SpainUDP: The Spanish Undiagnosed Rare Diseases Program

 , , , ,
, , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Access to the Program
2.2. Inclusion Criteria
- To show clinical signs and symptoms which are not clearly identifiable or attributable to a known rare disorder, despite extensive clinical investigations by specialists of the Spanish National Health System for a long time (diagnosis is still eluded).
- To provide the consent for registering in SpainRDR, for storing biological materials in BioNER, and for sharing de-identified clinical data and samples among undiagnosed RD experts from the UDNI [6] and other international networks.
2.3. Deep Phenotyping
2.4. Genomic Analysis
2.5. Data Sharing
2.6. Ethical Issues
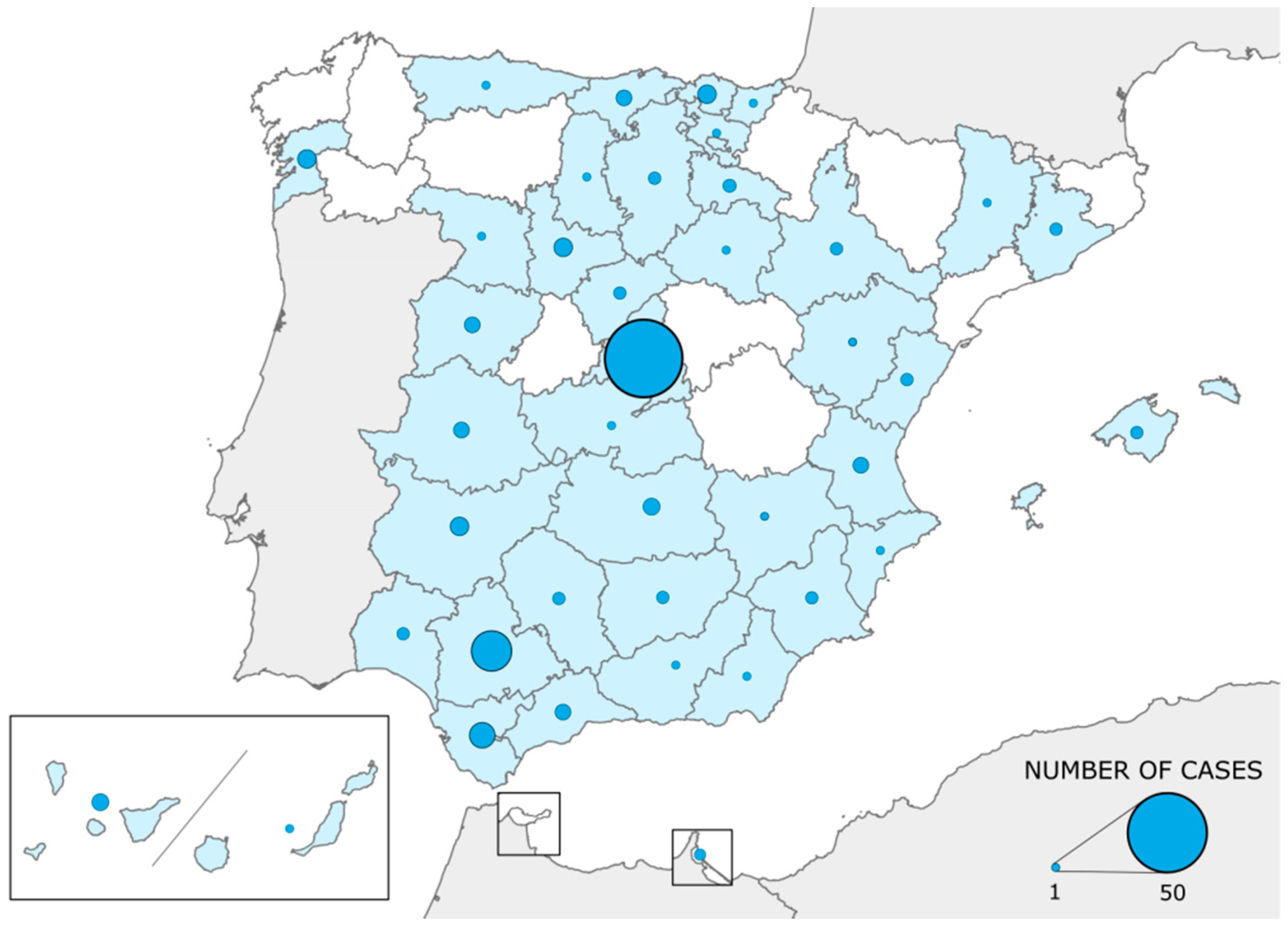
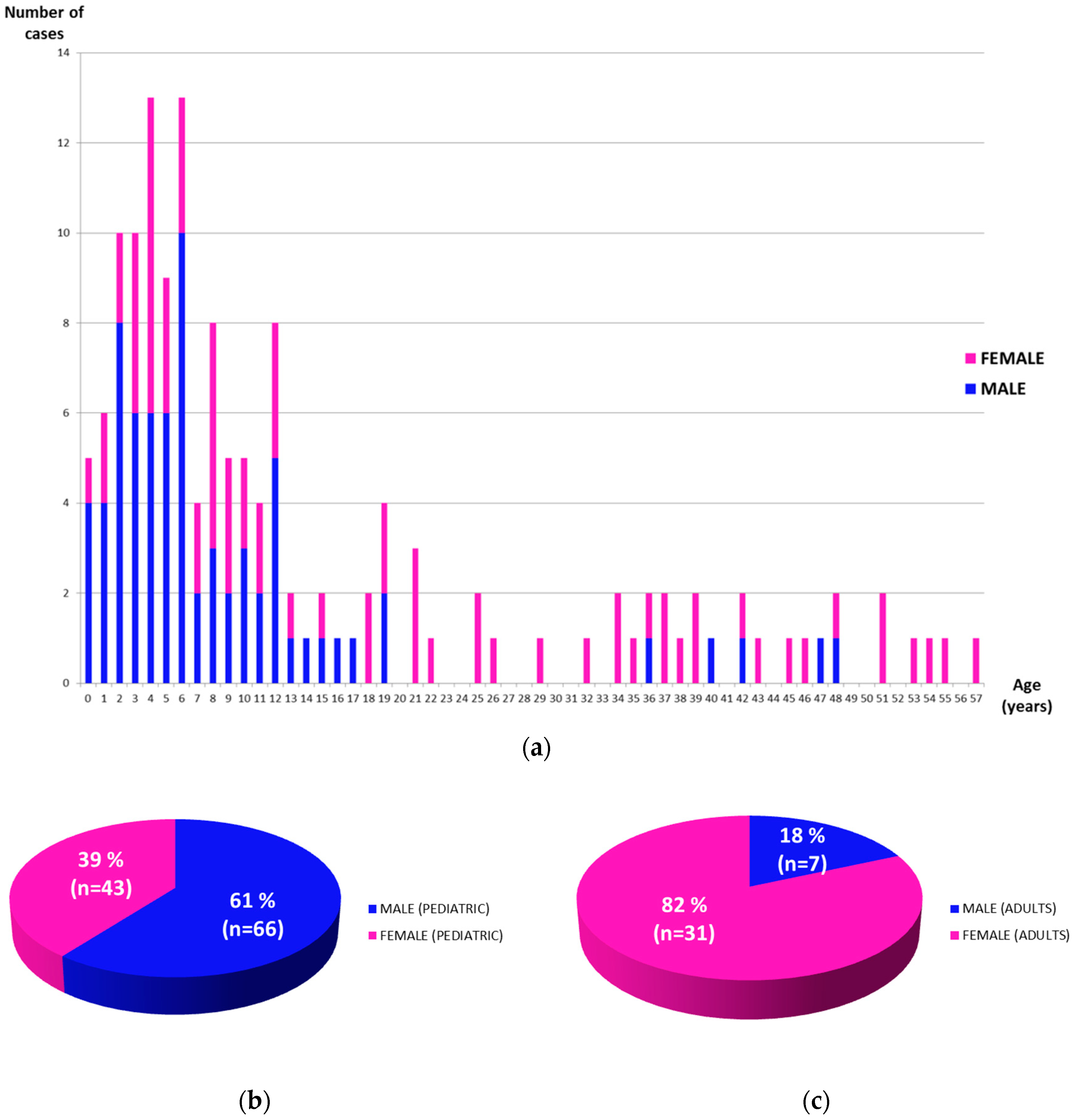
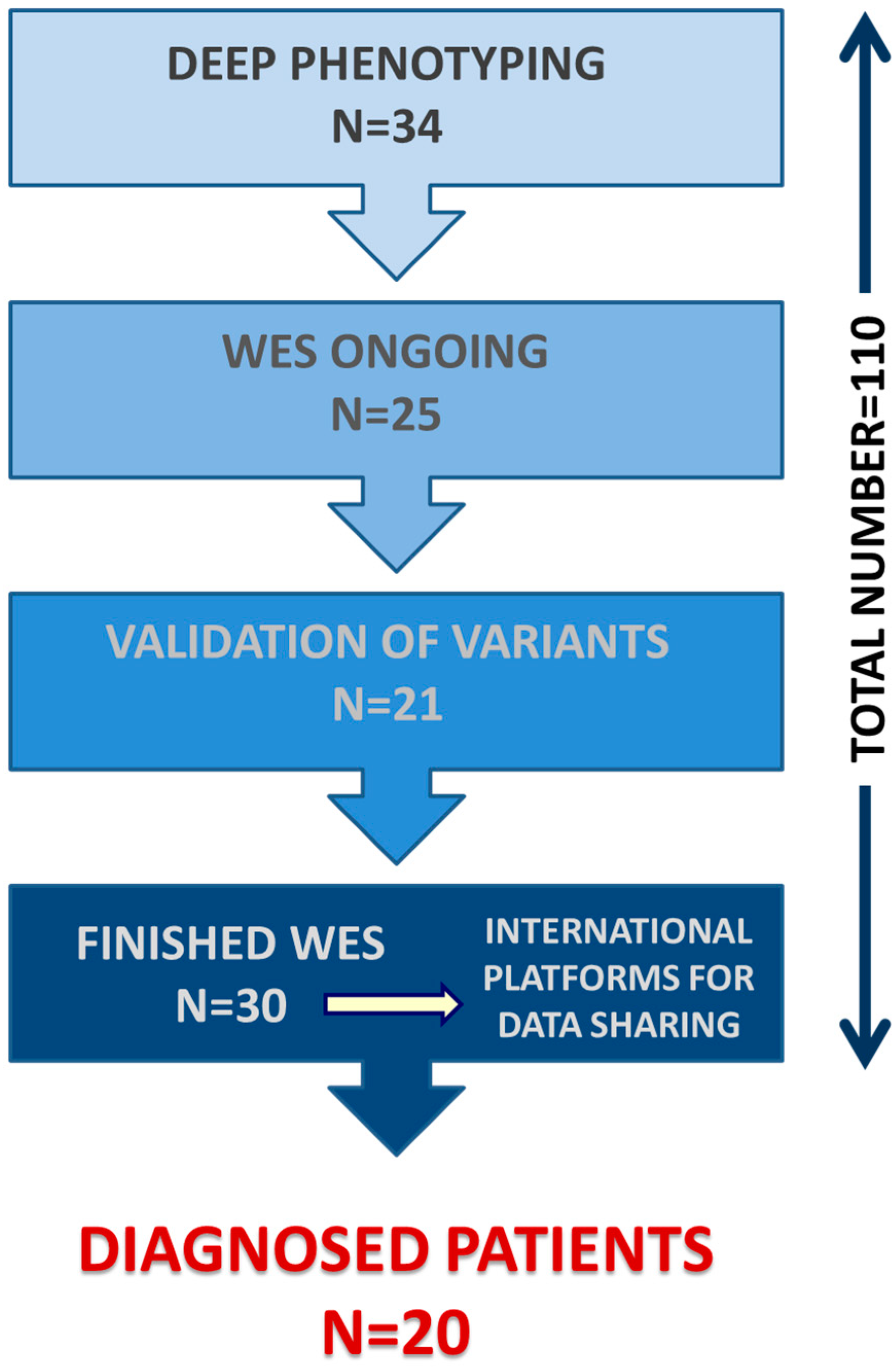
3. Results
- Case 1: a boy with a very complex phenotype including global developmental delay, osteoporosis and irregular hyperpigmentation.
- Case 2: a young woman with primary hyperparathyroidism, multiple bony cystic lesions and pathological fractures.
- Cases 3 and 4: two siblings with similar phenotypes characterized by global developmental delay, autistic features and hypotonia.
- Case 5: a boy with myoclonic epilepsy, autism spectrum disorder and frequent diarrheas.
- Case 6: a boy with severe lower limb amyotrophy, sensorimotor neuropathy and dysmorphic features.
- Case 7: a boy with a speech disorder and autistic behavior.
- Case 8: a girl with a systemic autoinflammatory disorder with a relevant digestive component (refractory to treatment).
- Case 9: a boy with remarkable dysmorphisms, such as prognathism, dental malocclusion and macrocephaly, and also absence crises, unexplained episodes of fever, intolerance to exercise and decreased muscle mass.
- Case 10: a boy who probably has two genetic diagnoses, as he has been diagnosed with epileptic encephalopathy early infantile type 4 (mutation of the STXBP1 gene) which only explains part of his phenotype, but he also has bilateral sensorineural hearing impairment of unexplained origin.
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A. SpainUDP Network
| Name | Institution | Department | City |
| Isabel Cuesta | Instituto de Salud Carlos III | Bioinformatics | Majadahonda (Madrid) |
| Sara Monzón | Instituto de Salud Carlos III | Bioinformatics | Majadahonda (Madrid) |
| Julián Lara | University Hospital Puerta de Hierro | Pediatrics | Majadahonda (Madrid) |
| Rosario Cazorla | University Hospital Puerta de Hierro | Pediatrics | Majadahonda (Madrid) |
| Gemma Iglesias | University Hospital Puerta de Hierro | Pediatrics | Majadahonda (Madrid) |
| Enriqueta Román | University Hospital Puerta de Hierro | Pediatrics | Majadahonda (Madrid) |
| Purificación Ros | University Hospital Puerta de Hierro | Pediatrics | Majadahonda (Madrid) |
| Pablo Tutor | University Hospital Puerta de Hierro | Internal Medicine | Majadahonda (Madrid) |
| Susana Mellor | University Hospital Puerta de Hierro | Internal Medicine | Majadahonda (Madrid) |
| Carlos Jiménez | University Hospital Puerta de Hierro | Neurology | Majadahonda (Madrid) |
| Mª José Cabrejas | University Hospital Puerta de Hierro | Clinical Biochemistry | Majadahonda (Madrid) |
| Emiliano González | University Hospital Puerta de Hierro | Clinical Biochemistry | Majadahonda (Madrid) |
| Luis González | University Children Hospital Niño Jesús | Pediatrics | Madrid |
| Marcos Madruga | University Hospital Virgen del Rocío | Pediatrics | Sevilla |
References
- Boycott, K.M.; Rath, A.; Chong, J.X.; Hartley, T.; Alkuraya, F.S.; Baynam, G.; Brookes, A.J.; Brudno, M.; Carracedo, A.; Den Dunnen, J.T.; et al. International cooperation to enable the diagnosis of all rare genetic diseases. Am. J. Hum. Genet. 2017, 100, 695–705. [Google Scholar] [CrossRef] [PubMed]
- Lochmüller, H.; Badowska, D.M.; Thompson, R.; Knoers, N.V.; Aartsma-Rus, A.; Gut, I.; Wood, L.; Harmuth, T.; Durudas, A.; Graessner, H.; et al. RD-Connect, NeurOmics and EURenOmics: Collaborative European initiative for rare diseases. Eur. J. Hum. Genet. 2018, 26, 778–785. [Google Scholar] [CrossRef] [PubMed]
- Posey, J.E.; Harel, T.; Liu, P.; Rosenfeld, J.A.; James, R.A.; Coban Akdemir, Z.H.; Walkiewicz, M.; Bi, W.; Xiao, R.; Ding, Y.; et al. Resolution of disease phenotypes resulting from multilocus genomic variation. N. Engl. J. Med. 2017, 376, 21–31. [Google Scholar] [CrossRef] [PubMed]
- Shashi, V.; McConkie-Rosell, A.; Rosell, B.; Schoch, K.; Vellore, K.; McDonald, M.; Jiang, Y.H.; Xie, P.; Need, A.; Goldstein, D.B. The utility of the traditional medical genetics diagnostic evaluation in the context of next-generation sequencing for undiagnosed genetic disorders. Genet. Med. 2014, 16, 176–182. [Google Scholar] [CrossRef] [PubMed]
- Gahl, W.A.; Mulvihill, J.J.; Toro, C.; Markello, T.C.; Wise, A.L.; Ramoni, R.B.; Adams, D.R.; Tifft, C.J. The NIH Undiagnosed Diseases Program and Network: Applications to modern medicine. Mol. Genet. Metab. 2016, 117, 393–400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- UDNI Website. Available online: http://www.udninternational.org (accessed on 17 May 2018).
- Taruscio, D.; Groft, S.C.; Cederroth, H.; Melegh, B.; Lasko, P.; Kosaki, K.; Baynam, G.; McCray, A.; Gahl, W.A. Undiagnosed Diseases Network International (UDNI): White paper for global actions to meet patient needs. Mol. Genet. Metab. 2015, 116, 223–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taruscio, D.; Floridia, G.; Salvatore, M.; Groft, S.C.; Gahl, W.A. Undiagnosed Diseases: Italy-US Collaboration and International Efforts to Tackle Rare and Common Diseases Lacking a Diagnosis. Adv. Exp. Med. Biol. 2017, 1031, 25–38. [Google Scholar] [CrossRef] [PubMed]
- Xue, Y.; Ankala, A.; Wilcox, W.R.; Hegde, M.R. Solving the molecular diagnostic testing conundrum for Mendelian disorders in the era of next-generation sequencing: Single-gene, gene panel, or exome/genome sequencing. Genet. Med. 2015, 17, 444–451. [Google Scholar] [CrossRef] [PubMed]
- FEDER Website. Available online: https://enfermedades-raras.org (accessed on 17 May 2018).
- Strategy for the Improvement of Health Care for Patients with Uncommon Diseases in the Community of Madrid (2016–2020). Available online: http://www.madrid.org/bvirtual/BVCM017933.pdf (accessed on 17 May 2018).
- Austin, C.P.; Dawkins, H.J.S. Medical research: Next decade’s goals for rare diseases. Nature 2017, 548, 158. [Google Scholar] [CrossRef] [PubMed]
- Asociación D’genes Website. Available online: http://www.dgenes.es/ (accessed on 17 May 2018).
- Laurie, S.; Fernandez-Callejo, M.; Marco-Sola, S.; Trotta, J.R.; Camps, J.; Chacón, A.; Espinosa, A.; Gut, M.; Gut, I.; Heath, S.; Beltran, S. From Wet-Lab to Variations: Concordance and Speed of Bioinformatics Pipelines for Whole Genome and Whole Exome Sequencing. Hum. Mutat. 2016, 37, 1263–1271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thompson, R.; Johnston, L.; Taruscio, D.; Monaco, L.; Béroud, C.; Gut, I.G.; Hansson, M.G.; ‘tHoen, P.B.; Patrinos, G.P.; Dawkins, H.; et al. RD-Connect: An integrated platform connecting databases, registries, biobanks and clinical bioinformatics for rare disease research. J. Gen. Intern. Med. 2014, 29, 780–787. [Google Scholar] [CrossRef] [PubMed]
- Köhler, S.; Vasilevsky, N.A.; Engelstad, M.; Foster, E.; McMurry, J.; Aymé, S.; Baynam, G.; Bello, S.M.; Boerkoel, C.F.; Boycott, K.M.; et al. The Human Phenotype Ontology in 2017. Nucleic Acids Res. 2017, 45, D865–D876. [Google Scholar] [CrossRef] [PubMed]
- Girdea, M.; Dumitriu, S.; Fiume, M.; Bowdin, S.; Boycott, K.M.; Chénier, S.; Chitayat, D.; Faghfoury, H.; Meyn, M.S.; Ray, P.N.; et al. PhenoTips: Patient phenotyping software for clinical and research use. Hum. Mutat. 2013, 34, 1057–1065. [Google Scholar] [CrossRef] [PubMed]
- Buske, O.J.; Girdea, M.; Dumitriu, S.; Gallinger, B.; Hartley, T.; Trang, H.; Misyura, A.; Friedman, T.; Beaulieu, C.; Bone, W.P.; et al. PhenomeCentral: A portal for phenotypic and genotypic matchmaking of patients with rare genetic diseases. Hum. Mutat. 2015, 36, 931–940. [Google Scholar] [CrossRef] [PubMed]
- Philippakis, A.A.; Azzariti, D.R.; Beltran, S.; Brookes, A.J.; Brownstein, C.A.; Brudno, M.; Brunner, H.G.; Buske, O.J.; Carey, K.; Doll, C.; et al. The Matchmaker Exchange: A platform for rare disease gene discovery. Hum. Mutat. 2015, 36, 915–921. [Google Scholar] [CrossRef] [PubMed]
- Zurynski, Y.; Deverell, M.; Dalkeith, T.; Johnson, S.; Christodoulou, J.; Leonard, H.; Elliott, E.J.; APSU Rare Diseases Impacts on Families Study group. Australian children living with rare diseases: Experiences of diagnosis and perceived consequences of diagnostic delays. Orphanet J. Rare Dis. 2017, 12, 68. [Google Scholar] [CrossRef] [PubMed]
- Fombonne, E. Epidemiology of pervasive developmental disorders. Pediatric Res. 2009, 65, 591–598. [Google Scholar] [CrossRef] [PubMed]
- Women and Men Face Different Chronic Disease Risks. Available online: https://www.paho.org (accessed on 17 May 2018).
- Posey, J.E.; Rosenfeld, J.A.; James, R.A.; Bainbridge, M.; Niu, Z.; Wang, X.; Dhar, S.; Wiszniewski, W.; Akdemir, Z.H.; Gambin, T.; et al. Molecular diagnostic experience of whole-exome sequencing in adult patients. Genet. Med. 2016, 18, 678–685. [Google Scholar] [CrossRef] [PubMed]
- Gahl, W.A.; Markello, T.C.; Toro, C.; Fajardo, K.F.; Sincan, M.; Gill, F.; Carlson-Donohoe, H.; Gropman, A.; Pierson, T.M.; Golas, G.; et al. The NIH Undiagnosed Diseases Program: Insights into Rare Diseases. Genet. Med. 2012, 14, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Muzny, D.M.; Xia, F.; Niu, Z.; Person, R.; Ding, Y.; Ward, P.; Braxton, A.; Wang, M.; Buhay, C.; et al. Molecular Findings Among Patients Referred for Clinical Whole-Exome Sequencing. JAMA 2014, 312, 1870–1879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farwell, K.D.; Shahmirzadi, L.; El-Khechen, D.; Powis, Z.; Chao, E.C.; Tippin-Davis, B.; Baxter, R.M.; Zeng, W.; Mroske, C.; Parra, M.C.; et al. Enhanced utility of family-centered diagnostic exome sequencing with inheritance model-based analysis: Results from 500 unselected families with undiagnosed genetic conditions. Genet. Med. 2015, 17, 578–586. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Muzny, D.M.; Reid, J.G.; Bainbridge, M.N.; Willis, A.; Ward, P.A.; Braxton, A.; Beuten, J.; Xia, F.; Niu, Z.; et al. Clinical whole-exome sequencing for the diagnosis of mendelian disorders. N. Engl. J. Med. 2013, 369, 1502–1511. [Google Scholar] [CrossRef] [PubMed]
- Meng, L.; Pammi, M.; Saronwala, A.; Magoulas, P.; Ghazi, A.R.; Vetrini, F.; Zhang, J.; He, W.; Dharmadhikari, A.V.; Qu, C.; et al. Use of Exome Sequencing for Infants in Intensive Care Units: Ascertainment of Severe Single-Gene Disorders and Effect on Medical Management. JAMA Pediatr. 2017, 171, e173438. [Google Scholar] [CrossRef] [PubMed]
- Retterer, K.; Juusola, J.; Cho, M.T.; Vitazka, P.; Millan, F.; Gibellini, F.; Vertino-Bell, A.; Smaoui, N.; Neidich, J.; Monaghan, K.G.; et al. Clinical application of whole-exome sequencing across clinical indications. Genet. Med. 2016, 18, 696–704. [Google Scholar] [CrossRef] [PubMed]
- Eldomery, M.K.; Coban-Akdemir, Z.; Harel, T.; Rosenfeld, J.A.; Gambin, T.; Stray-Pedersen, A.; Küry, S.; Mercier, S.; Lessel, D.; Denecke, J.; et al. Lessons learned from additional research analyses of unsolved clinical exome cases. Genome Med. 2017, 9, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.; Deignan, J.L.; Dorrani, N.; Strom, S.P.; Kantarci, S.; Quintero-Rivera, F.; Das, K.; Toy, T.; Harry, B.; Yourshaw, M.; et al. Clinical exome sequencing for genetic identification of rare Mendelian disorders. JAMA 2014, 312, 1880–1887. [Google Scholar] [CrossRef] [PubMed]
- IRDIRC State of Play of Research in the Field of Rare Diseases: 2014–2015. Available online: http://www.irdirc.org/wp-content/uploads/2015/09/IRDiRC_State-of-Play-2015.pdf (accessed on 17 May 2018).
- López, E.; Thompson, R.; Gainotti, S.; Wang, C.M.; Rubinstein, Y.; Taruscio, D.; Monaco, L.; Lochmüller, H.; Alonso, V.; Posada, M. Overview of existing initiatives to develop and improve access and data sharing in rare disease registries and biobanks worldwide. Expert Opin. Investig. Drugs. 2016, 4, 729–739. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Case | Molecular Testing before Entry in SpainUDP | Candidate Gene (Acronym & OMIM Number) | Mutation Effect | Mutation Type | Diagnosis (Disease Name & OMIM Number) | Inheritance 1 | Prevalence 2 |
|---|---|---|---|---|---|---|---|
| 1 | Karyotype, CGH array, single-gene testing & methylation study | MEF2C (OMIM 600662) | Stopgain | De novo | Mental retardation, autosomal dominant 20 (OMIM 613443) | AD | <1/1,000,000 |
| 2 | Karyotype & single-gene testing | VPS13B (OMIM 607817) | Stopgain/Splicing | Compound heterozygous | Cohen syndrome (OMIM 216550) | AR | Unknown 3 |
| 3 | Single-gene testing | SLC52A2 (OMIM 607882) | Missense | Homozygous | Brown-Vialetto-Van Laere syndrome 2 (OMIM 607882) | AR | <1/1,000,000 |
| 4 | Karyotype, CGH array & single-gene testing | AUTS2 (OMIM 607270) | Frameshift | De novo | Mental retardation, autosomal dominant 26 (OMIM 615834) | AD | <1/1,000,000 |
| 5 | Karyotype, CGH array & single-gene testing | CTNNB1 (OMIM 116806) | Frameshift | De novo | Mental retardation, autosomal dominant 19 (OMIM 615075) | AD | <1/1,000,000 |
| 6 | Karyotype & CGH array | EHMT1 (OMIM 607001) | Stopgain | De novo | Kleefstra syndrome 1 (OMIM 610253) | AD | <1/1,000,000 |
| 7 | CGH array | SCN2A (OMIM 182390) | Frameshift | De novo | Epileptic encephalopathy, early infantile, 11 (OMIM 613721) | AD | Unknown |
| 8 | Karyotype, CGH array & single-gene testing | KMT2D (OMIM 602113) | Frameshift | De novo | Kabuki syndrome 1 (OMIM 147920) | AD | 1–9/100,000 |
| 9 | Karyotype & single-gene testing | PIK3R1 (OMIM 171833) | Missense | De novo | Short syndrome (OMIM 269880) | AD | <1/1,000,000 |
| 10 | Karyotype, CGH array & single-gene testing | DDX3X (OMIM 300160) | Frameshift | De novo | Mental retardation, X-linked 102 (OMIM 300958) | XLD, XLR | <1/1,000,000 |
| 11 | CGH array and mitochondrial DNA study | PNPT1 (OMIM 610316) | Stopgain/Missense | Compound heterozygous | Combined oxidative phosphorylation deficiency 13 (OMIM 614932) | AR | <1/1,000,000 |
| 12 | Karyotype, CGH array & single-gene testing | KAT6A (OMIM 601408) | Frameshift | De novo | Mental retardation, autosomal dominant 32 (OMIM 616268) | AD | <1/1,000,000 |
| 13 | Single-gene testing | SLC6A1 (OMIM 137165) | Frameshift | De novo | Myoclonic-atonic epilepsy (OMIM 616421) | AD | Unknown |
| 14 | Karyotype, CGH array & single-gene testing | SYNGAP1 (OMIM 603384) | Frameshift | De novo | Mental retardation, autosomal dominant 5 (OMIM 612621) | AD | Unknown |
| 15 | Karyotype, CGH array & mitochondrial DNA study | SLC9A6 (OMIM 300231) | Frameshift | De novo | Mental retardation, X-linked, syndromic, Christianson type (OMIM 300243) | XLD | <1/1,000,000 |
| 16 | CGH array & single-gene testing | KCNQ2 (OMIM 602235) | Missense | De novo | Epileptic encephalopathy, early infantile, 7 (OMIM 613720) & Seizures, benign familial neonatal, 1 (OMIM 121200) | AD | Unknown |
| 17 | Single-gene testing & gene panel | NKX2-1 (OMIM 600635) | Frameshift | De novo | Choreoathetosis and congenital hypothyroidism with or without pulmonary dysfunction (OMIM 610978) | AD | <1/1,000,000 |
| 18 | Single-gene testing, mitochondrial DNA study, gene panel & single-WES in proband | VPS13D (OMIM 608877) | Missense/Missense | Compound heterozygous | Movement disorder with ataxia and spasticity(no OMIM code yet) | AR | Unknown |
| 19 | Karyotype, CGH array & single-gene testing | FRMPD4 (OMIM 300838) | Missense | Hemyzygous | Mental retardation, X-linked 104 (OMIM 300983) | XLR | Unknown |
| 20 | Karyotype, CGH array, single-gene testing & gene panel | KAT6A (OMIM 601408) | Missense | De novo | Mental retardation, autosomal dominant 32 (OMIM 616268) | AD | <1/1,000,000 |
| Case | Gender | Age at SpainUDP Entry (Years) | Age at Diagnosis (Years) | Time to Diagnosis (Months) |
|---|---|---|---|---|
| 1 | female | 5.3 | 6.8 | 18.3 |
| 2 | female | 3.1 | 3.2 | 1.8 |
| 3 | female | 26.8 | 27.7 | 10.6 |
| 4 | male | 2.2 | 3.0 | 9.4 |
| 5 | male | 4.6 | 5.3 | 8.3 |
| 6 | female | 11.1 | 12.5 | 16.6 |
| 7 | male | 2.9 | 4.1 | 13.5 |
| 8 | male | 6.0 | 7.2 | 14.4 |
| 9 | male | 8.5 | 9.9 | 16.0 |
| 10 | female | 9.9 | 11.4 | 18.7 |
| 11 | male | 1.5 | 3.0 | 18.1 |
| 12 | female | 6.4 | 8.1 | 19.5 |
| 13 | female | 6.3 | 7.3 | 11.4 |
| 14 | male | 14.0 | 14.9 | 11.2 |
| 15 | male | 7.2 | 8.1 | 10.5 |
| 16 | male | 6.1 | 6.9 | 10.1 |
| 17 | male | 12.7 | 13.6 | 10.8 |
| 18 | female | 8.5 | 9.7 | 13.5 |
| 19 | male | 5.6 | 7.6 | 23.9 |
| 20 | male | 6.6 | 8.1 | 17.8 |
| Mean ± SD | 7.8 ± 5.6 | 8.9 ± 5.6 | 13.7 ± 5.0 | |
| Median | 6.4 | 7.8 | 13.5 | |
| Minimum value | 1.5 | 3.0 | 1.8 | |
| Maximum value | 26.8 | 27.7 | 23.9 | |
| 10th percentile | 2.3 | 3.1 | 8.4 | |
| 90th percentile | 13.9 | 14.8 | 19.4 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
López-Martín, E.; Martínez-Delgado, B.; Bermejo-Sánchez, E.; Alonso, J.; The SpainUDP Network; Posada, M. SpainUDP: The Spanish Undiagnosed Rare Diseases Program. Int. J. Environ. Res. Public Health 2018, 15, 1746. https://doi.org/10.3390/ijerph15081746
López-Martín E, Martínez-Delgado B, Bermejo-Sánchez E, Alonso J, The SpainUDP Network, Posada M. SpainUDP: The Spanish Undiagnosed Rare Diseases Program. International Journal of Environmental Research and Public Health. 2018; 15(8):1746. https://doi.org/10.3390/ijerph15081746
Chicago/Turabian StyleLópez-Martín, Estrella, Beatriz Martínez-Delgado, Eva Bermejo-Sánchez, Javier Alonso, The SpainUDP Network, and Manuel Posada. 2018. "SpainUDP: The Spanish Undiagnosed Rare Diseases Program" International Journal of Environmental Research and Public Health 15, no. 8: 1746. https://doi.org/10.3390/ijerph15081746
APA StyleLópez-Martín, E., Martínez-Delgado, B., Bermejo-Sánchez, E., Alonso, J., The SpainUDP Network, & Posada, M. (2018). SpainUDP: The Spanish Undiagnosed Rare Diseases Program. International Journal of Environmental Research and Public Health, 15(8), 1746. https://doi.org/10.3390/ijerph15081746