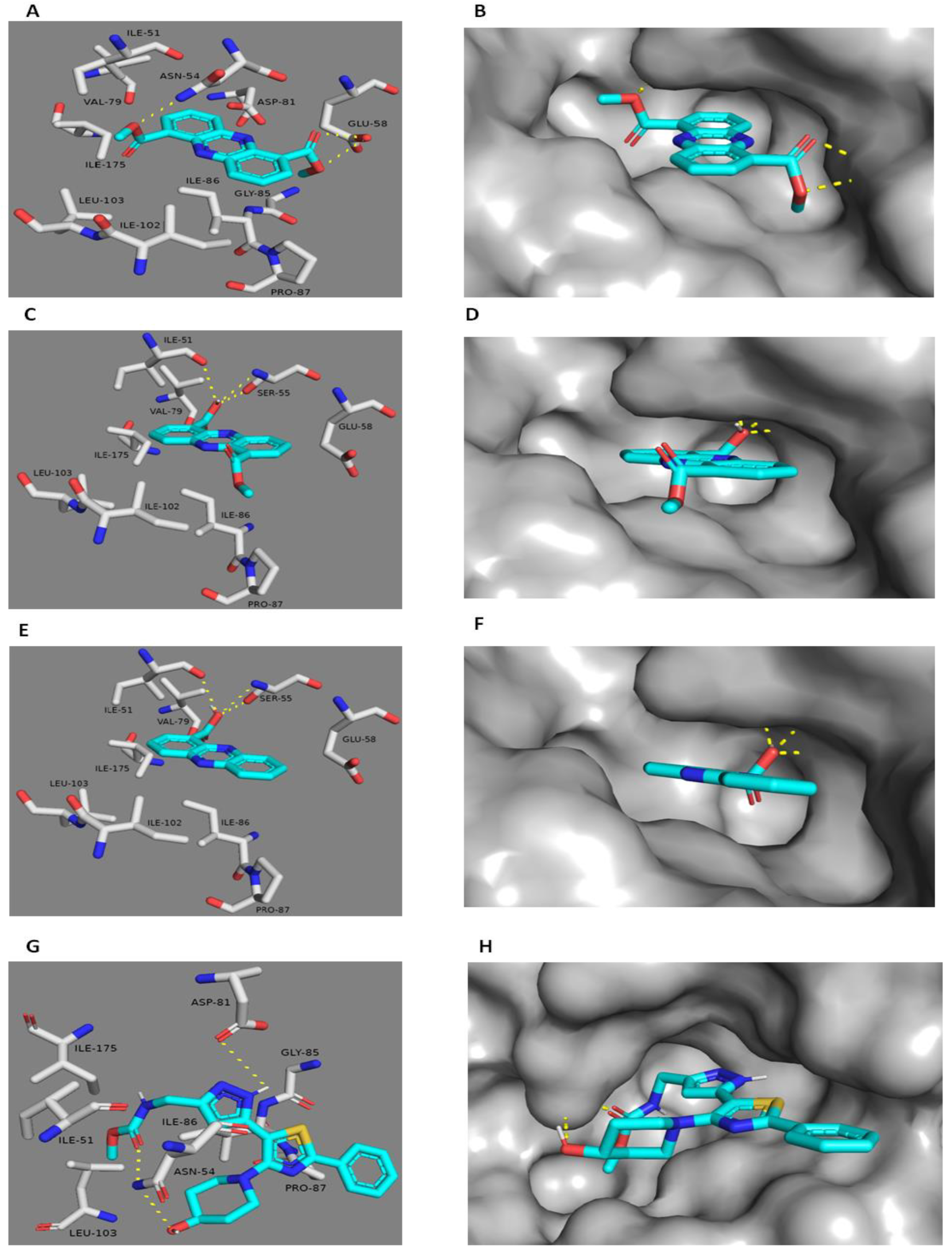
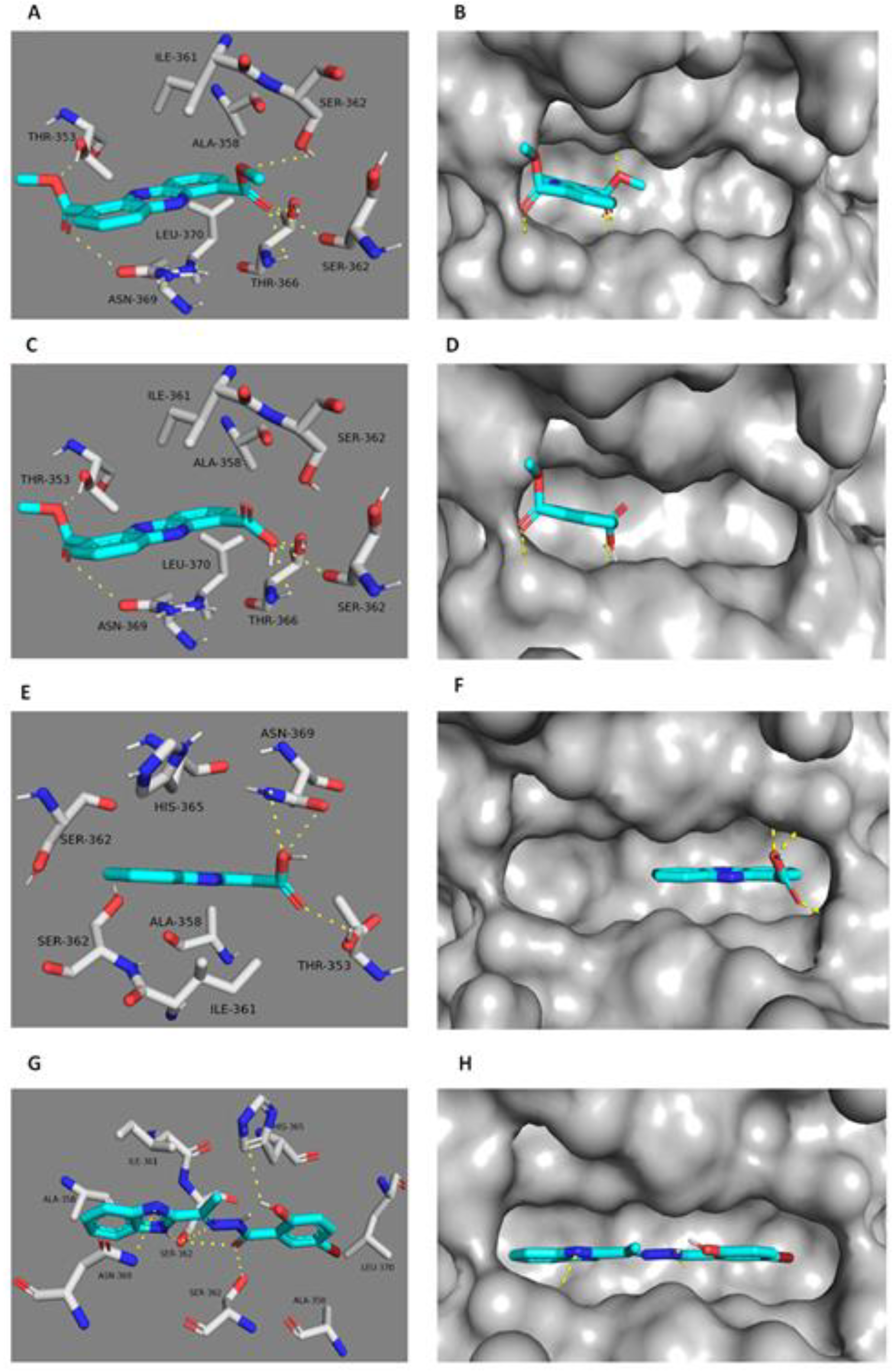
2.3. Docking Study
3. Material and Methods
3.1. General Experimental Procedures
3.2. Sponge Collection
3.3. Actinomycetes Isolation
3.4. Molecular Identification
3.5. Microbial Fermentation and Extract Preparation
3.6. LC-HR/MS Metabolomic Analysis
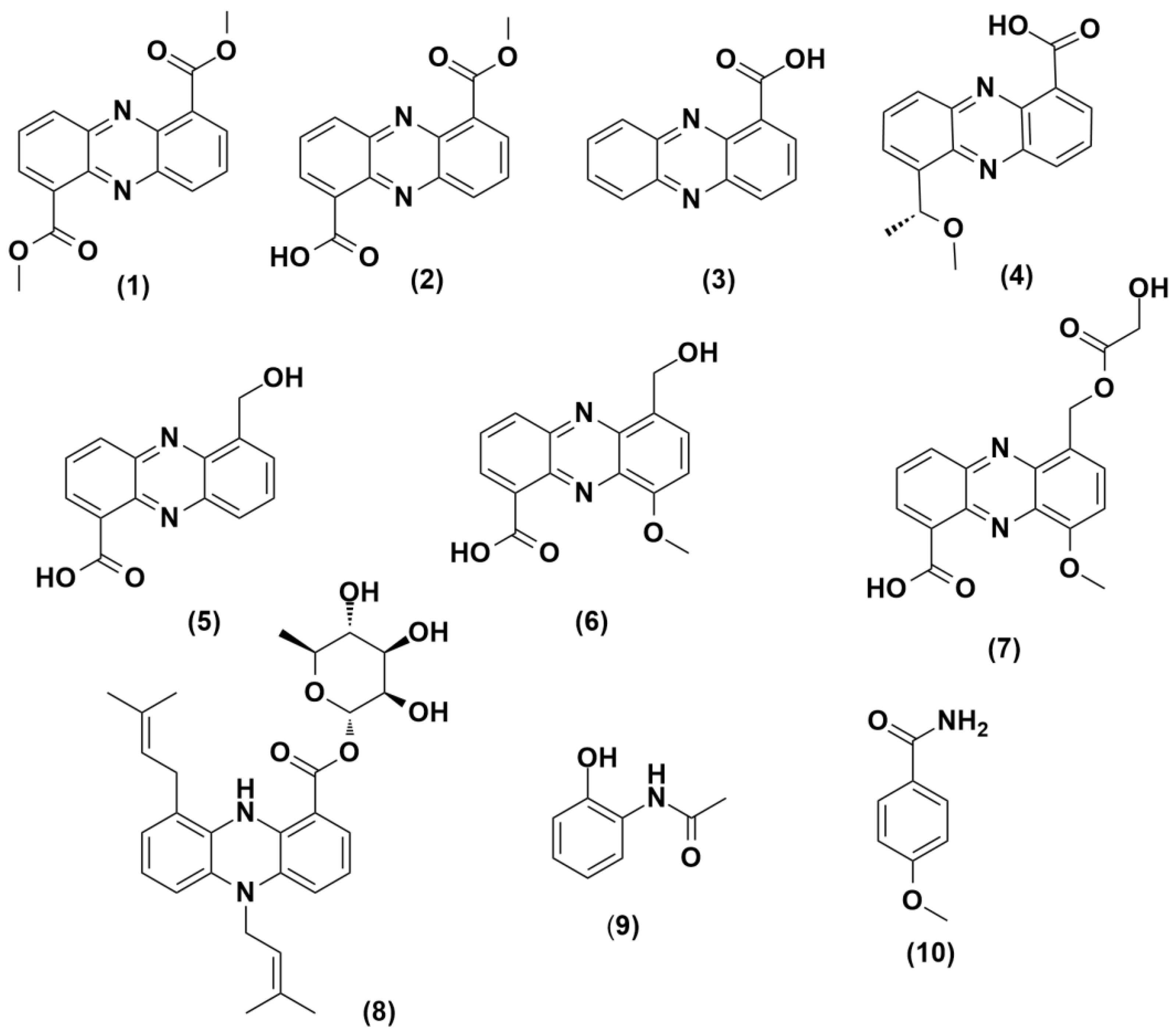
3.7. Isolation and Purification of Induced Metabolites
3.8. Assessment of Antibacterial Activity
3.9. Assessment of Antibiofilm Activity
3.10. Assessment of Cytotoxic Activity
MTT Assay
3.11. Enzyme Assays
3.12. Docking Analysis
3.13. Structuraal Elucidation of Isolated Compounds 1–3, 9, and 10.
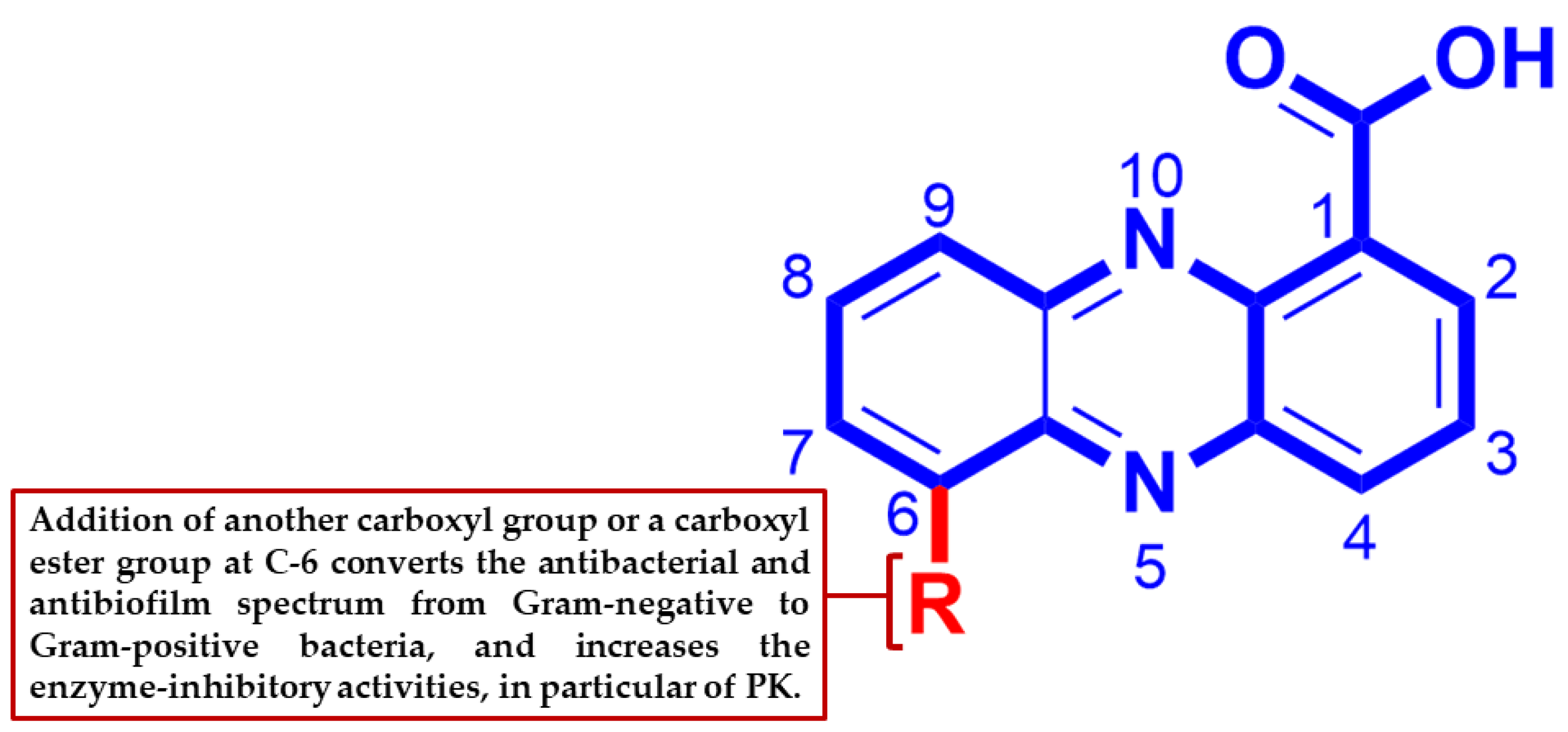
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Pettit, R.K. Mixed fermentation for natural product drug discovery. Appl. Microbiol. Biotechnol. 2009, 83, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Engelhardt, K.; Degnes, K.F.; Kemmler, M.; Bredholt, H.; Fjærvik, E.; Klinkenberg, G.; Sletta, H.; Ellingsen, T.E.; Zotchev, S.B. Production of a new thiopeptide antibiotic, TP-1161, by a marine Nocardiopsis species. Appl. Environ. Microbiol. 2010, 76, 4969–4976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olano, C.; Méndez, C.; Salas, J.A. Antitumor compounds from marine actinomycetes. Mar. Drugs 2009, 7, 210–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pimentel-Elardo, S.M.; Kozytska, S.; Bugni, T.S.; Ireland, C.M.; Moll, H.; Hentschel, U. Anti-parasitic compounds from Streptomyces sp. strains isolated from Mediterranean sponges. Mar. Drugs 2010, 8, 373–380. [Google Scholar] [CrossRef]
- Abdelmohsen, U.R.; Szesny, M.; Othman, E.M.; Schirmeister, T.; Grond, S.; Stopper, H.; Hentschel, U. Antioxidant and anti-protease activities of diazepinomicin from the sponge-associated Micromonospora strain RV115. Mar. Drugs 2012, 10, 2208–2221. [Google Scholar] [CrossRef]
- Wolfender, J.-L.; Marti, G.; Ferreira Queiroz, E. Advances in techniques for profiling crude extracts and for the rapid identificationof natural products: Dereplication, quality control and metabolomics. Curr. Org. Chem. 2010, 14, 1808–1832. [Google Scholar] [CrossRef]
- Wang, J.; Lin, W.; Wray, V.; Lai, D.; Proksch, P. Induced production of depsipeptides by co-culturing Fusarium tricinctum and Fusarium begoniae. Tetrahedron Lett. 2013, 54, 2492–2496. [Google Scholar] [CrossRef]
- Nett, M.; Ikeda, H.; Moore, B.S. Genomic basis for natural product biosynthetic diversity in the actinomycetes. Nat. Prod. Rep. 2009, 26, 1362–1384. [Google Scholar] [CrossRef]
- Winter, J.M.; Behnken, S.; Hertweck, C. Genomics-inspired discovery of natural products. Curr. Opin. Chem. Biol. 2011, 15, 22–31. [Google Scholar] [CrossRef]
- Oh, D.-C.; Jensen, P.R.; Kauffman, C.A.; Fenical, W. Libertellenones A–D: Induction of cytotoxic diterpenoid biosynthesis by marine microbial competition. Bioorg. Med. Chem. 2005, 13, 5267–5273. [Google Scholar] [CrossRef]
- Scherlach, K.; Hertweck, C. Triggering cryptic natural product biosynthesis in microorganisms. Org. Biomol. Chem. 2009, 7, 1753–1760. [Google Scholar] [CrossRef] [PubMed]
- Schroeckh, V.; Scherlach, K.; Nützmann, H.-W.; Shelest, E.; Schmidt-Heck, W.; Schuemann, J.; Martin, K.; Hertweck, C.; Brakhage, A.A. Intimate bacterial–fungal interaction triggers biosynthesis of archetypal polyketides in Aspergillus nidulans. Proc. Natl. Acad. Sci. USA 2009, 106, 14558–14563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiang, Y.-M.; Chang, S.-L.; Oakley, B.R.; Wang, C.C.C. Recent advances in awakening silent biosynthetic gene clusters and linking orphan clusters to natural products in microorganisms. Curr. Opin. Chem. Biol. 2011, 15, 137–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Hawary, S.S.; Sayed, A.M.; Mohammed, R.; Khanfar, M.; Rateb, M.E.; Mohammed, T.A.; Hajjar, D.; Hassan, H.M.; Gulder, T.A.M.; Abdelmohsen, U.R. New Pim-1 kinase inhibitor from the co-culture of two sponge-associated actinomycetes. Front. Chem. 2018, 6, 538. [Google Scholar] [CrossRef] [Green Version]
- Cueto, M.; Jensen, P.R.; Kauffman, C.; Fenical, W.; Lobkovsky, E.; Clardy, J. Pestalone, a new antibiotic produced by a marine fungus in response to bacterial challenge. J. Nat. Prod. 2001, 64, 1444–1446. [Google Scholar] [CrossRef]
- Thissera, B.; Alhadrami, H.A.; Hassan, M.H.A.; Hassan, H.M.; Bawazeer, M.; Yaseen, M.; Belbahri, L.; Rateb, M.E.; Behery, F.A. Induction of cryptic antifungal pulicatin derivatives from Pantoea agglomerans by microbial co-culture. Biomolecules 2020, 10, 268. [Google Scholar] [CrossRef] [Green Version]
- Oh, D.-C.; Kauffman, C.A.; Jensen, P.R.; Fenical, W. Induced production of emericellamides A and B from the marine-derived fungus Emericella sp. in competing co-culture. J. Nat. Prod. 2007, 70, 515–520. [Google Scholar] [CrossRef]
- McDonald, M.; Mavrodi, D.V.; Thomashow, L.S.; Floss, H.G. Phenazine biosynthesis in Pseudomonas f luorescens: Branchpoint from the primary shikimate biosynthetic pathway and role of phenazine-1, 6-dicarboxylic Acid. J. Am. Chem. Soc. 2001, 123, 9459–9460. [Google Scholar] [CrossRef]
- Liu, B.; Liu, K.; Lu, Y.; Zhang, D.; Yang, T.; Li, X.; Ma, C.; Zheng, M.; Wang, B.; Zhang, G. Systematic evaluation of structure-activity relationships of the riminophenazine class and discovery of a C2 pyridylamino series for the treatment of multidrug-resistant tuberculosis. Molecules 2012, 17, 4545–4559. [Google Scholar] [CrossRef]
- Kunz, A.; Labes, A.; Wiese, J.; Bruhn, T.; Bringmann, G.; Imhoff, J. Nature’s lab for derivatization: New and revised structures of a variety of streptophenazines produced by a sponge-derived Streptomyces strain. Mar. Drugs 2014, 12, 1699–1714. [Google Scholar] [CrossRef] [Green Version]
- Reddy, V.M.; O’Sullivan, J.F.; Gangadharam, P.R.J. Antimycobacterial activities of riminophenazines. J. Antimicrob. Chemother. 1999, 43, 615–623. [Google Scholar] [CrossRef] [PubMed]
- Mavrodi, D.V.; Blankenfeldt, W.; Thomashow, L.S. Phenazine compounds in fluorescent Pseudomonas spp. biosynthesis and regulation. Annu. Rev. Phytopathol. 2006, 44, 417–445. [Google Scholar] [CrossRef] [PubMed]
- Spicer, J.A.; Gamage, S.A.; Rewcastle, G.W.; Finlay, G.J.; Bridewell, D.J.A.; Baguley, B.C.; Denny, W.A. Bis (phenazine-1-carboxamides): Structure− activity relationships for a new class of dual topoisomerase I/II-directed anticancer drugs. J. Med. Chem. 2000, 43, 1350–1358. [Google Scholar] [CrossRef] [PubMed]
- Dashti, Y.; Grkovic, T.; Abdelmohsen, U.R.; Hentschel, U.; Quinn, R.J. Production of induced secondary metabolites by a co-culture of sponge-associated actinomycetes, Actinokineospora sp. EG49 and Nocardiopsis sp. RV163. Mar. Drugs 2014, 12, 3046–3059. [Google Scholar] [CrossRef]
- Boumehira, A.Z.; El-Enshasy, H.A.; Hacène, H.; Elsayed, E.A.; Aziz, R.; Park, E.Y. Recent progress on the development of antibiotics from the genus Micromonospora. Biotechnol. bioprocess Eng. 2016, 21, 199–223. [Google Scholar] [CrossRef] [Green Version]
- Kuncharoen, N.; Kudo, T.; Ohkuma, M.; Tanasupawat, S. Micromonospora azadirachtae sp. nov., isolated from roots of Azadirachta indica A. Juss. var. siamensis Valeton. Antonie van Leeuwenhoek, Int. J. Gen. Mol. Microbiol. 2019, 112, 253–262. [Google Scholar] [CrossRef]
- Borrero, N.V.; Bai, F.; Perez, C.; Duong, B.Q.; Rocca, J.R.; Jin, S.; Huigens, R.W., III. Phenazine antibiotic inspired discovery of potent bromophenazine antibacterial agents against Staphylococcus aureus and Staphylococcus epidermidis. Org. Biomol. Chem. 2014, 12, 881–886. [Google Scholar] [CrossRef]
- Zhuo, S.-T.; Li, C.-Y.; Hu, M.-H.; Chen, S.-B.; Yao, P.-F.; Huang, S.-L.; Ou, T.-M.; Tan, J.-H.; An, L.-K.; Li, D. Synthesis and biological evaluation of benzo [a] phenazine derivatives as a dual inhibitor of topoisomerase I and II. Org. Biomol. Chem. 2013, 11, 3989–4005. [Google Scholar] [CrossRef]
- Vicker, N.; Burgess, L.; Chuckowree, I.S.; Dodd, R.; Folkes, A.J.; Hardick, D.J.; Hancox, T.C.; Miller, W.; Milton, J.; Sohal, S. Novel angular benzophenazines: Dual topoisomerase I and topoisomerase II inhibitors as potential anticancer agents. J. Med. Chem. 2002, 45, 721–739. [Google Scholar] [CrossRef]
- Adjei, A.A.; Charron, M.; Rowinsky, E.K.; Svingen, P.A.; Miller, J.; Reid, J.M.; Sebolt-Leopold, J.; Ames, M.M.; Kaufmann, S.H. Effect of pyrazoloacridine (NSC 366140) on DNA topoisomerases I and II. Clin. cancer Res. 1998, 4, 683–691. [Google Scholar]
- Yi, L.; Lü, X. New strategy on antimicrobial-resistance: Inhibitors of DNA replication enzymes. Curr. Med. Chem. 2019, 26, 1761–1787. [Google Scholar] [CrossRef] [PubMed]
- Zoraghi, R.; Worrall, L.; See, R.H.; Strangman, W.; Popplewell, W.L.; Gong, H. Methicillin-resistant Staphylococcus aureus (MRSA) pyruvate kinase as a target for bis-indole alkaloids with antibacterial activities. J. Biol. Chem. 2011, 286, 44716–44725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohlsen, K.; Donat, S. The impact of serine/threonine phosphorylation in Staphylococcus aureus. Int. J. Med. Microbiol. 2010, 300, 137–141. [Google Scholar] [CrossRef] [PubMed]
- Vasu, D.; Sunitha, M.M.; Srikanth, L.; Swarupa, V.; Prasad, U.V.; Sireesha, K.; Yeswanth, S.; Kumar, P.S.; Venkatesh, K.; Chaudhary, A. In Staphylococcus aureus the regulation of pyruvate kinase activity by serine/threonine protein kinase favors biofilm formation. 3 Biotech 2015, 5, 505–512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Jiao, H.; Meng, J.; Qiao, M.; Du, H.; He, M.; Ming, K.; Liu, J.; Wang, D.; Wu, Y. Baicalin inhibits biofilm formation and the quorum-sensing system by regulating the MsrA drug efflux pump in Staphylococcus saprophyticus. Front. Microbiol. 2019, 10, 2800. [Google Scholar] [CrossRef]
- Benedetto Tiz, D.; Kikelj, D.; Zidar, N. Overcoming problems of poor drug penetration into bacteria: Challenges and strategies for medicinal chemists. Expert Opin. Drug Discov. 2018, 13, 497–507. [Google Scholar] [CrossRef]
- Ronkin, S.M.; Badia, M.; Bellon, S.; Grillot, A.-L.; Gross, C.H.; Grossman, T.H.; Mani, N.; Parsons, J.D.; Stamos, D.; Trudeau, M. Discovery of pyrazolthiazoles as novel and potent inhibitors of bacterial gyrase. Bioorg. Med. Chem. Lett. 2010, 20, 2828–2831. [Google Scholar] [CrossRef]
- Axerio-Cilies, P.; See, R.H.; Zoraghi, R.; Worral, L.; Lian, T.; Stoynov, N.; Jiang, J.; Kaur, S.; Jackson, L.; Gong, H. Cheminformatics-driven discovery of selective, nanomolar inhibitors for staphylococcal pyruvate kinase. ACS Chem. Biol. 2011, 7, 350–359. [Google Scholar] [CrossRef]
- Mincer, T.J.; Jensen, P.R.; Kauffman, C.A.; Fenical, W. Widespread and persistent populations of a major new marine actinomycete taxon in ocean sediments. Appl. Environ. Microbiol. 2002, 68, 5005–5011. [Google Scholar] [CrossRef] [Green Version]
- Shirling, E.B.T.; Gottlieb, D. Methods for characterization of Streptomyces species. Int. J. Syst. Bacteriol. 1966, 16, 313–340. [Google Scholar] [CrossRef] [Green Version]
- Olson, J.B.; Lord, C.C.; McCarthy, P.J. Improved recoverability of microbial colonies from marine sponge samples. Microb. Ecol. 2000, 40, 139–147. [Google Scholar] [CrossRef] [PubMed]
- Weiner, R.M.; Segall, A.M.; Colwell, R.R. Characterization of a marine bacterium associated with Crassostrea virginica (the eastern oyster). Appl. Environ. Microbiol. 1985, 49, 83–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Webster, N.S.; Wilson, K.J.; Blackall, L.L.; Hill, R.T. Phylogenetic diversity of bacteria associated with the marine sponge Rhopaloeides odorabile. Appl. Environ. Microbiol. 2001, 67, 434–444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lyman, J.; Fleming, R.H. Composition of sea water. J. mar. Res 1940, 3, 134–146. [Google Scholar]
- Hentschel, U.; Hopke, J.; Horn, M.; Friedrich, A.B.; Wagner, M.; Hacker, J.; Moore, B.S. Molecular evidence for a uniform microbial community in sponges from different oceans. Appl. Environ. Microbiol. 2002, 68, 4431–4440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashelford, K.E.; Chuzhanova, N.A.; Fry, J.C.; Jones, A.J.; Weightman, A.J. At least 1 in 20 16S rRNA sequence records currently held in public repositories is estimated to contain substantial anomalies. Appl. Environ. Microbiol. 2005, 71, 7724–7736. [Google Scholar] [CrossRef] [Green Version]
- Pruesse, E.; Peplies, J.; Glöckner, F.O. SINA: Accurate high-throughput multiple sequence alignment of ribosomal RNA genes. Bioinformatics 2012, 28, 1823–1829. [Google Scholar] [CrossRef]
- Abdelmohsen, U.; Cheng, C.; Viegelmann, C.; Zhang, T.; Grkovic, T.; Ahmed, S.; Quinn, R.; Hentschel, U.; Edrada-Ebel, R. Dereplication strategies for targeted isolation of new antitrypanosomal actinosporins A and B from a marine sponge associated-Actinokineospora sp. EG49. Mar. Drugs 2014, 12, 1220–1244. [Google Scholar] [CrossRef] [Green Version]
- Ingebrigtsen, R.A.; Hansen, E.; Andersen, J.H.; Eilertsen, H.C. Light and temperature effects on bioactivity in diatoms. J. Appl. Phycol. 2016, 28, 939–950. [Google Scholar] [CrossRef] [Green Version]
- Antunes, A.L.S.; Trentin, D.S.; Bonfanti, J.W.; PINTO, C.C.F.; PEREZ, L.R.R.; Macedo, A.J.; Barth, A.L. Application of a feasible method for determination of biofilm antimicrobial susceptibility in staphylococci. Apmis 2010, 118, 873–877. [Google Scholar] [CrossRef]
- Durcik, M.; Tammela, P.; Barančoková, M.; Tomašič, T.; Ilaš, J.; Kikelj, D.; Zidar, N. Synthesis and evaluation of N-phenylpyrrolamides as DNA gyrase B Inhibitors. ChemMedChem 2018, 13, 186–198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, K.; Ito, S.; Shimizu-Ibuka, A.; Sakai, H. Crystal structure of pyruvate kinase from Geobacillus stearothermophilus. J. Biochem. 2008, 144, 305–312. [Google Scholar] [CrossRef] [PubMed]
- El-Sayed, M.T.; Zoraghi, R.; Reiner, N.; Suzen, S.; Ohlsen, K.; Lalk, M.; Altanlar, N.; Hilgeroth, A. Novel inhibitors of the methicillin-resistant Staphylococcus aureus (MRSA)-pyruvate kinase. J. Enzyme Inhib. Med. Chem. 2016, 31, 1666–1671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sayed, A.M.; Alhadrami, H.A.; El-Hawary, S.S.; Mohammed, R.; Hassan, H.M.; Rateb, M.E.; Abdelmohsen, U.R.; Bakeer, W. Discovery of two brominated oxindole alkaloids as Staphylococcal DNA gyrase and pyruvate kinase inhibitors via inverse virtual screening. Microorganisms 2020, 8, 293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chatterjee, S.; Vijayakumar, E.K.S.; Franco, C.M.M.; Maurya, R.; Blumbach, J.; Ganguli, B.N. Phencomycin, a new antibiotic from a Streptomyces species HIL Y-9031725. J. Antibiot. (Tokyo). 1995, 48, 1353–1354. [Google Scholar] [CrossRef] [Green Version]
- Pusecker, K.; Laatsch, H.; Helmke, E.; Weyland, H. Dihydrophencomycin methyl ester, a new phenazine derivative from a marine streptomycete. J. Antibiot. (Tokyo). 1997, 50, 479–483. [Google Scholar] [CrossRef] [Green Version]
- Cheng, C.; Othman, E.M.; Fekete, A.; Krischke, M.; Stopper, H.; Edrada-Ebel, R.A.; Mueller, M.J.; Hentschel, U.; Abdelmohsen, U.R. Strepoxazine A, a new cytotoxic phenoxazin from the marine sponge-derived bacterium Streptomyces sp. SBT345. Tetrahedron Lett. 2016, 57, 4196–4199. Available online: http://dx.doi.org/10.1016/j.tetlet.2016.08.005. [CrossRef] [Green Version]
- Geiger, A.; Keller-Schierlein, W.; Brandl, M.; Zahner, H. Metabolites of microorganisms. 247 phenazines from Streptomyces antibioticus, strain Tü 2706. J. Antibiot. 1988, 41, 1542–1551. [Google Scholar] [CrossRef] [Green Version]
- Gebhardt, K.; Schimana, J.; Krastel, P.; Dettner, K.; Rheinheimer, J.; Zeeck, A.; Fiedler, H.-P. Endophenazines AD, new phenazine antibiotics from the Arthropod associated endosymbiont Streptomyces anulatus. J. Antibiot. 2002, 55, 794–800. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.-S.; Kang, J.S.; Choi, B.-K.; Lee, H.-S.; Lee, Y.-J.; Lee, J.; Shin, H.J. Phenazine derivatives with anti-inflammatory activity from the deep-sea sediment-derived yeast-like fungus Cystobasidium laryngis IV17-028. Mar. Drugs. 2019, 17, 482. [Google Scholar] [CrossRef] [Green Version]
- National Center for Biotechnology Information.pub Chemdatabase. 4-Methoxy Benzamide. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/4-Methoxybenzamide (accessed on 15 April 2020).







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tested Compounds | Staphylococcus aureus | Bacillus subtilis | Escherichia coli | Pseudomonas aeruginosa |
|---|---|---|---|---|
| 1 | 57.19 ± 1.2 | 19.61 ± 1.5 | 9.93 ± 1.3 | 23.41 ± 1.7 |
| 2 | 68.87 ± 0.8 | 32.13 ± 1.3 | 11.15 ± 1.4 | 23.25 ± 2.2 |
| 3 | NA | 1.69 ± 1.2 | 24.34 ± 1.8 | 94.17 ± 2.8 |
| 9 | 1.1 ± 0.9 | NA | NA | 4.74 ± 1.6 |
| 10 | 53.17 ± 1.2 | 42.16 ± 1.9 | 19.27 ± 2.5 | 70.23 ± 1.1 |
| Gentamicin | 99.1 ± 0.7 | 97 ± 1.6 | 99 ± 0.6 | 99.7 ± 0.2 |
| Tested Compounds | S. aureus | B. subtilis | E. coli | P. aeruginosa |
|---|---|---|---|---|
| 1 | 50.26 ± 0.4 | 12.10 ± 3.2 | 36.66 ± 2.9 | 18.36 ± 0.9 |
| 2 | 75.10 ± 2.4 | 18.65 ± 1.6 | 54.07 ± 2.5 | 22.28 ± 1.5 |
| 3 | NA | NA | 54.67 ± 1.4 | 93.98 ± 2.2 |
| 9 | 11.47 ± 2.9 | 4.91 ± 1.8 | 34.55 ± 2.6 | 7.39 ± 1.9 |
| 10 | 61.20 ± 3.7 | 20.29 ± 1.1 | 57.47 ± 3.1 | 73.52 ± 1.3 |
| Tested Compounds | WI38 | HCT116 | HePG-2 | MCF7 |
|---|---|---|---|---|
| 1 | 63.18 ± 3.6 | 85.04 ± 3.9 | 92.06 ± 4.7 | >100 |
| 2 | 76.30 ± 3.9 | 60.81 ± 3.5 | 76.11 ± 3.9 | 82.24 ± 4.4 |
| 3 | 51.22 ± 3.2 | 91.27 ± 4.6 | >100 | >100 |
| 9 | 36.47 ± 2.3 | 14.56 ± 1.2 | 10.16 ± 0.9 | 12.65 ± 1.1 |
| 10 | >100 | >100 | >100 | >100 |
| Doxorubicin | 6.72 ± 0.5 | 5.23 ± 0.3 | 4.50 ± 0.2 | 4.17±0.2 |
| Protein Target | Ligand | IC50 (µM) * | Binding Energy (kcal/mol) | Hydrogen Bonding Interactions | Hydrophobic Interactions |
|---|---|---|---|---|---|
| Gyr-B | 1 | 19.18 ± 1.69 | −7.6 | ASN-54, GLU-58 | ILE-51, VAL-79 PRO-87, ILE-86 ILE-102, ILE-103 ILE-175 |
| 2 | 21.28 ± 2.36 | −7.5 | SER-55, ILE-51 | ILE-51, VAL-79 PRO-87, ILE-86 ILE-102, ILE-103 ILE-175 | |
| 3 | 27.69 ± 1.08 | −7.2 | SER-55, ILE-51 | ILE-51, VAL-79 PRO-87, ILE-86 ILE-102, ILE-103 ILE-175 | |
| Co-crystallized ligand | 0.091 | - | ASP-81, ASN-54 | ILE-51, VAL-79 PRO-87, ILE-86 ILE-102, ILE-103 ILE-175 | |
| PK | 1 | 7.2 ± 0.07 | −7.7 | SER-362A, SER-362B THR-366A, THR-353B, ASN-369A | ALA-358B, ILE-361B, LEU-370A |
| 2 | 9.3 ± 0.03 | −7.5 | SER-362A, THR-366A, THR-353B, ASN-369A | ALA-358B, ILE-361B, LEU-370A | |
| 3 | 22.5 ± 0.04 | −6.5 | ASN-369B, THR-353A | ALA-358A, ILE-361A | |
| Co-crystallized ligand | 0.24 | - | SER-362A, SER-362B, ASN-369B, HIS-365A | ALA-358A, ALA-358B ILE-361B, LEU-370A |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
S. Hifnawy, M.; Hassan, H.M.; Mohammed, R.; M. Fouda, M.; Sayed, A.M.; A. Hamed, A.; F. AbouZid, S.; Rateb, M.E.; Alhadrami, H.A.; Abdelmohsen, U.R. Induction of Antibacterial Metabolites by Co-Cultivation of Two Red-Sea-Sponge-Associated Actinomycetes Micromonospora sp. UR56 and Actinokinespora sp. EG49. Mar. Drugs 2020, 18, 243. https://doi.org/10.3390/md18050243
S. Hifnawy M, Hassan HM, Mohammed R, M. Fouda M, Sayed AM, A. Hamed A, F. AbouZid S, Rateb ME, Alhadrami HA, Abdelmohsen UR. Induction of Antibacterial Metabolites by Co-Cultivation of Two Red-Sea-Sponge-Associated Actinomycetes Micromonospora sp. UR56 and Actinokinespora sp. EG49. Marine Drugs. 2020; 18(5):243. https://doi.org/10.3390/md18050243
Chicago/Turabian StyleS. Hifnawy, Mohamed, Hossam M. Hassan, Rabab Mohammed, Mohamed M. Fouda, Ahmed M. Sayed, Ahmed A. Hamed, Sameh F. AbouZid, Mostafa E. Rateb, Hani A. Alhadrami, and Usama Ramadan Abdelmohsen. 2020. "Induction of Antibacterial Metabolites by Co-Cultivation of Two Red-Sea-Sponge-Associated Actinomycetes Micromonospora sp. UR56 and Actinokinespora sp. EG49" Marine Drugs 18, no. 5: 243. https://doi.org/10.3390/md18050243
APA StyleS. Hifnawy, M., Hassan, H. M., Mohammed, R., M. Fouda, M., Sayed, A. M., A. Hamed, A., F. AbouZid, S., Rateb, M. E., Alhadrami, H. A., & Abdelmohsen, U. R. (2020). Induction of Antibacterial Metabolites by Co-Cultivation of Two Red-Sea-Sponge-Associated Actinomycetes Micromonospora sp. UR56 and Actinokinespora sp. EG49. Marine Drugs, 18(5), 243. https://doi.org/10.3390/md18050243