
Cytosporin Derivatives from Arctic-Derived Fungus Eutypella sp. D-1 via the OSMAC Approach

 , ,
, ,
Abstract
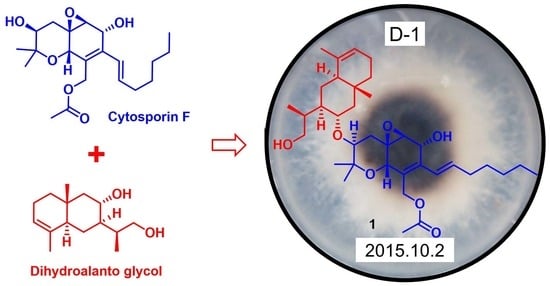
:
1. Introduction
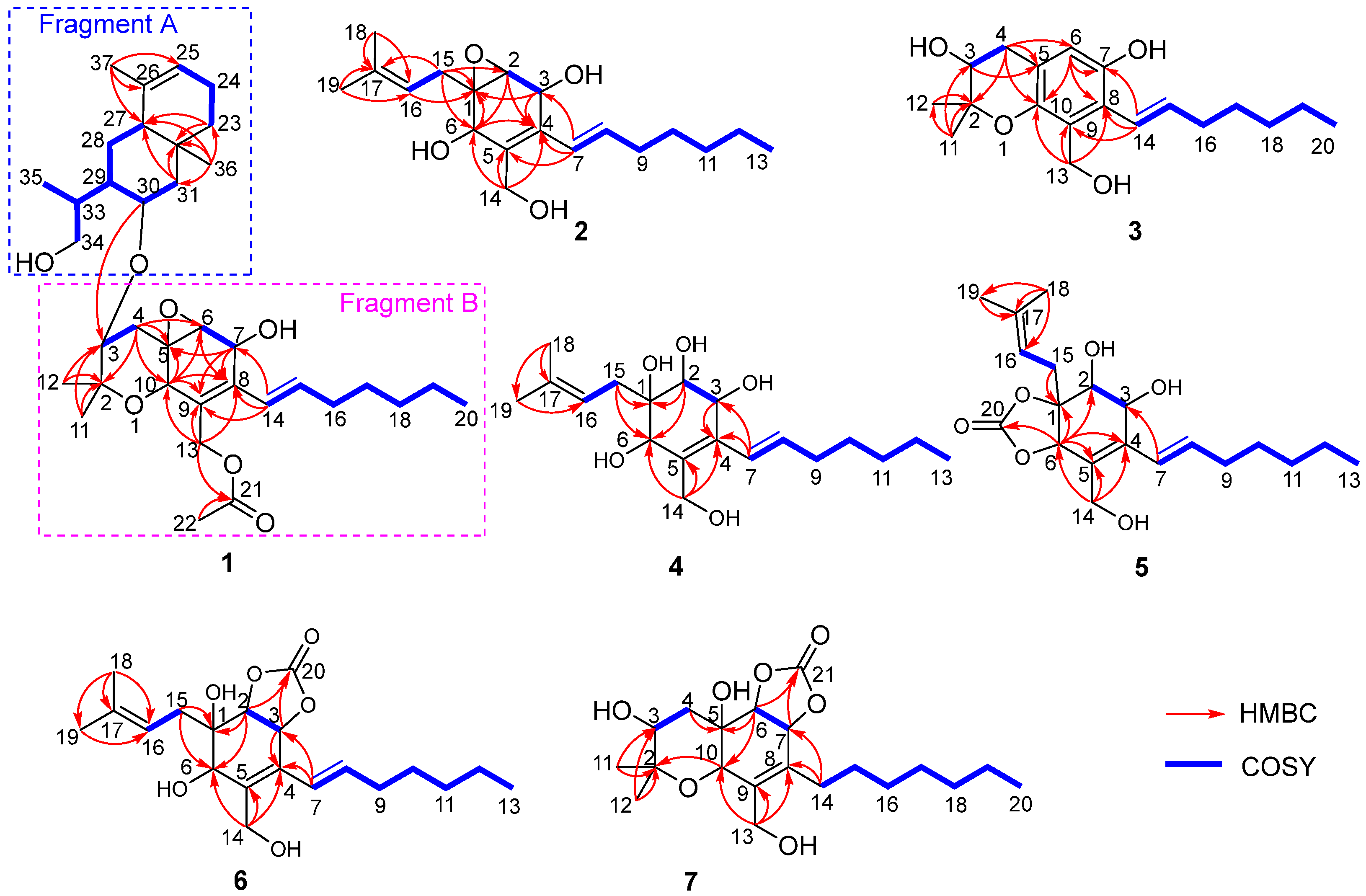
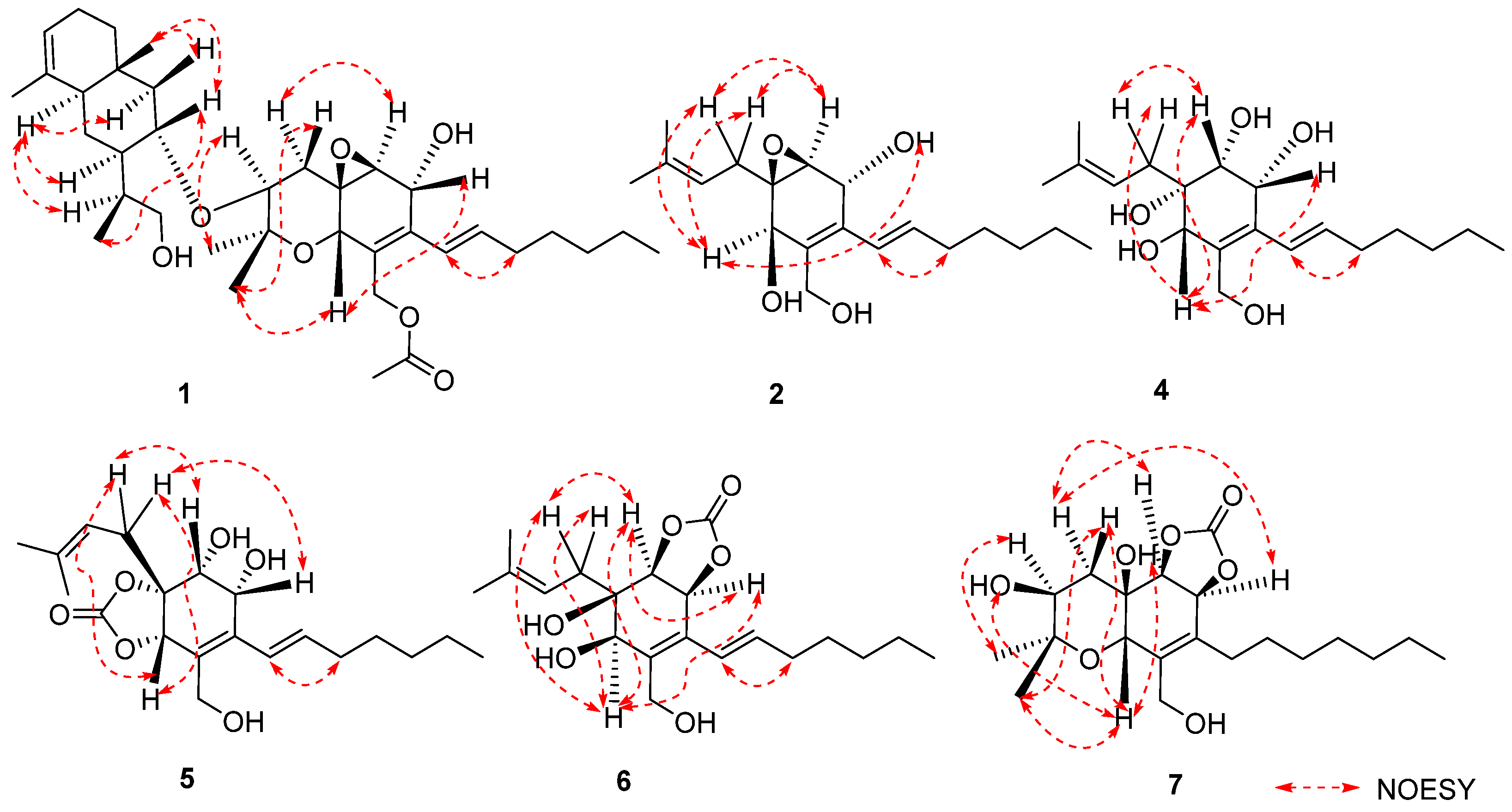
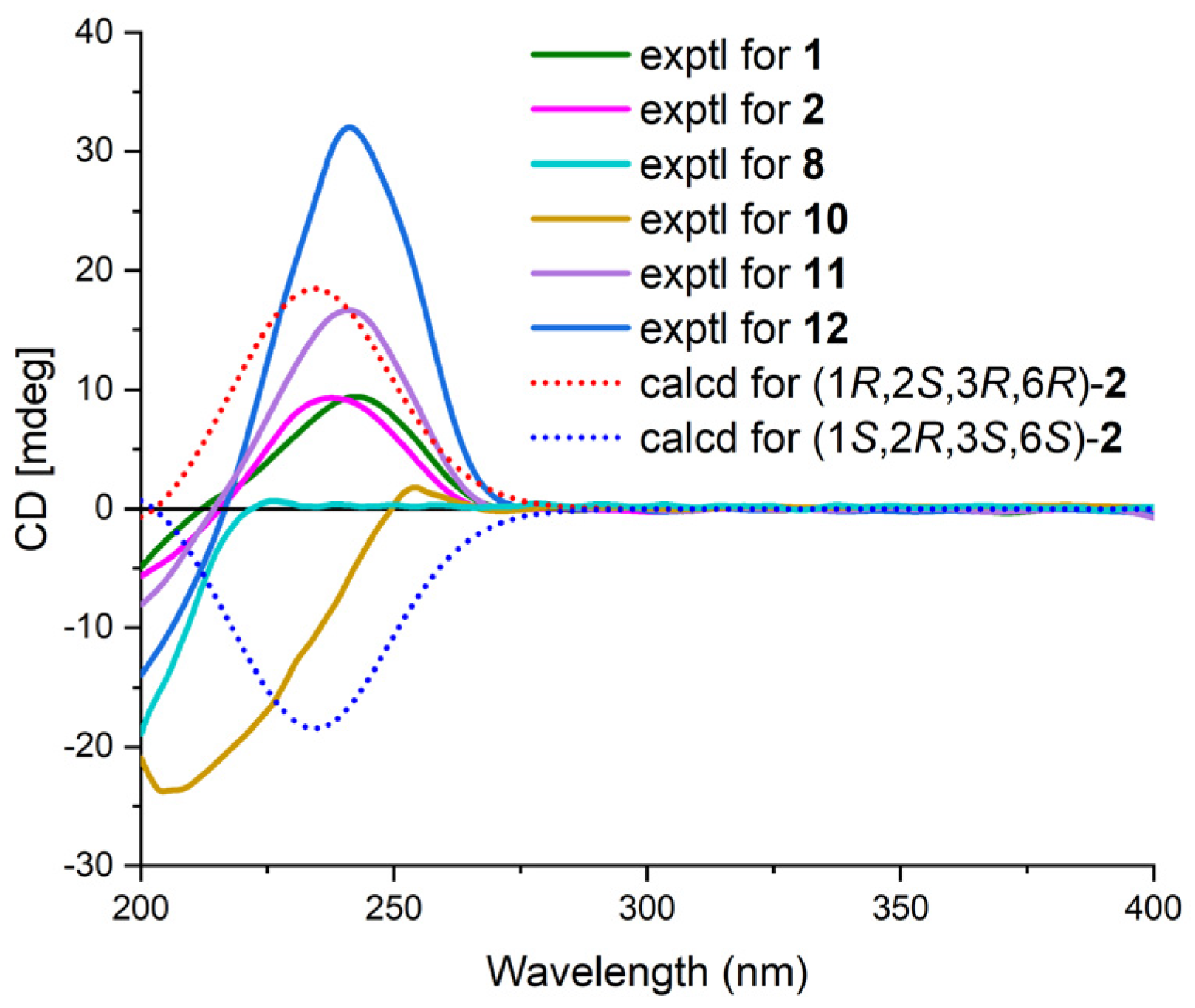
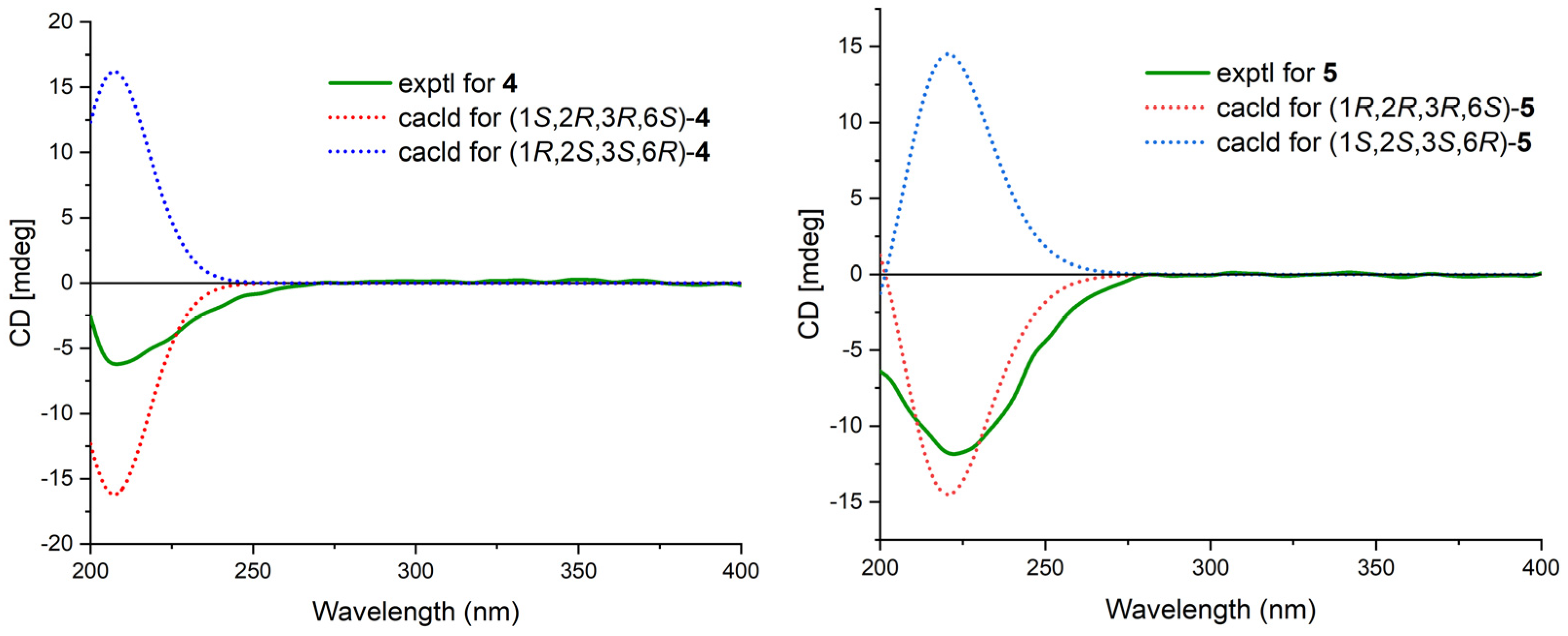
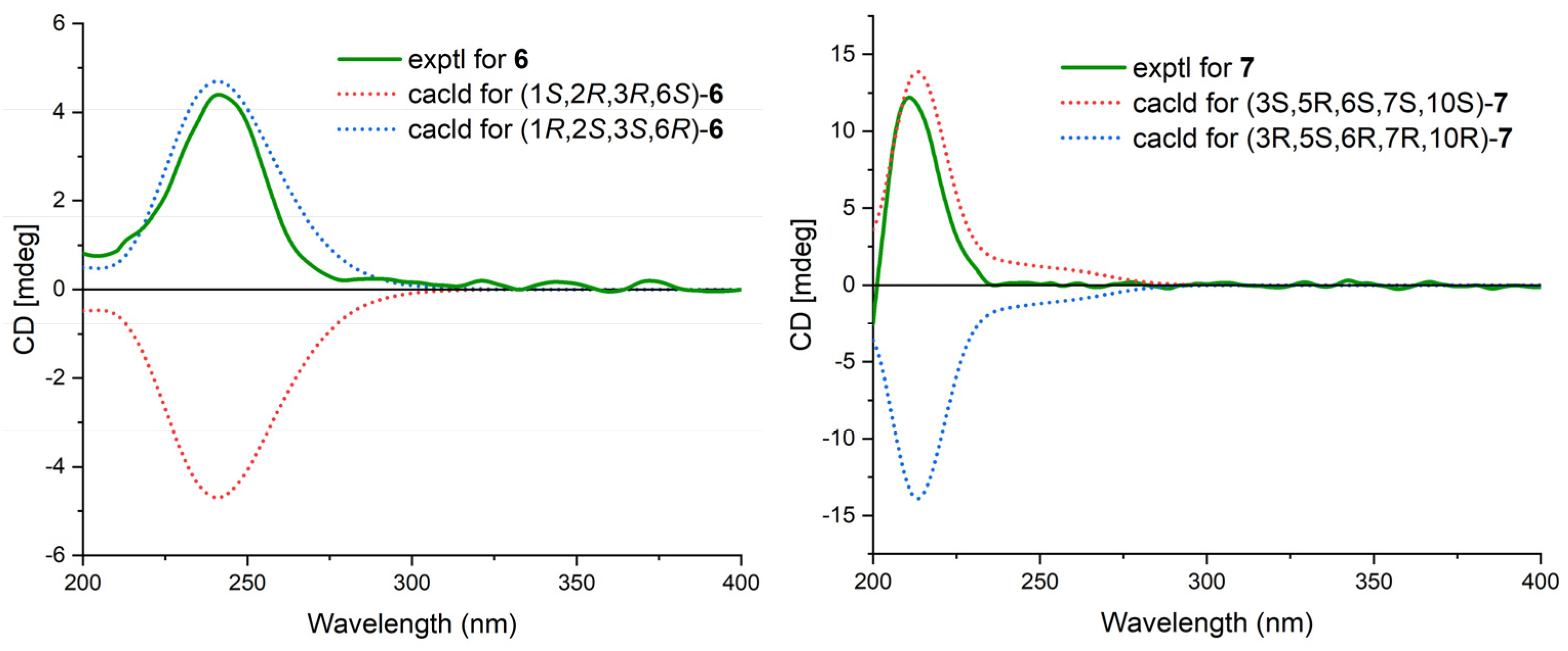
2. Results
3. Materials and Methods
3.1. General Experimental Procedures
3.2. Fungal Material
3.3. Fermentation, Extraction, and Isolation
3.4. Biological Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Arrieche, D.; Cabrera-Pardo, J.R.; San-Martin, A.; Carrasco, H.; Taborga, L. Natural Products from Chilean and Antarctic Marine Fungi and Their Biomedical Relevance. Mar. Drugs 2023, 21, 98. [Google Scholar] [CrossRef] [PubMed]
- Stevens-Miles, S.; Goetz, M.A.; Bills, G.F.; Giacobbe, R.A.; Tkacz, J.S.; Chang, R.S.; Mojena, M.; Martin, I.; Diez, M.T.; Pelaez, F. Discovery of an angiotensin II binding inhibitor from a Cytospora sp. using semi-automated screening procedures. J. Antibiot. 1996, 49, 119–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akone, S.H.; El Amrani, M.; Lin, W.; Lai, D.; Proksch, P. Cytosporins F–K, new epoxyquinols from the endophytic fungus Pestalotiopsis theae. Tetrahedron Lett. 2013, 54, 6751–6754. [Google Scholar] [CrossRef]
- Ciavatta, M.L.; Lopez-Gresa, M.P.; Gavagnin, M.; Nicoletti, R.; Manzo, E.; Mollo, E.; Guo, Y.-W.; Cimino, G. Cytosporin-related compounds from the marine-derived fungus Eutypella scoparia. Tetrahedron 2008, 64, 5365–5369. [Google Scholar] [CrossRef]
- Zhang, Y.-X.; Yu, H.-B.; Xu, W.-H.; Hu, B.; Guild, A.; Zhang, J.-P.; Lu, X.-L.; Liu, X.-Y.; Jiao, B.-H. Eutypellacytosporins A–D, Meroterpenoids from the Arctic Fungus Eutypella sp. D-1. J. Nat. Prod. 2019, 82, 3089–3095. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Müller, W.E.G.; Meier, D.; Kalscheuer, R.; Guo, Z.; Zou, K.; Umeokoli, B.O.; Liu, Z.; Proksch, P.J.M.D. Polyketide Derivatives from Mangrove Derived Endophytic Fungus Pseudopestalotiopsis theae. Mar. Drugs 2020, 18, 129–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, H.-B.; Wang, X.-L.; Zhang, Y.-X.; Xu, W.-H.; Zhang, J.-P.; Zhou, X.-Y.; Lu, X.-L.; Liu, X.-Y.; Jiao, B.-H. Libertellenones O–S and Eutypellenones A and B, Pimarane Diterpene Derivatives from the Arctic Fungus Eutypella sp. D-1. J. Nat. Prod. 2018, 81, 1553–1560. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.J.; Zhang, J.; Li, Y.M. Secondary metabolites from Eutypella species and their bioactivities. Mycosystema 2017, 36, 1181–1191. [Google Scholar]
- Yu, H.-B.; Wang, X.-L.; Xu, W.-H.; Zhang, Y.-X.; Qian, Y.-S.; Zhang, J.-P.; Lu, X.-L.; Liu, X.-Y. Eutypellenoids A–C, New Pimarane Diterpenes from the Arctic Fungus Eutypella sp. D-1. Mar. Drugs 2018, 16, 284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romano, S.; Jackson, S.A.; Patry, S.; Dobson, A.D.W. Extending the “One Strain Many Compounds” (OSMAC) Principle to Marine Microorganisms. Mar. Drugs 2018, 16, 244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, X.-D.; Zhao, N.; Xu, S.; Lü, H.-N.; Ma, S.-G.; Liu, Y.-B.; Li, Y.; Qu, J.; Yu, S.-S. Total synthesis of illicidione A and illihendione A. Tetrahedron 2015, 71, 4821–4829. [Google Scholar] [CrossRef]
- Lim, J.; Kim, I.-H.; Kim, H.H.; Ahn, K.-S.; Han, H. Enantioselective syntheses of decursinol angelate and decursin. Tetrahedron Lett. 2001, 42, 4001–4003. [Google Scholar] [CrossRef]
- Zhang, Y.-H.; Du, H.-F.; Gao, W.-B.; Li, W.; Cao, F.; Wang, C.-Y. Anti-inflammatory Polyketides from the Marine-Derived Fungus Eutypella scoparia. Mar. Drugs 2022, 20, 486. [Google Scholar] [CrossRef] [PubMed]
- Liao, H.-X.; Sun, D.-W.; Zheng, C.-J.; Wang, C.-Y. A new hexahydrobenzopyran derivative from the gorgonian-derived Fungus Eutypella sp. Nat. Prod. Res. 2017, 31, 1640–1646. [Google Scholar] [CrossRef] [PubMed]
- Allemann, R.K. Chemical wizardry? The generation of diversity in terpenoid biosynthesis. Pure Appl. Chem. 2008, 80, 1791–1798. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.-B.; Jiao, H.; Zhu, Y.-P.; Zhang, J.-P.; Lu, X.-L.; Liu, X.-Y. Bioactive metabolites from the Arctic fungus Nectria sp. B-13. J. Asian Nat. Prod. Res. 2019, 21, 961–969. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.-B.; Gu, B.-B.; Iwasaki, A.; Jiang, W.-L.; Ecker, A.; Wang, S.-P.; Yang, F.; Lin, H.-W. Dactylospenes A–E, Sesterterpenes from the Marine Sponge Dactylospongia elegans. Mar. Drugs 2020, 18, 491. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Zhang, X.; Huang, F.; Li, G.; Leadlay, P.F. Efophylins A and B, Two C2-Asymmetric Macrodiolide Immunosuppressants from Streptomyces malaysiensis. J. Nat. Prod. 2021, 84, 1579–1586. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position | δC | δH, mult. (J in Hz) | Position | δC | δH, mult. (J in Hz) |
|---|---|---|---|---|---|
| 2 | 76.6, C | 20 | 14.0, CH3 | 0.89, t (7.0) | |
| 3 | 73.8, CH | 3.72, m | 21 | 171.0, C | |
| 4α | 35.5, CH2 | 1.72, dd (12.5, 5.0) | 22 | 20.9, CH3 | 2.08, s |
| 4β | 2.27, t (12.0) | 23 | 37.5, CH2 | 1.35, m | |
| 5 | 55.7, C | 24 | 22.7, CH2 | 1.99, m | |
| 6 | 59.7, CH | 3.32, s | 25 | 121.2, CH | 5.31, brs |
| 7 | 64.6, CH | 4.73, s | 26 | 134.1, C | |
| 8 | 135.6, C | 27 | 46.3, CH | 1.97, m | |
| 9 | 125.1, C | 28α | 28.5, CH2 | 1.66, m | |
| 10 | 67.5, CH | 4.38, s | 28β | 1.23, m | |
| 11 | 16.0, CH3 | 1.30, s | 29 | 49.7, CH | 1.50, m |
| 12 | 27.7, CH3 | 1.29, s | 30 | 66.7, CH | 3.87, td (11.0, 5.0) |
| 13a | 61.5, CH2 | 4.67, d (12.5) | 31α | 49.4, CH2 | 1.15, m |
| 13b | 4.81, d (12.5) | 31β | 1.79, dd (12.5, 5.0) | ||
| 14 | 124.7, CH | 6.33, d (16.0) | 32 | 33.7, C | |
| 15 | 136.4, CH | 6.16, m | 33 | 39.0, CH | 1.87, m |
| 16 | 33.5, CH2 | 2.17, m | 34 | 67.5, CH2 | 3.67, dd (7.5, 4.0) |
| 17 | 28.8, CH2 | 1.42, m | 35 | 11.9, CH3 | 1.01, d (7.0) |
| 18 | 31.4, CH2 | 1.27, m | 36 | 16.5, CH3 | 0.79, s |
| 19 | 22.5, CH2 | 1.28, m | 37 | 21.2, CH3 | 1.61, s |
| 2 | 3 | ||||
|---|---|---|---|---|---|
| Position | δC | δH, mult. (J in Hz) | Position | δC | δH, mult. (J in Hz) |
| 1 | 59.3, C | 2 | 77.2, C | ||
| 2 | 57.5, CH | 3.29, s | 3 | 69.7, CH | 3.79, t (5.0) |
| 3 | 64.3, CH2 | 4.72, s | 4α | 31.5, CH2 | 3.03, dd (17.0, 5.0) |
| 4 | 131.6, C | 4β | 2.72, dd (17.0, 5.0) | ||
| 5 | 131.4, C | 5 | 118.8, C | ||
| 6 | 69.5, CH | 4.45, s | 6 | 115.1, CH | 6.61, s |
| 7 | 124.6, CH | 6.28, d (16.0) | 7 | 146.7, C | |
| 8 | 135.4, CH | 6.05, m | 8 | 123.6, C | |
| 9 | 33.5, CH2 | 2.15, m | 9 | 126.8, C | |
| 10 | 28.9, CH2 | 1.41, m | 10 | 144.8, C | |
| 11 | 31.5, CH2 | 1.28, m | 11 | 22.4, CH3 | 1.36, s |
| 12 | 22.5, CH2 | 1.29, m | 12 | 24.9, CH3 | 1.32, s |
| 13 | 14.0, CH3 | 0.88, t (6.0) | 13 | 58.8, CH2 | 4.66, s |
| 14a | 62.2, CH2 | 4.57, d (12.0) | 14 | 122.7, CH | 6.35, d (16.5) |
| 14b | 4.06, d (12.0) | 15 | 140.0, CH | 5.95, m | |
| 15α | 29.7, CH2 | 2.82, dd (15.0, 8.0) | 16 | 33.4, CH2 | 2.27, m |
| 15β | 2.30, dd (15.0, 8.0) | 17 | 29.0, CH2 | 1.50, m | |
| 16 | 117.3, CH | 5.20, t (7.0) | 18 | 31.5, CH2 | 1.35, m |
| 17 | 135.9, C | 19 | 22.6, CH2 | 1.35, m | |
| 18 | 18.0, CH3 | 1.66, s | 20 | 14.1, CH3 | 0.91, t (7.0) |
| 19 | 25.9, CH3 | 1.73, s | |||
| 4 | 5 | |||
|---|---|---|---|---|
| Position | δC | δH, mult. (J in Hz) | δC | δH, mult. (J in Hz) |
| 1 | 74.3, C | 84.4, C | ||
| 2 | 75.0, CH | 3.76, d (4.5) | 71.3, CH | 3.94, d (4.5) |
| 3 | 69.6, CH | 4.49, d (4.5) | 68.6, CH | 4.41, d (4.5) |
| 4 | 135.8, C | 138.3, C | ||
| 5 | 134.5, C | 127.3, C | ||
| 6 | 73.6, CH | 3.88, s | 76.9, CH | 5.15, s |
| 7 | 126.6, CH | 6.24, d (16.0) | 124.8, CH | 6.41, d (16.0) |
| 8 | 137.2, CH | 6.01, m | 136.7, CH | 6.12, dt (16.0, 7.0) |
| 9 | 35.0, CH2 | 2.17, m | 33.1, CH2 | 2.21, m |
| 10 | 30.5, CH2 | 1.46, m | 28.6, CH2 | 1.47, m |
| 11 | 32.9, CH2 | 1.28, m | 31.2, CH2 | 1.33, m |
| 12 | 23.9, CH2 | 1.34, m | 22.2, CH2 | 1.33, m |
| 13 | 14.7, CH3 | 0.91, t (7.0) | 13.0, CH3 | 0.91, t (7.0) |
| 14a | 61.1, CH2 | 4.25, d (13.0) | 58.1, CH2 | 4.13, d (13.0) |
| 14b | 4.38, d (13.0) | 4.51, d (13.0) | ||
| 15a | 35.7, CH2 | 2.60, m | 31.8, CH2 | 2.53, dd (15.0, 8.5) |
| 15b | 2.66, dd (15.0, 7.0) | |||
| 16 | 119.8, CH | 5.40, m | 115.3, CH | 5.25, m |
| 17 | 136.1, C | 137.7, C | ||
| 18 | 26.7, CH3 | 1.76, s | 24.9, CH3 | 1.77, s |
| 19 | 18.6, CH3 | 1.70, s | 16.9, CH3 | 1.68, s |
| 20 | 154.8, C | |||
| 6 | 7 | ||||
|---|---|---|---|---|---|
| Position | δC | δH, mult. (J in Hz) | Position | δC | δH, mult. (J in Hz) |
| 1 | 72.3, C | 2 | 77.7, C | ||
| 2 | 79.2, CH | 4.71, dd (8.0, 2.0) | 3 | 71.6, CH | 3.96, dd (12.0, 5.0) |
| 3 | 75.2, CH | 5.55, d (8.0) | 4α | 42.7, CH2 | 1.89, dd (12.0, 5.0) |
| 4 | 129.1, C | 4β | 2.25, d (12.0) | ||
| 5 | 139.4, C | 5 | 68.0, C | ||
| 6 | 71.5, CH | 4.14, d (2.0) | 6 | 81.3, CH | 4.66, dd (8.0, 2.0) |
| 7 | 126.1, CH | 6.52, d (16.0) | 7 | 78.5, CH | 5.23, d (8.0) |
| 8 | 136.6, CH | 6.03, dt (16.0, 7.0) | 8 | 133.3, C | |
| 9 | 34.9, CH2 | 2.21, dd (14.0, 7.0) | 9 | 135.6, C | |
| 10 | 30.4, CH2 | 1.45, m | 10 | 69.7, CH | 4.17, d (2.0) |
| 11 | 32.9, CH2 | 1.34, m | 11 | 16.9, CH3 | 1.26, s |
| 12 | 23.9, CH2 | 1.34, m | 12 | 28.5, CH3 | 1.21, s |
| 13 | 14.7, CH3 | 0.91, t (7.0) | 13a | 60.2, CH2 | 4.09, d (12.0) |
| 14a | 60.8, CH2 | 4.29, d (12.5) | 13b | 4.29, d, (12.0) | |
| 14b | 4.43, d (12.5) | 14 | 31.0, CH2 | 2.26, m | |
| 15a | 35.0, CH2 | 2.51, dd (15.0, 6.5) | 15 | 30.0, CH2 | 1.49, m |
| 15b | 2.71, dd (15.0, 8.5) | 16 | 31.1, CH2 | 2.25, m | |
| 16 | 118.8, CH | 5.38, m | 17 | 30.5, CH2 | 1.35, m |
| 17 | 136.9, C | 18 | 33.3, CH2 | 1.31, m | |
| 18 | 26.7, CH3 | 1.77, s | 19 | 24.0, CH2 | 1.32, m |
| 19 | 18.6, CH3 | 1.71, s | 20 | 14.7, CH3 | 0.9, t (7.0) |
| 20 | 156.8, C | 21 | 156.3, C | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, H.-B.; Ning, Z.; Hu, B.; Zhu, Y.-P.; Lu, X.-L.; He, Y.; Jiao, B.-H.; Liu, X.-Y. Cytosporin Derivatives from Arctic-Derived Fungus Eutypella sp. D-1 via the OSMAC Approach. Mar. Drugs 2023, 21, 382. https://doi.org/10.3390/md21070382
Yu H-B, Ning Z, Hu B, Zhu Y-P, Lu X-L, He Y, Jiao B-H, Liu X-Y. Cytosporin Derivatives from Arctic-Derived Fungus Eutypella sp. D-1 via the OSMAC Approach. Marine Drugs. 2023; 21(7):382. https://doi.org/10.3390/md21070382
Chicago/Turabian StyleYu, Hao-Bing, Zhe Ning, Bo Hu, Yu-Ping Zhu, Xiao-Ling Lu, Ying He, Bing-Hua Jiao, and Xiao-Yu Liu. 2023. "Cytosporin Derivatives from Arctic-Derived Fungus Eutypella sp. D-1 via the OSMAC Approach" Marine Drugs 21, no. 7: 382. https://doi.org/10.3390/md21070382
APA StyleYu, H. -B., Ning, Z., Hu, B., Zhu, Y. -P., Lu, X. -L., He, Y., Jiao, B. -H., & Liu, X. -Y. (2023). Cytosporin Derivatives from Arctic-Derived Fungus Eutypella sp. D-1 via the OSMAC Approach. Marine Drugs, 21(7), 382. https://doi.org/10.3390/md21070382