Is the Fischer-Tropsch Conversion of Biogas-Derived Syngas to Liquid Fuels Feasible at Atmospheric Pressure?


Abstract
:1. Introduction
2. Materials and Methods
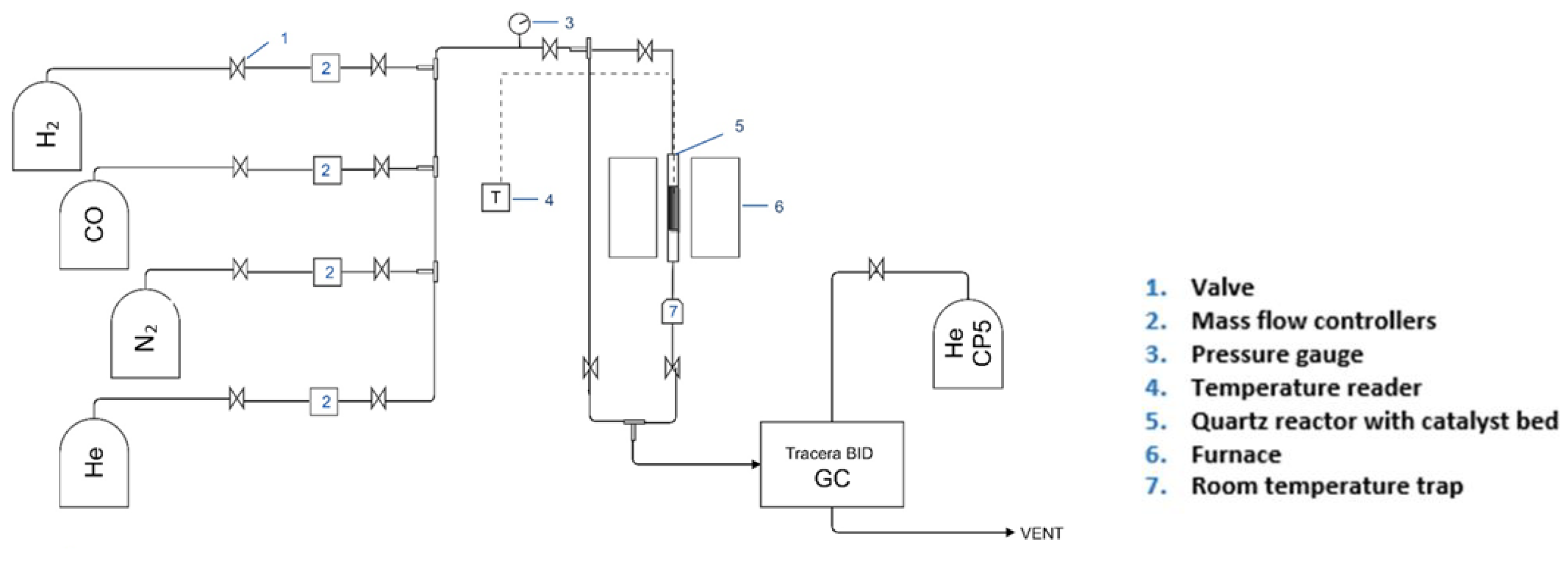
2.1. Experimental Rig Setup and Reaction Refinement
2.2. Testing the FTS System’s Robustness by the Introduction of Interruption Factors
2.3. Economic Feasibility of the Fischer-Tropsch Conversion of Biogas-Derived Syngas to Liquid Fuels
- Scenario H1/T1: AD » biogas » reforming » syngas » Fischer-Tropsch » liquid fuel » heat/transport
- Scenario H2/T2: AD » biogas » upgrading » biomethane » compression » CNG » heat/transport
- Scenario H3/T3: AD » biogas » upgrading » biomethane » liquefaction » LNG » heat/transport
3. Results and Discussion
3.1. Iterative Refinement of the FT Reaction at 0.1 MPa (1 bar) (R1, R2, and R3)
3.2. Optimising the Final FTS Refined System Setup (R4)
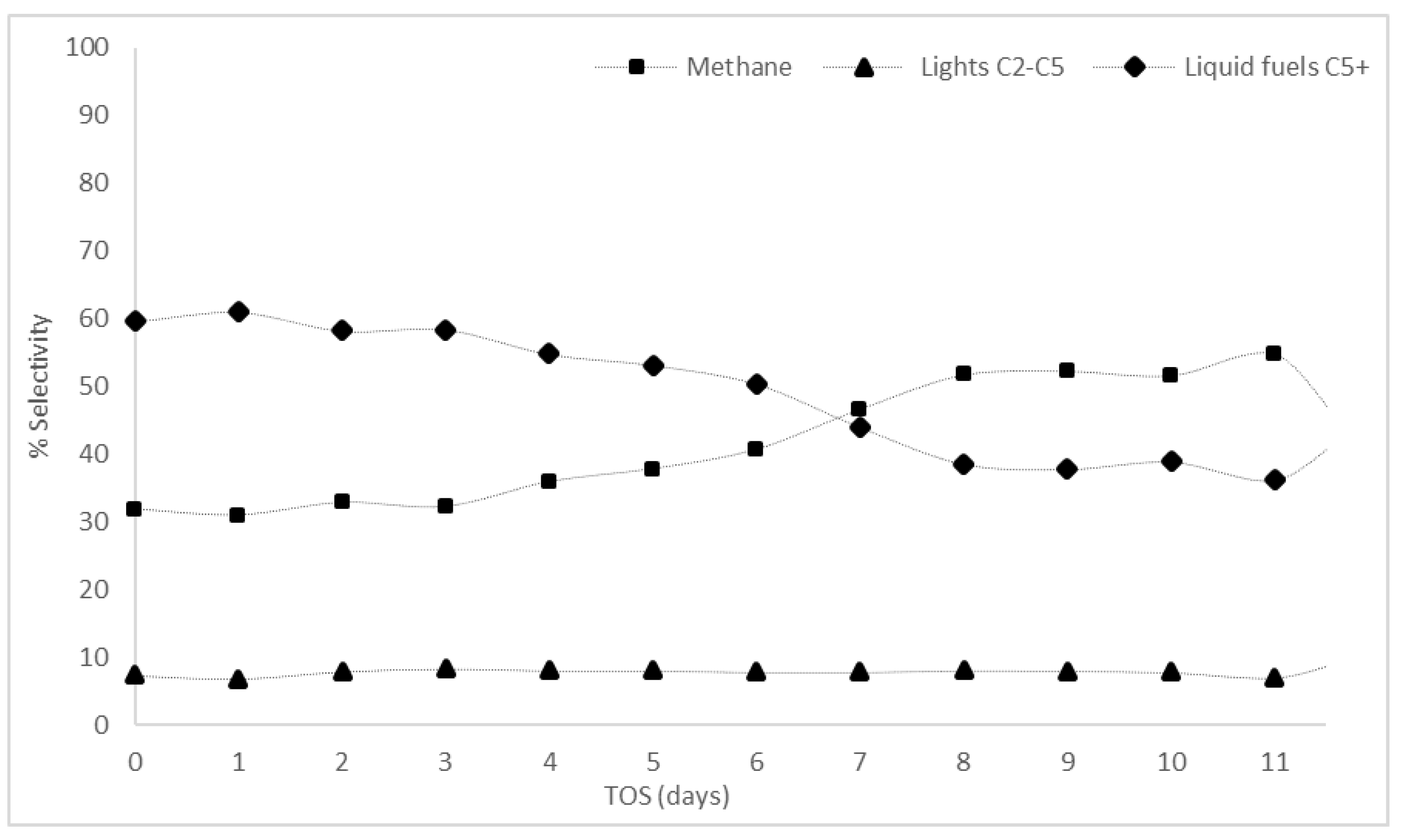
3.2.1. Conversion and Selectivity Results
3.2.2. Possibility of a Dual α Mechanism
3.3. Testing the FTS System’s Robustmess by the Introduction of Interruption Factors
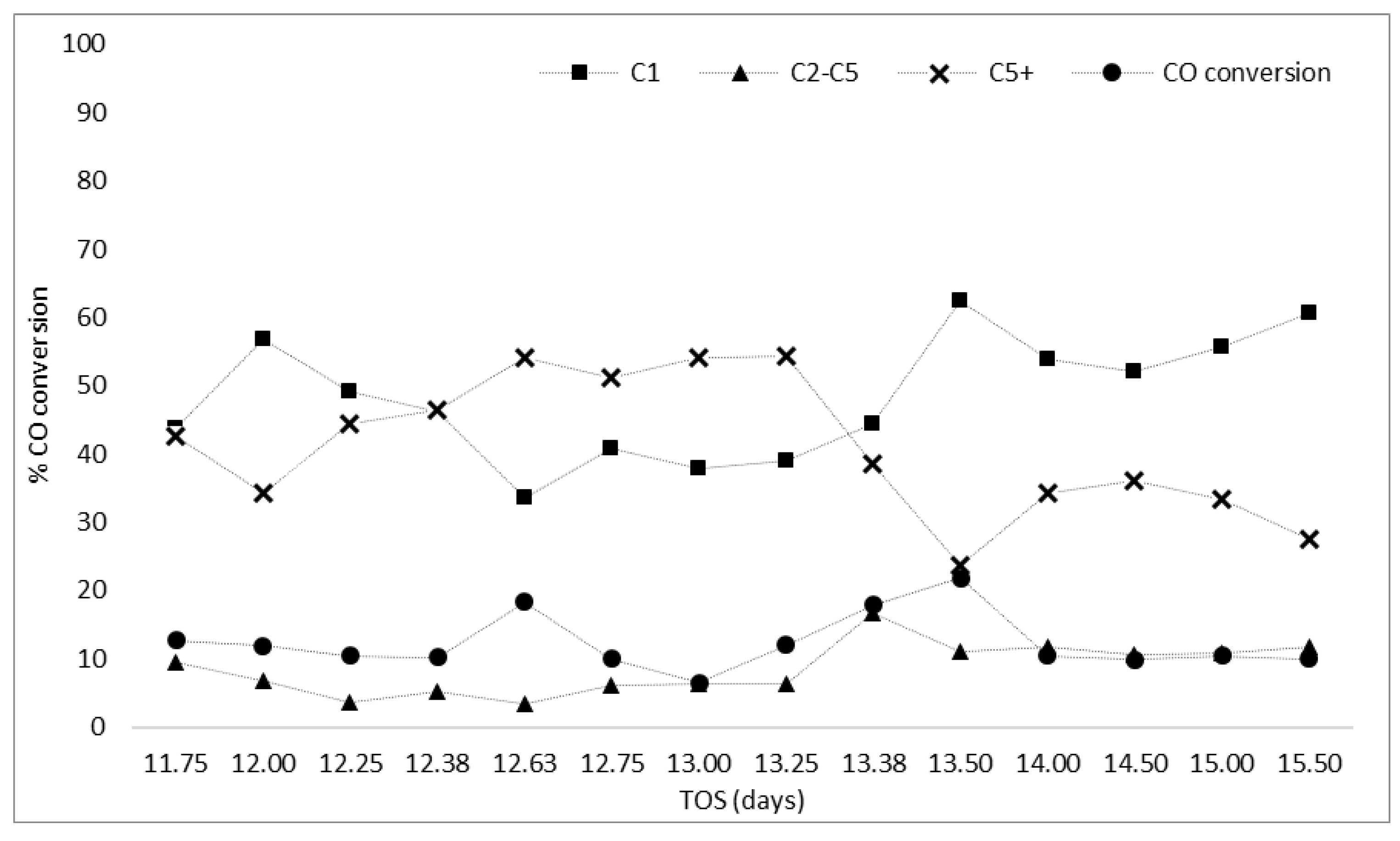
3.3.1. Fluctuation in Feed Gas Flow and Reaction Temperature (Zones A, B, and C)
3.3.2. Effect of Feed Ratio on Catalyst Activity (Zone D)
3.4. Overall Findings for the Technical Feasibility of Liquid Fuel Production from Biogas at Atmospheric Pressure
3.5. Economic Feasibility of the Fischer-Tropsch Conversion of Biogas-Derived Syngas to Liquid Fuels
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AD | anaerobic digestion |
| ASF | Anderson Schulz Flory |
| BID | barrier discharge ionization detector |
| CNG | compressed natural gas |
| FID | flame ionization detector |
| FT | Fischer–Tropsch |
| FTS | Fischer–Tropsch synthesis |
| GC | gas chromatograph(y) |
| GC–MS | gas chromatography–mass spectrometry |
| GHG | greenhouse gas |
| GHSV | gas hourly space velocity |
| ID | internal diameter |
| LNG | liquefied natural gas |
| RHI | Renewable Heat Incentive |
| St | Sterling |
| TD | thermal desorption |
| TGA | thermogravimetric analysis |
| TOS | time on stream |
| TPD | temperature programmed desorption |
| VOC | volatile organic compounds |
| wt | weight |
Appendix A. Additional Information on Testing and Characterization
Appendix A.1. Thermogravimetric Analysis



Appendix A.2. Thermal Desorption (TD)



Appendix A.3. BID
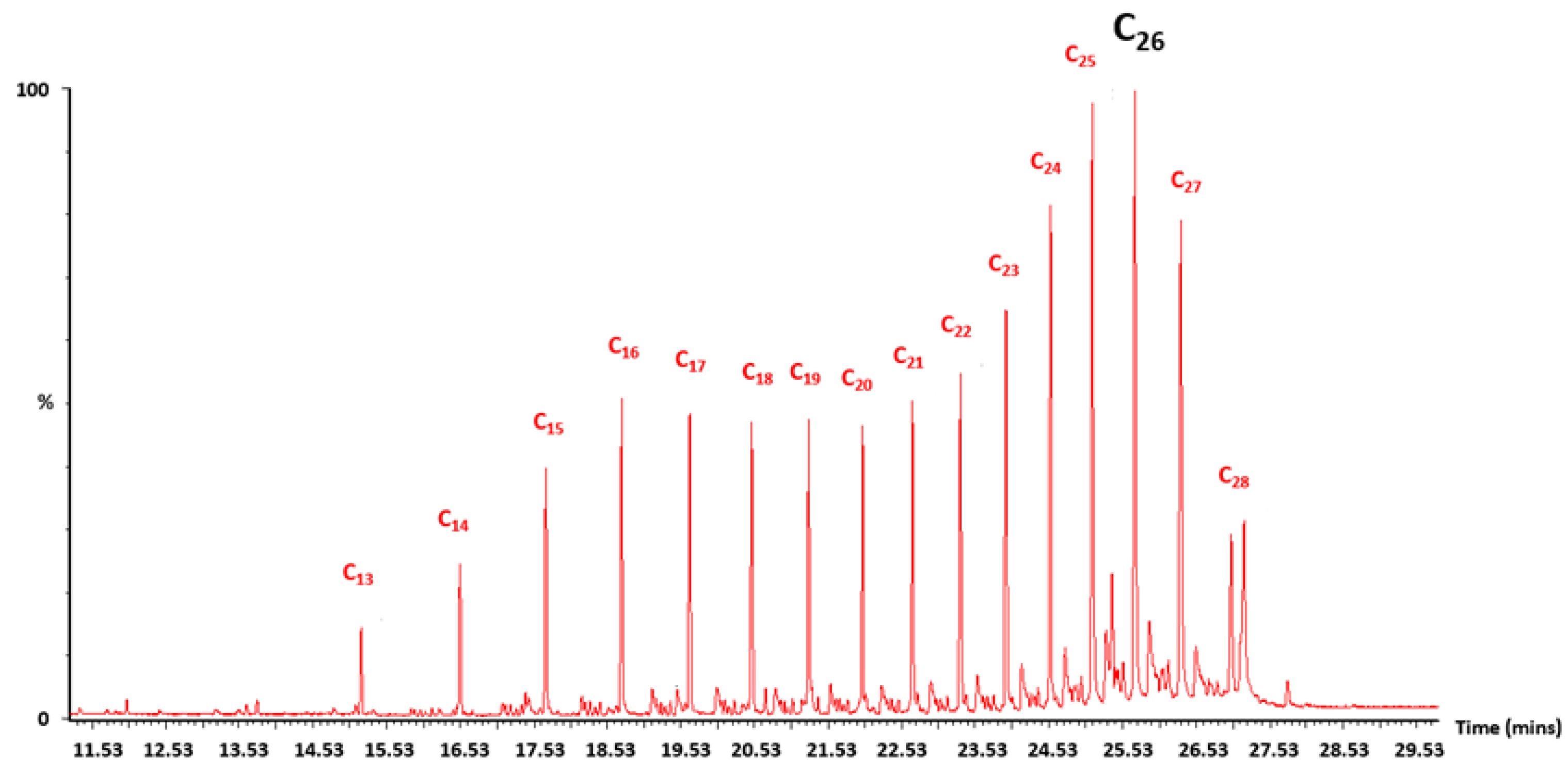
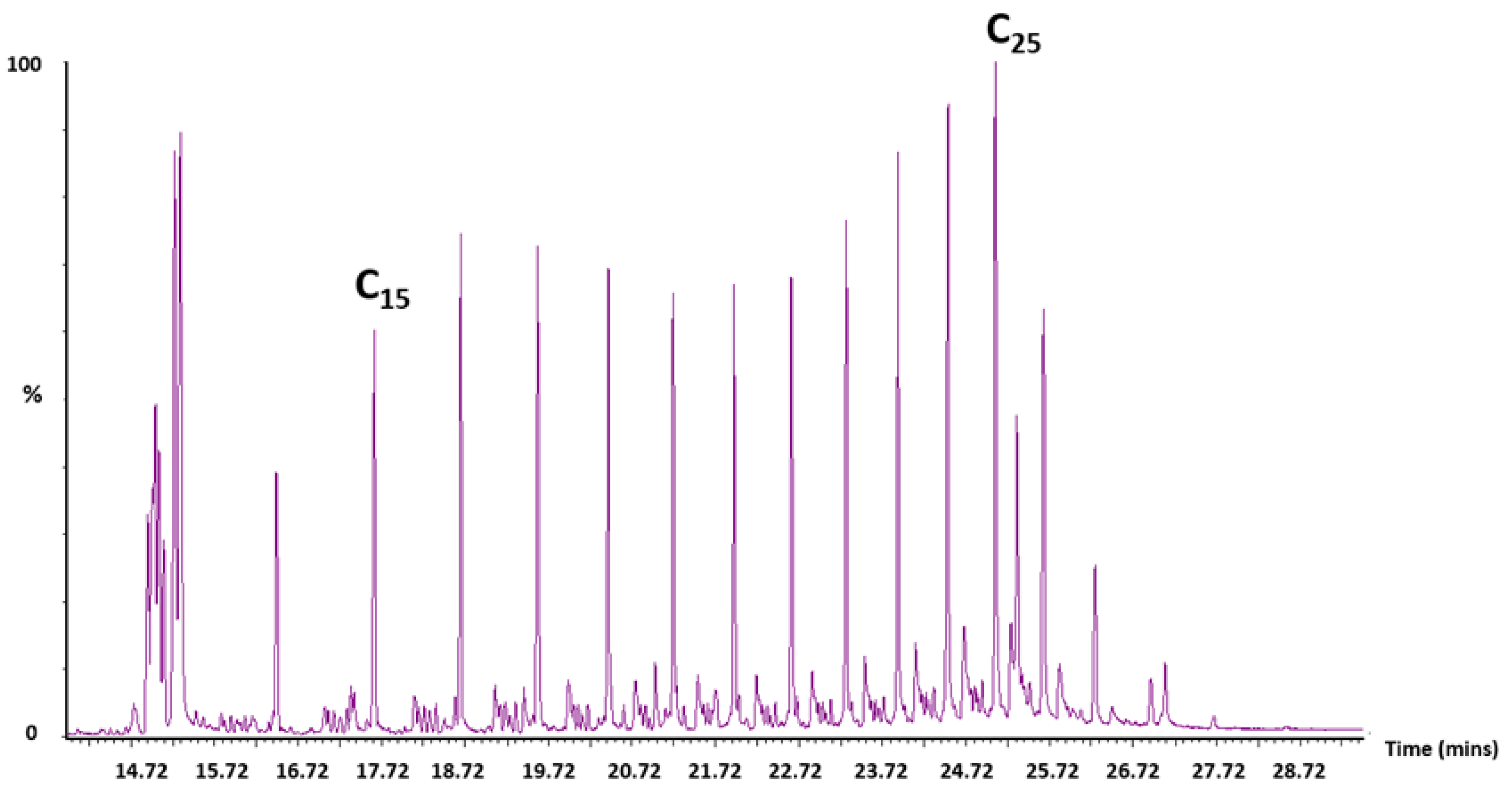
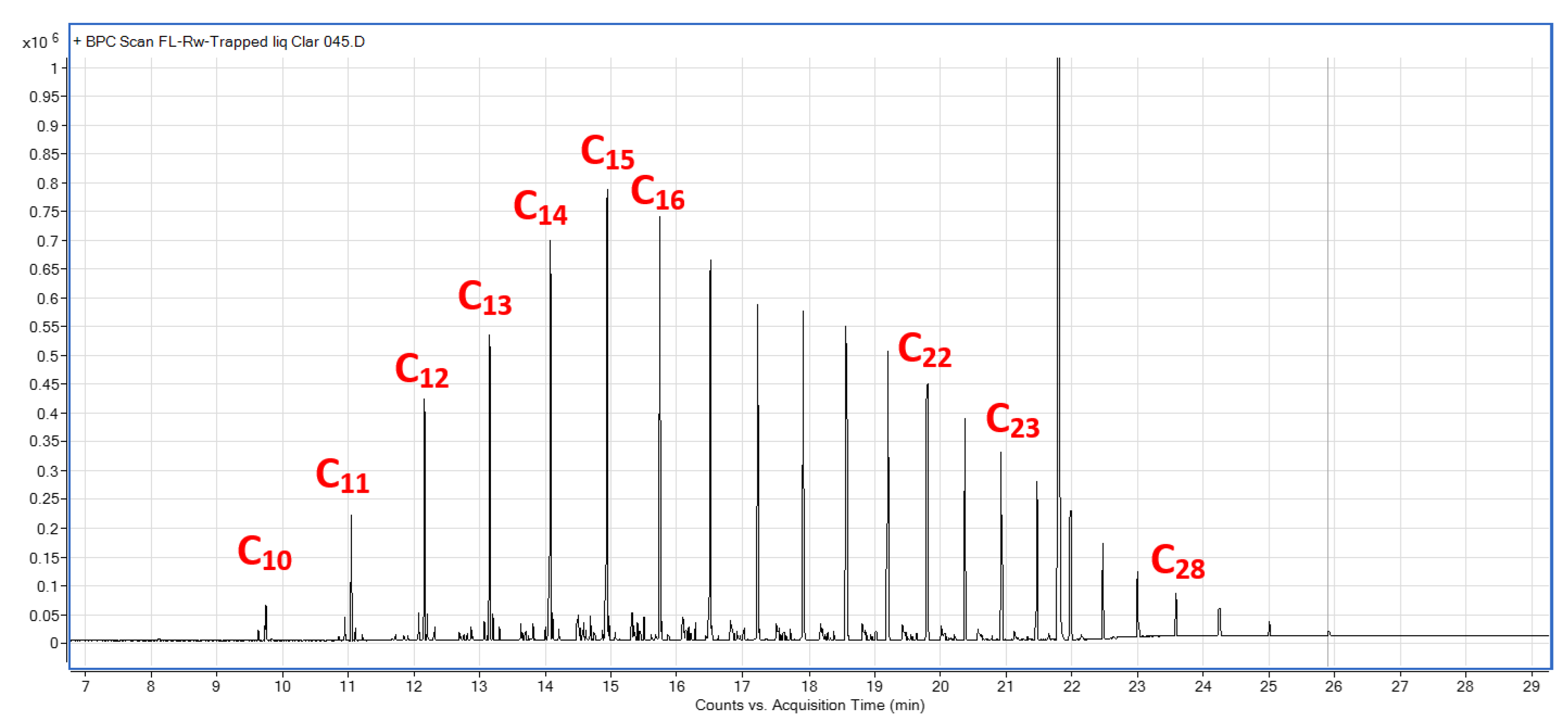
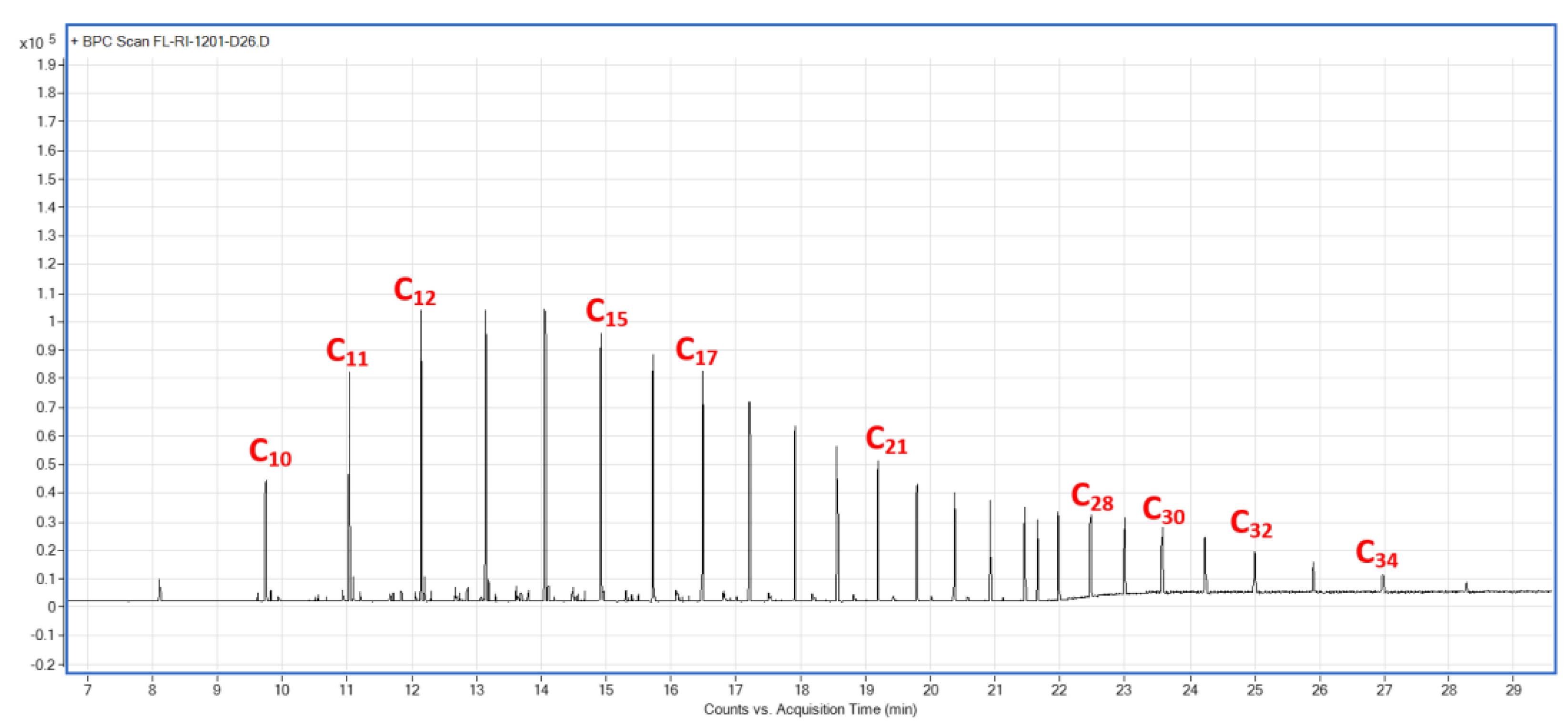
Appendix A.4. GC–MS





Appendix B. Additional Information on Economic Calculations

| Details | Capital Expenditure (St£) | Annual Expenditure (St£/year) |
|---|---|---|
| Capital costs | - | - |
| Reforming plant | 1,332,645 | 137,213 |
| FT plant | 3,285,500 | 338,284 |
| Total | 4,618,142 | 475,497 |
| Fixed costs | - | - |
| Reforming operation and maintenance | - | 133,264 |
| FT operation and maintenance | - | 131,420 |
| Depreciation | - | 307,876 |
| Biogas production | - | 334,892 |
| Total | - | 907,452 |
| Combined total cash expenditure | - | 1,382,949 |
| Details | Value |
|---|---|
| Income | - |
| Liquid fuels sold (£/year) | 149,740 |
| Total cash income (£/year) | 149,740 |
| Profit (£/year) | −1,233,209 |
| Profit (£/m3 biogas) | −1.44 |
| ROCE (%) | −89.17 |
| Details | Capital Expenditure (St£) | Annual Expenditure (St£/year) |
|---|---|---|
| Capital costs | - | - |
| Conventional upgrading plant | 341,021 | 35,112 |
| Connection to gas grid | 213,000 | 21,931 |
| Total | 554,021 | 57,044 |
| Fixed costs | - | - |
| Upgrading operation and maintenance | - | 57,056 |
| Depreciation | - | 36,935 |
| Biogas production | - | 334,892 |
| Total | - | 428,883 |
| Combined total cash expenditure | - | 485,926 |
| Details | Including RHI | Excluding RHI |
|---|---|---|
| Income (H2) | - | - |
| Grid injection | 491,812 | 95,713 |
| Total cash income | - | 95,713 |
| Income (T2) | - | - |
| Grid injection | - | 210,902 |
| Total cash income | - | 210,902 |
| Profit (H2) (£/year) | 5886 | −390,214 |
| Profit (£/m3 biogas) | 0.01 | −0.45 |
| ROCE (%) | 1.21 | −80.30 |
| Profit (T2) (£/year) | - | −275,025 |
| Profit (£/m3 biogas) | - | −0.32 |
| ROCE (%) | - | −56.60 |
| Details | Capital Expenditure (St£) | Annual Expenditure (St£/year) |
|---|---|---|
| Capital costs | - | - |
| Conventional upgrading plant | 341,021 | 35,112 |
| LNG plant | 200,000 | 20,593 |
| Total | 541,021 | 55,705 |
| Fixed costs | - | - |
| Upgrading operation and maintenance | - | 57,056 |
| LNG operation and maintenance | - | 30,000 |
| Transportation of LNG | - | 5639 |
| Depreciation | - | 36,068 |
| Biogas production | - | 334,892 |
| Total | - | 463,655 |
| Combined total cash expenditure | - | 519,360 |
| Details | Including RHI | Excluding RHI |
|---|---|---|
| Income (H3) | - | - |
| LNG sold | 491,812 | 95,713 |
| Total cash income | - | 95,713 |
| Income (T3) | - | - |
| LNG sold | - | 210,902 |
| Total cash income | - | 210,902 |
| Profit (H3) (£/year) | −27,548 | −423,647 |
| Profit (£/m3 biogas) | −0.03 | −0.49 |
| ROCE (%) | −5.30 | −81.57 |
| Profit (T3) (£/year) | - | −308,458 |
| Profit (£/m3 biogas) | - | −0.36 |
| ROCE (%) | - | −59.39 |
References
- Xu, J.; Zhou, W.; Li, Z.; Wang, J.; Ma, J. Biogas reforming for hydrogen production over a Ni–Co bimetallic catalyst: Effect of operating conditions. Int. J. Hydrogen Energy 2010, 35, 13013–13020. [Google Scholar] [CrossRef]
- BP. BP Statistical Review of World Energy. London, UK. 2018. Available online: https://www.bp.com/en/global/corporate/energy-economics/statistical-review-of-world-energy.html (accessed on 20 December 2018).
- Ashraf, M.T.; Bastidas-Oyanedel, J.R.; Schmidt, J.E. Conversion efficiency of biogas to liquids fuels through Fischer-Tropsch process. In Proceedings of the 23rd European Biomass Conference and Exhibition, Vienna, Austria, 1–4 June 2015. [Google Scholar]
- Lee, C.-Y.; Huh, S.-Y. Forecasting the diffusion of renewable electricity considering the impact of policy and oil prices: The case of South Korea. Appl. Energy 2017, 197, 29–39. [Google Scholar] [CrossRef]
- Nuttall, W.J.; Manz, D.L. A new energy security paradigm for the twenty-first century. Technol. Forecast. Soc. Chang. 2008, 75, 1247–1259. [Google Scholar] [CrossRef]
- Dorian, J.P.; Franssen, H.T.; Simbeck, D.R. Global challenges in energy. Energy Policy 2006, 34, 1984–1991. [Google Scholar] [CrossRef]
- Scholten, D.; Bosman, R. The geopolitics of renewables; exploring the political implications of renewable energy systems. Technol. Forecast. Soc. Chang. 2016, 103, 273–283. [Google Scholar] [CrossRef] [Green Version]
- Scarlat, N.; Dallemand, J.-F.; Fahl, F. Biogas: Developments and perspectives in Europe. Renew. Energy 2018, 129, 457–472. [Google Scholar] [CrossRef]
- EurObserv’ER. Biogas Barometer. Available online: https://www.eurobserv-er.org/biogas-barometer-2017/ (accessed on 10 December 2018).
- European Biogas Association (EBA). EBA Statistical Report 2017; European Biogas Association: Bruxelles, Belgium, 2017. [Google Scholar]
- De Rosa, F.; Smyth, B.M.; McCullough, G.; Goguet, A. Using multi-criteria and thermodynamic analysis to optimize process parameters for mixed reforming of biogas. Int. J. Hydrogen Energy 2018, 43, 18801–18813. [Google Scholar] [CrossRef]
- Siedlecki, M.; De Jong, W.; Verkooijen, A.H.M. Fluidized Bed Gasification as a Mature and Reliable Technology for the Production of Bio-Syngas and Applied in the Production of Liquid Transportation Fuels—A Review. Energies 2011, 4, 389–434. [Google Scholar] [CrossRef]
- Rafati, M.; Wang, L.; Dayton, D.C.; Schimmel, K.; Kabadi, V.; Shahbazi, A. Techno-economic analysis of production of Fischer-Tropsch liquids via biomass gasification: The effects of Fischer-Tropsch catalysts and natural gas co-feeding. Energy Convers. Manag. 2017, 133, 153–166. [Google Scholar] [CrossRef]
- Ail, S.S.; Dasappa, S. Investigations into enhanced wax production with combustion synthesized Fischer–Tropsch catalysts. Energy Convers. Manag. 2016, 116, 80–90. [Google Scholar] [CrossRef]
- Eurostat. Oil Statistics 2016 Update. Available online: https://ec.europa.eu/eurostat/statistics-explained/index.php?title=Oil_and_petroleum_products_-_a_statistical_overview#Use_of_petroleum_products (accessed on 10 December 2018).
- Bolder, F.H.A. Fischer-Tropsch wax hydrogenation over a sulfided nickel-molybdenum catalyst. Energy Fuels 2007, 21, 1396–1399. [Google Scholar] [CrossRef]
- Kumar, A.; Jones, D.D.; Hanna, A.M. Thermochemical Biomass Gasification: A Review of the Current Status of the Technology. Energies 2009, 2, 556–581. [Google Scholar] [CrossRef] [Green Version]
- Gorimbo, J.; Lu, X.; Liu, X.; Hildebrandt, D.; Glasser, D. A long term study of the gas phase of low pressure Fischer-Tropsch products when reducing an iron catalyst with three different reducing gases. Appl. Catal. A Gen. 2017, 534, 1–11. [Google Scholar] [CrossRef]
- van Vliet, O.P.R.; Faaij, A.P.C.; Turkenburg, W.C. Fischer–Tropsch diesel production in a well-to-wheel perspective: A carbon, energy flow and cost analysis. Energy Convers. Manag. 2009, 50, 855–876. [Google Scholar] [CrossRef]
- Choi, Y.H.; Jang, Y.J.; Park, H.; Kim, W.Y.; Lee, Y.H.; Choi, S.H.; Lee, J.S. Carbon dioxide Fischer-Tropsch synthesis: A new path to carbon-neutral fuels. Appl. Catal. B Environ. 2017, 202, 605–610. [Google Scholar] [CrossRef]
- Aluha, J.; Abatzoglou, N. Synthetic fuels from 3-φ Fischer-Tropsch synthesis using syngas feed and novel nanometric catalysts synthesised by plasma. Biomass Bioenergy 2016, 95, 330–339. [Google Scholar] [CrossRef]
- Chein, R.-Y.; Hsu, W.-H. Analysis of Syngas Production from Biogas via the Tri-Reforming Process. Energies 2018, 11, 1075. [Google Scholar] [CrossRef]
- Yang, J.; Ma, W.; Chen, D.; Holmen, A.; Davis, B.H. Fischer–Tropsch synthesis: A review of the effect of CO conversion on methane selectivity. Appl. Catal. A Gen. 2014, 470, 250–260. [Google Scholar] [CrossRef]
- Dinse, A.; Aigner, M.; Ulbrich, M.; Johnson, G.R.; Bell, A.T. Effects of Mn promotion on the activity and selectivity of Co/SiO2 for Fischer–Tropsch Synthesis. J. Catal. 2012, 288, 104–114. [Google Scholar] [CrossRef]
- Tavasoli, A.; Khodadadi, A.; Mortazavi, Y.; Sadaghiani, K.; Ahangari, M.G. Lowering methane and raising distillates yields in Fischer–Tropsch synthesis by using promoted and unpromoted cobalt catalysts in a dual bed reactor. Fuel Process. Technol. 2006, 87, 641–647. [Google Scholar] [CrossRef]
- Mirzaei, A.A.; Vahid, S.; Feyzi, M. Fischer-Tropsch synthesis over iron manganese catalysts: Effect of preparation and operating conditions on catalyst performance. Adv. Phys. Chem. 2009, 2009, 151489. [Google Scholar] [CrossRef]
- Savost’yanov, A.P.; Yakovenko, R.E.; Sulima, S.I.; Bakun, V.G.; Narochnyi, G.B.; Chernyshev, V.M.; Mitchenko, S.A. The impact of Al2O3 promoter on an efficiency of C5+ hydrocarbons formation over Co/SiO2 catalysts via Fischer-Tropsch synthesis. Catal. Today 2017, 279, 107–114. [Google Scholar] [CrossRef]
- Dixit, J.B. Structured System Analysis and Design; Firewall Media, Laxmi Publications: New Delhi, India, 2007; ISBN 8131802663. [Google Scholar]
- Xiang, D.; Yang, S.; Qian, Y. Techno-economic analysis and comparison of coal based olefins processes. Energy Convers. Manag. 2016, 110, 33–41. [Google Scholar] [CrossRef]
- Broberg Viklund, S.; Johansson, M.T. Technologies for utilization of industrial excess heat: Potentials for energy recovery and CO2 emission reduction. Energy Convers. Manag. 2014, 77, 369–379. [Google Scholar] [CrossRef] [Green Version]
- Moret, S.; Peduzzi, E.; Gerber, L.; Maréchal, F. Integration of deep geothermal energy and woody biomass conversion pathways in urban systems. Energy Convers. Manag. 2016, 129, 305–318. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Wang, Y.; Liu, T.; Chen, J. Theoretical basis and performance optimization analysis of a solid oxide fuel cell–gas turbine hybrid system with fuel reforming. Energy Convers. Manag. 2014, 86, 1102–1109. [Google Scholar] [CrossRef]
- Shimura, K.; Miyazawa, T.; Hanaoka, T.; Hirata, S. Fischer–Tropsch synthesis over alumina supported cobalt catalyst: Effect of promoter addition. Appl. Catal. A Gen. 2015, 494, 1–11. [Google Scholar] [CrossRef]
- Chu, W.; Xu, J.; Hong, J.; Lin, T.; Khodakov, A. Design of efficient Fischer–Tropsch cobalt catalysts via plasma enhancement: Reducibility and performance (Review). Catal. Today 2015, 256, 41–48. [Google Scholar] [CrossRef]
- Khodakov, A.Y.; Chu, W.; Fongarland, P. Advances in the Development of Novel Cobalt Fischer−Tropsch Catalysts for Synthesis of Long-Chain Hydrocarbons and Clean Fuels. Chem. Rev. 2007, 107, 1692–1744. [Google Scholar] [CrossRef]
- Rodrigues, J.J.; Lima, L.A.; Lima, W.S.; Rodrigues, M.G.F.; Fernandes, F.A.N. Fischer-Tropsch Synthesis in slurry-phase Reactors using Co/SBA-15 catalysts. Braz. J. Pet. Gas 2011, 5. [Google Scholar] [CrossRef]
- Eurostat. SHARES (RENEWABLES). Available online: https://ec.europa.eu/eurostat/web/energy/data/shares (accessed on 19 February 2019).
- DAERA. The Agricultural Census in Northern Ireland Results for June 2018. 2019. Available online: https://www.daera-ni.gov.uk/publications/agricultural-census-northern-ireland-2018 (accessed on 28 February 2019).
- Smyth, B.M.; Murphy, J.D.; O’Brien, C.M. What is the energy balance of grass biomethane in Ireland and other temperate northern European climates? Renew. Sustain. Energy Rev. 2009, 13, 2349–2360. [Google Scholar] [CrossRef]
- NNFCC Biogas Map (AD Portal Map Site List). Available online: http://www.biogas-info.co.uk/resources/biogas-map/ (accessed on 18 February 2019).
- Smyth, B.M.; Smyth, H.; Murphy, J.D. Can grass biomethane be an economically viable biofuel for the farmer and the consumer? Biofuels Bioprod. Biorefining 2010, 4, 519–537. [Google Scholar] [CrossRef]
- Keatley, P. Farm Business Data 2015; Department of Agriculture and Rural Development (DARD): Belfast, UK, 2015. [Google Scholar]
- Agersborg, J.; Linghed, E. Integration of Power-to-Gas in Gasendal and GoBiGas. Master’s Thesis, Chalmers University of Technology, Göteborg, Sweden, 2013. [Google Scholar]
- Global Capital Finance; Clean Energy Pipeline. The European Renewable Energy Investor Landscape. 2014. Available online: http://cleanenergypipeline.com/Resources/CE/ResearchReports/The European Renewable Energy Investor Landscape.pdf (accessed on 28 February 2019).
- Paturska, A.; Repele, M.; Bazbauers, G. Economic Assessment of Biomethane Supply System based on Natural Gas Infrastructure. Energy Procedia 2015, 72, 71–78. [Google Scholar] [CrossRef] [Green Version]
- Blesl, M.; Bruchof, D. Liquid Fuels Production from Coal & Gas (Technology Brief P06). 2010. Available online: https://iea-etsap.org/E-TechDS/PDF/P06-CTLGTL-GS-gct-AD_gs.pdf (accessed on 5 December 2016).
- Songhurst, B. LNG Price Cost Escalation. Oxford, UK. 2014. Available online: https://www.oxfordenergy.org/wpcms/wp-content/uploads/2014/02/NG-83.pdf (accessed on 2 February 2017).
- Energy Solutions Historical Natural Gas Prices. Available online: www.energybrokers.co.uk/gas/historic-price-data-graph.htm (accessed on 25 January 2017).
- CNG Europe Great Britain CNG Filling Stations. Available online: http://cngeurope.com/countries/great-britain/ (accessed on 12 December 2015).
- Petrol Prices UK Petrol Prices. Available online: www.petrolprices.com (accessed on 1 February 2017).
- UK Government Tax on Shopping and Services. Available online: www.gov.uk/tax-on-shopping/fuel-duty (accessed on 12 February 2017).
- Dransfield, R.; Needham, D. Advanced Business; Heinemann Educational Publishers: Oxford, UK, 2000. [Google Scholar]
- Ofgem The Renewables Obligation (RO) Buy-Out Price (£44.33) and Mutualisation Ceilings 2015-16. Available online: https://www.ofgem.gov.uk/publications-and-updates/renewables-obligation-ro-buy-out-price-44-33-and-mutualisation-ceilings-2015-16 (accessed on 2 March 2019).
- Petersson, A.; Wellinger, A. Biogas Upgrading Technologies—Developments and Innovations. 2009. Available online: https://www.iea-biogas.net/files/daten-redaktion/download/publi-task37/upgrading_rz_low_final.pdf (accessed on 10 December 2016).
- Maitlis, P.M. Greener Fischer-Tropsch Processes for Fuels and Feedstocks; Maitlis, P.M., de Klerk, A., Eds.; Wiley-VCH: Weinheim, Germany, 2013; ISBN 9783527329458. [Google Scholar]
- Storsæter, S.; Borg, Ø.; Blekkan, E.A.; Holmen, A. Study of the effect of water on Fischer–Tropsch synthesis over supported cobalt catalysts. J. Catal. 2005, 231, 405–419. [Google Scholar] [CrossRef]
- Gholami, R.; Alyani, M.; Smith, J.K. Deactivation of Pd Catalysts by Water during Low Temperature Methane Oxidation Relevant to Natural Gas Vehicle Converters. Catalysts 2015, 5, 561–594. [Google Scholar] [CrossRef] [Green Version]
- Marwaha, B.; Luss, D. Hot zones formation in packed bed reactors. Chem. Eng. Sci. 2003, 58, 733–738. [Google Scholar] [CrossRef]
- van der Laan, G.P.; Beenackers, A.A.C.M. Kinetics and Selectivity of the Fischer–Tropsch Synthesis: A Literature Review. Catal. Rev. 1999, 41, 255–318. [Google Scholar] [CrossRef]
- Iglesia, E. Design, synthesis, and use of cobalt-based Fischer-Tropsch synthesis catalysts. Appl. Catal. A Gen. 1997, 161, 59–78. [Google Scholar] [CrossRef]
- Zhan, X.; Davis, B.H. Two alpha f1scher-tropsch product distribution. A role for vapor-liquid equilibrium? Pet. Sci. Technol. 2000, 18, 1037–1053. [Google Scholar] [CrossRef]
- Bukur, D.B.; Todic, B.; Elbashir, N. Role of water-gas-shift reaction in Fischer–Tropsch synthesis on iron catalysts: A review. Catal. Today 2016, 275, 66–75. [Google Scholar] [CrossRef] [Green Version]
- Tavasoli, A.; Nakhaei Pour, A.; Ahangari, M.G. Kinetics and product distribution studies on ruthenium-promoted cobalt/alumina Fischer-Tropsch synthesis catalyst. J. Nat. Gas Chem. 2010, 19, 653–659. [Google Scholar] [CrossRef]
- Argyle, M.; Bartholomew, C.; Argyle, M.D.; Bartholomew, C.H. Heterogeneous Catalyst Deactivation and Regeneration: A Review. Catalysts 2015, 5, 145–269. [Google Scholar] [CrossRef] [Green Version]
- Rytter, E.; Holmen, A. Deactivation and regeneration of commercial type Fischer-Tropsch Co-catalysts—A mini-review. Catalysts 2015, 5, 478–499. [Google Scholar] [CrossRef]
- Calleja, G.; de Lucas, A.; van Grieken, R. Co/HZSM-5 catalyst for syngas conversion: Influence of process variables. Fuel 1995, 74, 445–451. [Google Scholar] [CrossRef]
- Tristantini, D.; Lögdberg, S.; Gevert, B.; Borg, Ø.; Holmen, A. The effect of synthesis gas composition on the Fischer–Tropsch synthesis over Co/γ-Al2O3 and Co–Re/γ-Al2O3 catalysts. Fuel Process. Technol. 2007, 88, 643–649. [Google Scholar] [CrossRef]
- Yates, I.C.; Satterfield, C.N. Hydrocarbon selectivity from cobalt Fischer-Tropsch catalysts. Energy Fuels 1992, 6, 308–314. [Google Scholar] [CrossRef]
- Filot, I.A.W.; Zijlstra, B.; Broos, R.J.P.; Chen, W.; Pestman, R.; Hensen, E.J.M. Kinetic aspects of chain growth in Fischer–Tropsch synthesis. Faraday Discuss. 2017, 197, 153–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Wang, J.; Chen, C.; Jia, L.; Hou, B.; Li, D. Effects of macropores on reducing internal diffusion limitations in Fischer–Tropsch synthesis using a hierarchical cobalt catalyst. RSC Adv. 2017, 7, 9436–9445. [Google Scholar] [CrossRef] [Green Version]
- Lualdi, M.; Lögdberg, S.; Boutonnet, M.; Järås, S. On the effect of water on the Fischer–Tropsch rate over a Co-based catalyst: The influence of the H2/CO ratio. Catal. Today 2013, 214, 25–29. [Google Scholar] [CrossRef]
- Mansouri, M.; Atashi, H.; Tabrizi, F.F.; Mansouri, G.; Setareshenas, N. Fischer–Tropsch synthesis on cobalt–manganese nanocatalyst: Studies on rate equations and operation conditions. Int. J. Ind. Chem. 2014, 5, 14. [Google Scholar] [CrossRef]
- Ma, W.; Jacobs, G.; Sparks, D.E.; Spicer, R.L.; Davis, B.H.; Klettlinger, J.L.S.; Yen, C.H. Fischer–Tropsch synthesis: Kinetics and water effect study over 25% Co/Al2O3 catalysts. Catal. Today 2014, 228, 158–166. [Google Scholar] [CrossRef]
- Hakawati, R.; Smyth, B.M.; McCullough, G.; De Rosa, F.; Rooney, D. What is the most energy efficient route for biogas utilization: Heat, electricity or transport? Appl. Energy 2017, 206, 1076–1087. [Google Scholar] [CrossRef]
- Samavati, M.; Martin, A.; Santarelli, M.; Nemanova, V. Synthetic Diesel Production as a Form of Renewable Energy Storage. Energies 2018, 11, 1223. [Google Scholar] [CrossRef]
- Simonsen, M.; Walnum, H.J. Energy chain analysis of passenger car transport. Energies 2011, 4, 324–351. [Google Scholar] [CrossRef]
- Stefanakis, A.; Akratos, C.S.; Tsihrintzis, V.A. Vertical Flow Constructed Wetlands, 1st ed.; Elsevier: Amsterdam, The Netherlands, 2014; ISBN 9780124046122. [Google Scholar]
- OECD. Crude Oil Import Prices. Available online: https://data.oecd.org/energy/crude-oil-import-prices.htm (accessed on 2 March 2019).
- Couto, R.A. Regional Development within National Industrial Policies: An Analysis of the British Coal Industry. Growth Chang. 1990, 21, 51–68. [Google Scholar] [CrossRef]
- Hossein-Zadeh, I. The Political Economy of US Wars of Choice: Are They Really Oil Wars? In The Nation in the Global Era; Brill: Leiden, The Netherlands, 2009; pp. 185–204. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}




| Reaction | Purpose of Test |
|---|---|
| R1 | Preliminary setup to test if the commercial catalyst was active for reaction at atmospheric pressure |
| R2 | To test the impact of catalyst quantity and to verify that the catalyst’s activity and conversion could be boosted by increasing the quantity of catalyst |
| R3 | To test the impact of reactor dimensions and system configuration on conversion and activity |
| Parameters | R1 | R2 | R3 |
|---|---|---|---|
| Reactor length (cm) | 50 | 50 | 50 |
| Reactor internal diameter (ID) (cm) | 0.6 | 0.6 | 1.2 |
| Catalyst mass (g) | 0.2 | 0.44 | 0.44 |
| SiC mass (g) | 0.4 | 0.88 | 0.88 |
| Total catalyst bed height (cm) | 8 | 16 | 4 |
| Feed flow rate (mL/min) | 32.5 | 32.5 | 32.5 |
| Gas hourly space velocity (GHSV) 1 (hr−1) | 862.83 | 430.46 | 463.46 |
| H2 in He reduction gas flow rate (mL/min) | 26 | 13 | 13 |
| Pressure increase during reduction (MPa) | 0.2 | 0.1 | 0.05 |
| Pressure increase during reaction (MPa) | 0.15 | 0.1 | 0.05 |
| System configuration | No trap | No trap | Added trap 2 |
| Parameters | R4 |
|---|---|
| Reactor length (cm) | 50 |
| Reactor internal diameter (cm) | 1.2 |
| Unsieved catalyst mass (g) | 1 |
| SiC mass (g) | 2 |
| Feed flow rate (mL/min) | 32.5 |
| H2 in He reduction gas flow rate (mL/min) | 13 |
| Pressure increase during reduction (MPa) | 0.02 |
| Pressure increase during reaction (MPa) | 0.02 |
| Zone | Parameter Investigated | Experimental Steps |
|---|---|---|
| A | Feed gas interruption of supply | Reaction was stopped at day 11.5, then cooled in He from 488 K (215 °C) to 373 K (100 °C) at 10 K/min (10 °C/min). Reactor was kept under He flow for 30 min. Reacting feed was introduced at 373 K (100 °C). Temperature was increased under feed conditions from 373 K (100 °C) to 488 K (215 °C) at 1 K/min (1 °C/min). |
| B | Technical problems that could interrupt the reaction 1 | Reaction was stopped at day 12.3. Gas supply was shut down and the reactor cooled to 303 K (30 °C) (by turning off the furnace). Temperature was increased under feed conditions from 303 K (30 °C) to 488 K (215 °C) at 10 K/min (10 °C/min). |
| C | The feasibility of regenerating and reusing the catalyst | Reaction was stopped at day 13.5, then cooled in He (by turning off the furnace) until temperature of 303 K (30 °C) was recorded. Re-reduced in H2 (as per prereaction process). |
| D | The catalyst’s behavior if the feed ratio was interrupted or altered during the reaction | At day 16, after 9 h of reaction, the H2:CO feed ratio was altered from 2:1 to 3:1 for 15 h. The ratio was then changed to 1:1 until day 19. |
| Technology | Capital Expenditure (St£) | Operation and Maintenance (St£/yr) | Source |
|---|---|---|---|
| Conventional upgrading (membrane separation) | 341,021 | 57,055 | Average calculated from Smyth et al. and Paturska et al. [41,45] |
| Reforming | 1,332,642 | 133,264 | Estimated from Blesl and Bruchof [46] using Equation (6) |
| Fischer-Tropsch | 3,285,500 | 131,420 | Estimated from Blesl and Bruchof [46] using Equation (6) |
| LNG | 200,000 | 30,000 | Estimated from Songhurst [47] using Equation (4) |
| Details | Value 1 | Source |
|---|---|---|
| Wholesale gas price | St£0.0181/kWh | Calculated from Energy Solutions [48] |
| Average CNG price | St£0.89/kg | [49] |
| Average diesel price | St£1.222/litre | [50] |
| Fuel tax duty (diesel, petrol and biodiesel) | St£0.5795 | [51] |
| Fuel tax duty (CNG) | St£0.247/kg | [51] |
| Connection to gas grid | St£213,000 | [41] |
| Cost for transporting LNG per km 2 | St£1.86/km for 36,370 l lorry | [52] |
| Biomethane grid injection RHI | St£0.075/kWh | [53] |
| Average density of liquid fuel | 874.74 kg/m3 | - |
| Methane slip (conventional upgrading) | 0.04% | [54] |
| Liquid fuel conversion efficiency via FT | 33% | [55] |
| CH4 density | 0.69 kg/m3 | - |
| CH4 thermal energy | 11.19 kWh/m3 | - |
| H2 molecular weight | 2 g/mol | - |
| CH4 molecular weight | 18 g/mol | - |
| CO molecular weight | 28 g/mol | - |
| H2 to CH4 molar ratio 3 | 3 | - |
| CO to CH4 molar ratio 3 | 1 | - |
| Steam reforming reaction conversion efficiency (decimal) | 1 | Best case scenario |
| Liquid fuels produced 4 | 233.06 m3 | - |
| Scenario | Calculation Steps |
|---|---|
| T1/H1 | Biogas » reforming » syngas » Fischer-Tropsch » liquid fuel » heat/transport |
| CH4 mass (kg) = biogas production (m3/year) × biogas purity × CH4 density (kg/m3) Mass of H2 produced (kg) = (H2 molecular weight/CH4 molecular weight) × (H:CH4 molar ratio) × [mass CH4 (kg)] × (reaction conversion efficiency) Mass of CO produced (kg) = (CO molecular weight/CH4 molecular weight) × (CO:CH4 molar ratio) × [mass CH4 (kg)] × (reaction conversion efficiency) Liquid fuel produced (m3) = [mass of CO produced (kg) + mass of H2 produced (kg)] × (Fischer-Tropsch conversion efficiency to liquid fuels) × [liquid fuel average density (kg/m3)] Income = (Liquid fuel produced (m3)/1000) × [liquid fuel sale price (St£/l) − diesel fuel tax duty (St£/l)] | |
| T2/H2 and T3/H3 | Biogas » upgrading » biomethane » compression/liquefaction » CNG/LNG » heat/transport |
| CH4 produced (m3) = [biogas production (m3/yr) × CH4 purity] − [CH4 slip during upgrading (m3)] Heating income = CH4 produced (m3) × CH4 energy density (kWh/m3) × wholesale gas price (St£/kWh) Transport income = CH4 produced (m3) × methane density (kg/m3) × [CNG filling station price (St£/kg) − CNG fuel tax duty (St£/kg)] |
| Parameters Investigated | Conclusions |
|---|---|
| Iterative refinement | |
| R1 | Commercial catalyst was active at 0.1 MPa (1 bar) producing 96% methane |
| R2 | An increase in the amount of catalyst increased the activity and selectivity to liquid fuel production which reached 20% |
| R3 | Doubling the reactor’s internal diameter, while using the same amount of catalyst as R2, increased the catalyst’s activity to an average of 12%, compared to 7% in R2 |
| Optimization | |
| R4 | Using the same reactor as R3 but further increasing the amount of catalyst boosted conversion to an average of 18% within the first week with liquid fuel selectivity reaching 60% |
| System robustness (R4) | |
| Zone A | Cooling the reactor to 373 K (100 °C) in the presence of He helped regain catalyst activity at FT conditions, as CO conversion increased to 20% with higher selectivity to liquid fuels than methane |
| Zone B | Complete shutdown of gas supply and cooling to 303 K (30 °C) caused CO conversion to drop (<10% at FT conditions) but higher selectivity to liquid fuels (53%) was maintained |
| Zone C | Catalyst re-reduction with H2 helped eliminate waxy products stuck on the catalyst but enhanced methane selectivity with a drop in CO conversion (from 23% to 10%) |
| Zone D | H2/CO feed ratio of 2:1 was the most suitable for FT reaction at 0.1 MPa (1 bar) |
| Scenario 1 | Total Annual Expenditure (St£/year) | Total Capital Expenditure (St£) | Profit Ex RHI (St£/year) | Profit (St£/m3 biogas) | ROCE (%) | Appropriate Profit Level (St£/m3 biogas) 2 | Required Subsidy (St£/m3 biogas) 3 |
|---|---|---|---|---|---|---|---|
| T1/H1 | 1,382,949 | 4,618,142 | −1,233,209 | −1.44 | −89.17 | 0.12 | 1.55 |
| H2 | 485,926 | 554,021 | −390,213 | −0.45 | −80.30 | 0.04 | 0.50 |
| T2 | 485,926 | 554,021 | −275,025 | −0.32 | −56.60 | 0.04 | 0.36 |
| H3 | 519,360 | 541,021 | −423,647 | −0.49 | −81.57 | 0.04 | 0.54 |
| T3 | 519,360 | 541,021 | −308,458 | −0.36 | −59.39 | 0.04 | 0.4 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hakawati, R.; Smyth, B.; Daly, H.; McCullough, G.; Rooney, D. Is the Fischer-Tropsch Conversion of Biogas-Derived Syngas to Liquid Fuels Feasible at Atmospheric Pressure? Energies 2019, 12, 1031. https://doi.org/10.3390/en12061031
Hakawati R, Smyth B, Daly H, McCullough G, Rooney D. Is the Fischer-Tropsch Conversion of Biogas-Derived Syngas to Liquid Fuels Feasible at Atmospheric Pressure? Energies. 2019; 12(6):1031. https://doi.org/10.3390/en12061031
Chicago/Turabian StyleHakawati, Rawan, Beatrice Smyth, Helen Daly, Geoffrey McCullough, and David Rooney. 2019. "Is the Fischer-Tropsch Conversion of Biogas-Derived Syngas to Liquid Fuels Feasible at Atmospheric Pressure?" Energies 12, no. 6: 1031. https://doi.org/10.3390/en12061031
APA StyleHakawati, R., Smyth, B., Daly, H., McCullough, G., & Rooney, D. (2019). Is the Fischer-Tropsch Conversion of Biogas-Derived Syngas to Liquid Fuels Feasible at Atmospheric Pressure? Energies, 12(6), 1031. https://doi.org/10.3390/en12061031