Growing Teratoma Syndrome in the Setting of Sarcoidosis: A Case Report and Literature Review

 ,
, {kind=link}
{kind=link}
Abstract
:1. Introduction
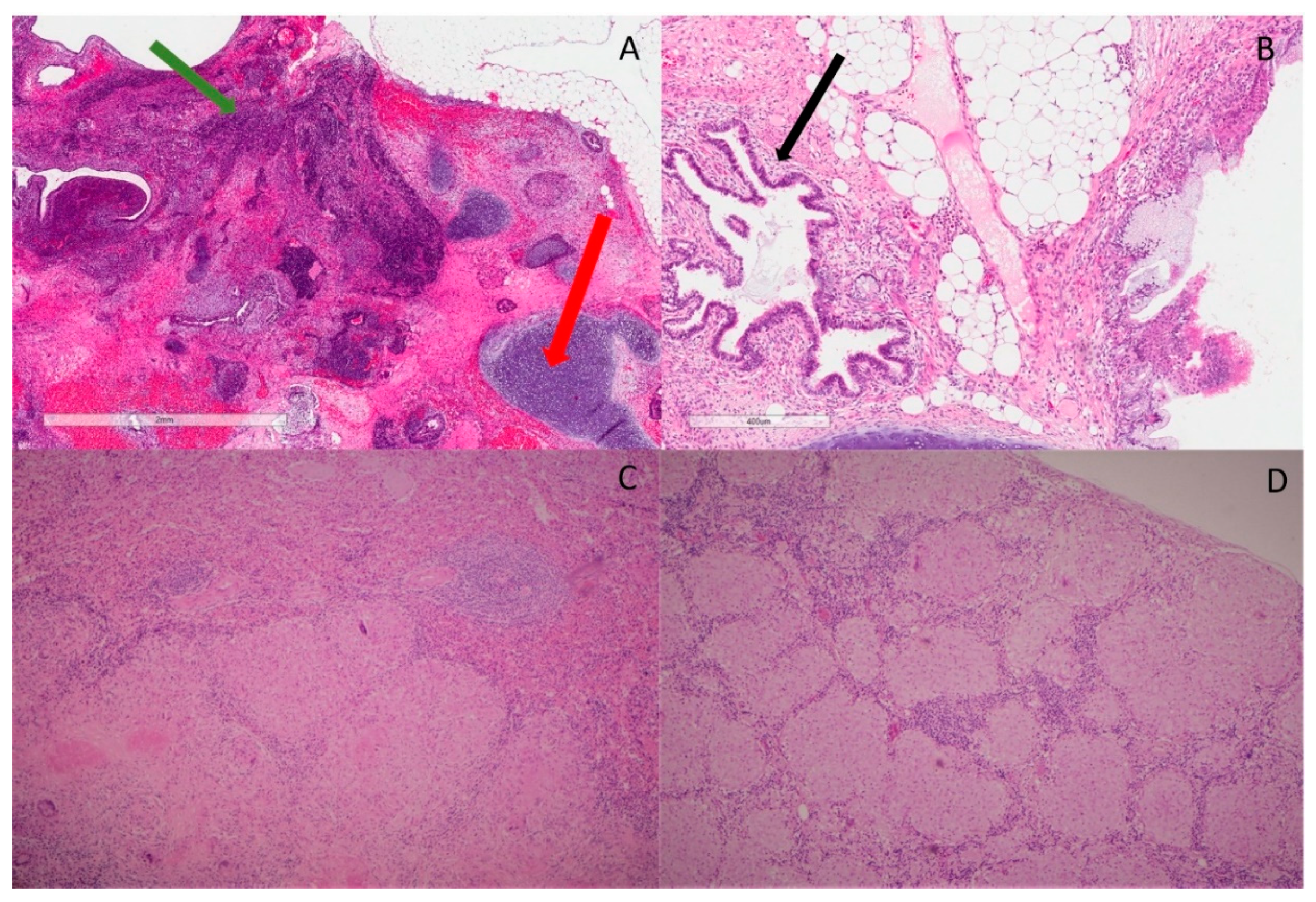
2. Case Report/Case Presentation
3. Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Outwater, E.K.; Siegelman, E.S.; Hunt, J.L. Ovarian teratomas: Tumor types and imaging characteristics. Radiographics 2001, 21, 475–490. [Google Scholar] [CrossRef] [Green Version]
- Heifetz, S.A.; Cushing, B.; Giller, R.; Shuster, J.J.; Stolar, C.J.H.; Vinocur, C.D.; Hawkins, E.P. Immature teratomas in children: Pathologic considerations: A report from the combined Pediatric Oncology Group/Children’s Cancer Group. Am. J. Surg. Pathol. 1998, 22, 1115–1124. [Google Scholar] [CrossRef]
- Gershenson, D.M. Management of Ovarian Germ Cell Tumors. J. Clin. Oncol. 2007, 25, 2938–2943. [Google Scholar] [CrossRef]
- Williams, S.D.; Blessing, J.A.; Hatch, K.D.; Homesley, H.D. Chemotherapy of advanced dysgerminoma: Trials of the Gynecologic Oncology Group. J. Clin. Oncol. 1991, 9, 1950–1955. [Google Scholar] [CrossRef] [PubMed]
- Gershenson, D.M. Treatment of ovarian cancer in young women. Clin. Obstet. Gynecol. 2012, 55, 65–74. [Google Scholar] [CrossRef]
- Salani, R.; Khanna, N.; Frimer, M.; Bristow, R.E.; Chen, L.M. An update on post-treatment surveillance and diagnosis of recurrence in women with gynecologic malignancies: Society of Gynecologic Oncology (SGO) recommendations. Gynecol. Oncol. 2017, 146, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Messing, M.J.; Gershenson, D.M.; Morris, M.; Burke, T.W.; Kavanagh, J.J.; Wharton, J.T. Primary treatment failure in patients with malignant ovarian germ cell neoplasms. Int. J. Gynecol. Cancer 1992, 2, 295–300. [Google Scholar] [CrossRef]
- Abdul Razak, A.R.; Li, L.; Bryant, A.; Diaz-Padilla, I. Chemotherapy for malignant germ cell ovarian cancer in adult patients with early stage, advanced and recurrent disease. Cochrane Database Syst. Rev. 2011, 3, CD007584. [Google Scholar] [CrossRef] [Green Version]
- Adra, N.; Abonour, R.; Althouse, S.K.; Albany, C.; Hanna, N.H.; Einhorn, L.H. High-Dose Chemotherapy and Autologous Peripheral-Blood Stem-Cell Transplantation for Relapsed Metastatic Germ Cell Tumors: The Indiana University Experience. J. Clin. Oncol. 2017, 35, 1096–1102. [Google Scholar] [CrossRef]
- Logothetis, C.J.; Samuels, M.L.; Trindade, A.; Johnson, D.E. The growing teratoma syndrome. Cancer 1982, 50, 1629–1635. [Google Scholar] [CrossRef]
- Amsalem, H.; Nadjari, M.; Prus, D.; Hiller, N.; Benshushan, A. Growing teratoma syndrome vs. chemotherapeutic retroconversion: Case report and review of the literature. Gynecol. Oncol. 2004, 92, 357–360. [Google Scholar] [CrossRef] [PubMed]
- Spiess, P.E.; Kassouf, W.; Brown, G.A.; Kamat, A.M.; Liu, P.; Gomez, J.A.; Tu, S.-M.; Tannir, N.M.; Pisters, L.L. Surgical management of growing teratoma syndrome: The M. D. Anderson cancer center experience. J. Urol. 2007, 1, 1330–1334; discussion 4. [Google Scholar] [CrossRef]
- Wang, D.; Zhu, S.; Jia, C.; Cao, D.; Wu, M.; Shen, K.; Yang, J.; Pan, L.; Cheng, N.; Xiang, Y. Diagnosis and management of growing teratoma syndrome after ovarian immature teratoma: A single center experience. Gynecol. Oncol. 2020, 157, 94–100. [Google Scholar] [CrossRef]
- Bentivegna, E.; Azaïs, H.; Uzan, C.; Leary, A.; Pautier, P.; Gonthier, C.; Genestie, C.; Balleyguier, C.; Lhomme, C.; Duvillard, P.; et al. Surgical Outcomes After Debulking Surgery for Intraabdominal Ovarian Growing Teratoma Syndrome: Analysis of 38 Cases. Ann. Surg. Oncol. 2015, 22 (Suppl. 3), S964–S970. [Google Scholar] [CrossRef] [PubMed]
- André, F.; Fizazi, K.; Culine, S.; Droz, J.; Taupin, P.; Lhommé, C.; Terrier-Lacombe, M.-J.; Theodore. The growing teratoma syndrome: Results of therapy and long-term follow-up of 33 patients. Eur. J. Cancer 2000, 36, 1389–1394. [Google Scholar] [CrossRef]
- Tangjitgamol, S.; Manusirivithaya, S.; Leelahakorn, S.; Thawaramara, T.; Suekwatana, P.; Sheanakul, C. The growing teratoma syndrome: A case report and a review of the literature. Int. J. Gynecol. Cancer 2006, 16 (Suppl. 1), 384–390. [Google Scholar] [CrossRef] [PubMed]
- Shibata, K.; Kajiyama, H.; Kikkawa, F. Growing Teratoma Syndrome of the Ovary Showing Three Patterns of Metastasis: A Case Report. Case Rep. Oncol. 2013, 6, 544–549. [Google Scholar] [CrossRef] [PubMed]
- Moskovic, E.; Jobling, T.; Fisher, C.; Wiltshaw, E.; Parsons, C. Retroconversion of immature teratoma of the ovary: CT appearances. Clin. Radiol. 1991, 43, 402–408. [Google Scholar] [CrossRef]
- Wang, M.; Jiang, S.; Zhang, Y.; Jiang, C.; Xia, F.; Lyu, W.; Ma, X. The application of 18F-FDG PET/CT in ovarian immature teratomas when pathological examination results contradict clinical observations: A case report. Medicine 2017, 96, e9171. [Google Scholar] [CrossRef] [PubMed]
- Zeman, M.N.; Green, C.; Akin, E.A. Spectrum of [(18)F]FDG-PET/CT] Findings in Benign Lymph Node Pathology. Mol. Imaging Biol. 2021, 23, 469–480. [Google Scholar] [CrossRef]
- Valeyre, D.; Jeny, F.; Rotenberg, C.; Bouvry, D.; Uzunhan, Y.; Seve, P.; Nunes, H.; Bernaudin, J.-F. How to Tackle the Diagnosis and Treatment in the Diverse Scenarios of Extrapulmonary Sarcoidosis. Adv. Ther. 2021, 38, 4605–4627. [Google Scholar] [CrossRef]
- Arkema, E.V.; Cozier, Y.C. Sarcoidosis epidemiology: Recent estimates of incidence, prevalence and risk factors. Curr. Opin. Pulm. Med. 2020, 26, 527–534. [Google Scholar] [CrossRef]
- Koyama, T.; Ueda, H.; Togashi, K.; Umeoka, S.; Kataoka, M.; Nagai, S. Radiologic manifestations of sarcoidosis in various organs. Radiographics 2004, 24, 87–104. [Google Scholar] [CrossRef] [PubMed]
- Baughman, R.P.; Teirstein, A.S.; Judson, M.A.; Rossman, M.D.; Yeager, H., Jr.; Bresnitz, E.A.; DePalo, L.; Hunninghake, G.; Iannuzzi, M.C.; Johns, C.J.; et al. Clinical characteristics of patients in a case control study of sarcoidosis. Am. J. Respir. Crit. Care Med. 2001, 164 Pt 10, 1885–1889. [Google Scholar] [CrossRef] [Green Version]
- Iannuzzi, M.C.; Rybicki, B.A.; Teirstein, A.S. Sarcoidosis. N. Engl. J. Med. 2007, 357, 2153–2165. [Google Scholar] [CrossRef]
- Teirstein, A.S.; Machac, J.; Almeida, O.; Lu, P.; Padilla, M.L.; Iannuzzi, M.C. Results of 188 whole-body fluorodeoxyglucose positron emission tomography scans in 137 patients with sarcoidosis. Chest 2007, 132, 1949–1953. [Google Scholar] [CrossRef]
- Kennedy, P.T.; Zakaria, N.; Modawi, S.B.; Papadopoulou, A.M.; Murray-Lyon, I.; du Bois, R.M.; Jervoise, N.A.; Devlin, J. Natural history of hepatic sarcoidosis and its response to treatment. Eur. J. Gastroenterol. Hepatol. 2006, 18, 721–726. [Google Scholar] [CrossRef] [PubMed]
- Hunt, B.M.; Vallieres, E.; Buduhan, G.; Aye, R.; Louie, B. Sarcoidosis as a benign cause of lymphadenopathy in cancer patients. Am. J. Surg. 2009, 197, 629–632. [Google Scholar] [CrossRef]
- Reich, J.M. Neoplasia in the etiology of sarcoidosis. Eur. J. Intern. Med. 2006, 17, 81–87. [Google Scholar] [CrossRef]
- Brincker, H. Sarcoid reactions and sarcoidosis in Hodgkin’s disease and other malignant lymphomata. Br. J. Cancer 1972, 26, 120–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merchant, T.E.; Filippa, D.A.; Yahalom, J. Sarcoidosis following chemotherapy for Hodgkin’s disease. Leuk Lymphoma 1994, 13, 339–347. [Google Scholar] [CrossRef] [PubMed]
- Hurst, E.A.; Mauro, T. Sarcoidosis associated with pegylated interferon alfa and ribavirin treatment for chronic hepatitis C: A case report and review of the literature. Arch. Dermatol. 2005, 141, 865–868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chanson, N.; Ramos-Casals, M.; Pundole, X.; Suijkerbuijk, K.; e Silva, M.J.d.B.; Lidar, M.; Benesova, K.; Leipe, J.; Acar-Denizli, N.; Pradere, P.; et al. Immune checkpoint inhibitor–associated sarcoidosis: A usually benign disease that does not require immunotherapy discontinuation. Eur. J. Cancer 2021, 158, 208–216. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shahnam, A.; Sayer, R.; Herbst, U.; Sharma, R.; Yoon, W.-h.; Dinihan, T.; Gao, B. Growing Teratoma Syndrome in the Setting of Sarcoidosis: A Case Report and Literature Review. Curr. Oncol. 2022, 29, 4148-4154. https://doi.org/10.3390/curroncol29060331
Shahnam A, Sayer R, Herbst U, Sharma R, Yoon W-h, Dinihan T, Gao B. Growing Teratoma Syndrome in the Setting of Sarcoidosis: A Case Report and Literature Review. Current Oncology. 2022; 29(6):4148-4154. https://doi.org/10.3390/curroncol29060331
Chicago/Turabian StyleShahnam, Adel, Robyn Sayer, Unine Herbst, Raghwa Sharma, Won-hee Yoon, Tim Dinihan, and Bo Gao. 2022. "Growing Teratoma Syndrome in the Setting of Sarcoidosis: A Case Report and Literature Review" Current Oncology 29, no. 6: 4148-4154. https://doi.org/10.3390/curroncol29060331
APA StyleShahnam, A., Sayer, R., Herbst, U., Sharma, R., Yoon, W. -h., Dinihan, T., & Gao, B. (2022). Growing Teratoma Syndrome in the Setting of Sarcoidosis: A Case Report and Literature Review. Current Oncology, 29(6), 4148-4154. https://doi.org/10.3390/curroncol29060331