
Study of the Critical Pore Radius That Results in Critical Gas Saturation during Methane Hydrate Dissociation at the Single-Pore Scale: Analytical Solutions for Small Pores and Potential Implications to Methane Production from Geological Media




Abstract
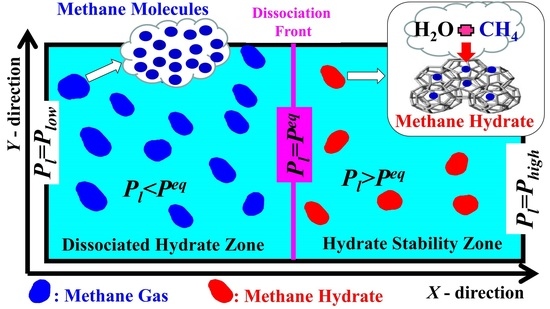
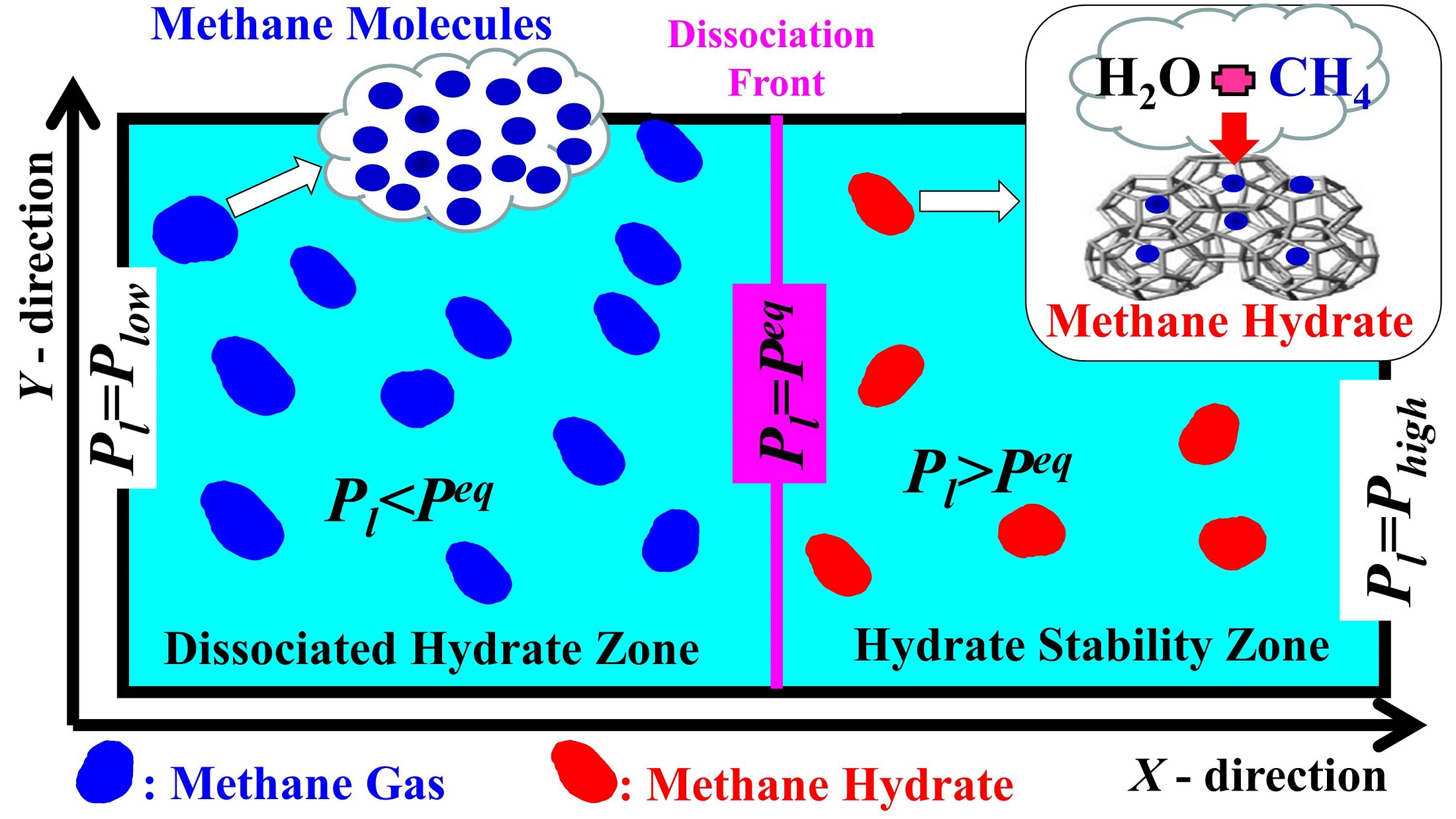
:
1. Introduction
2. Background
3. Mathematical Modeling
3.1. Case A: Ignoring the Effect of Small Pores on the Hydrate Equilibrium Conditions
3.2. Case B: Incorporating the Effect of Small Pores on the Hydrate Equilibrium Conditions
4. Results and Discussion
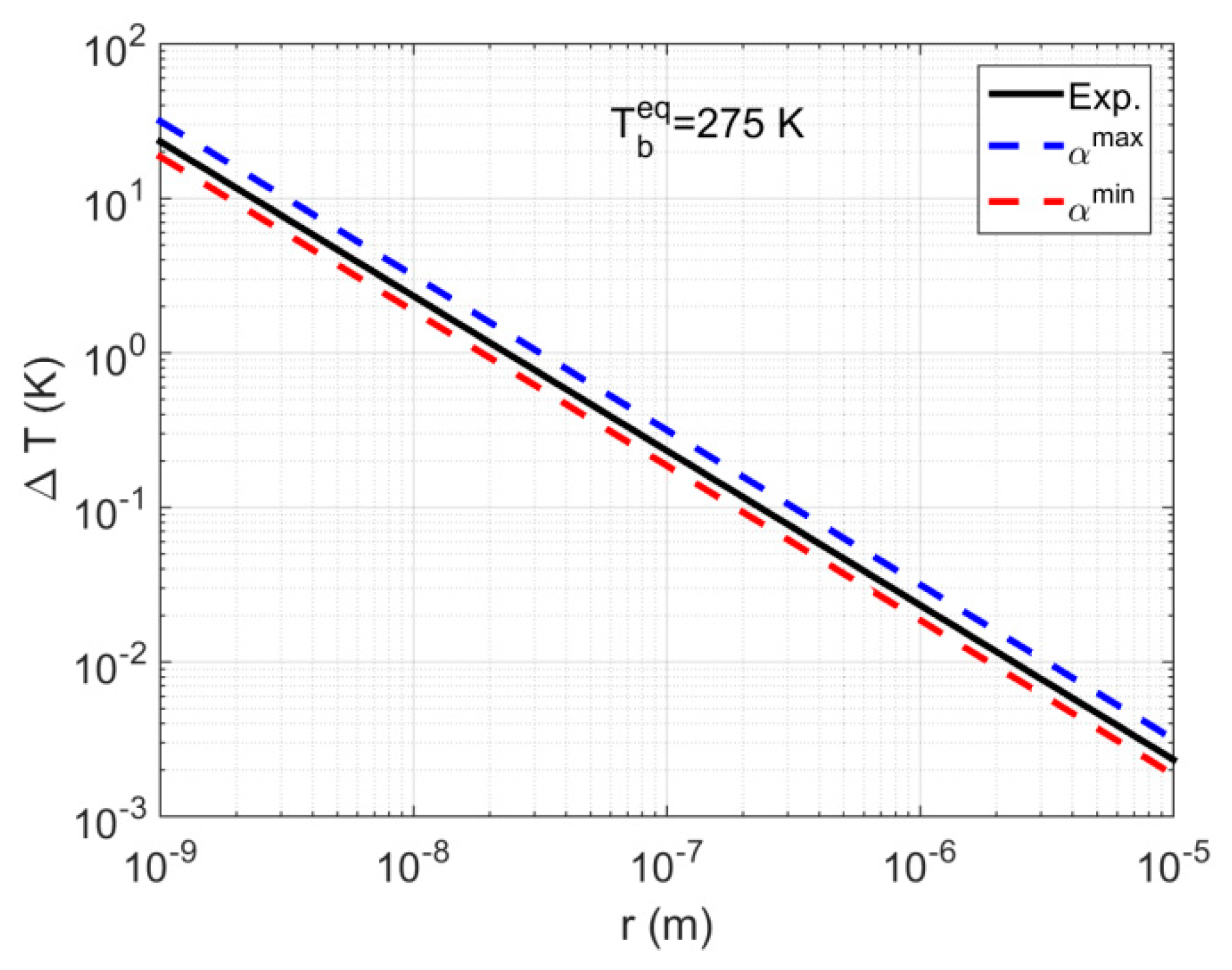
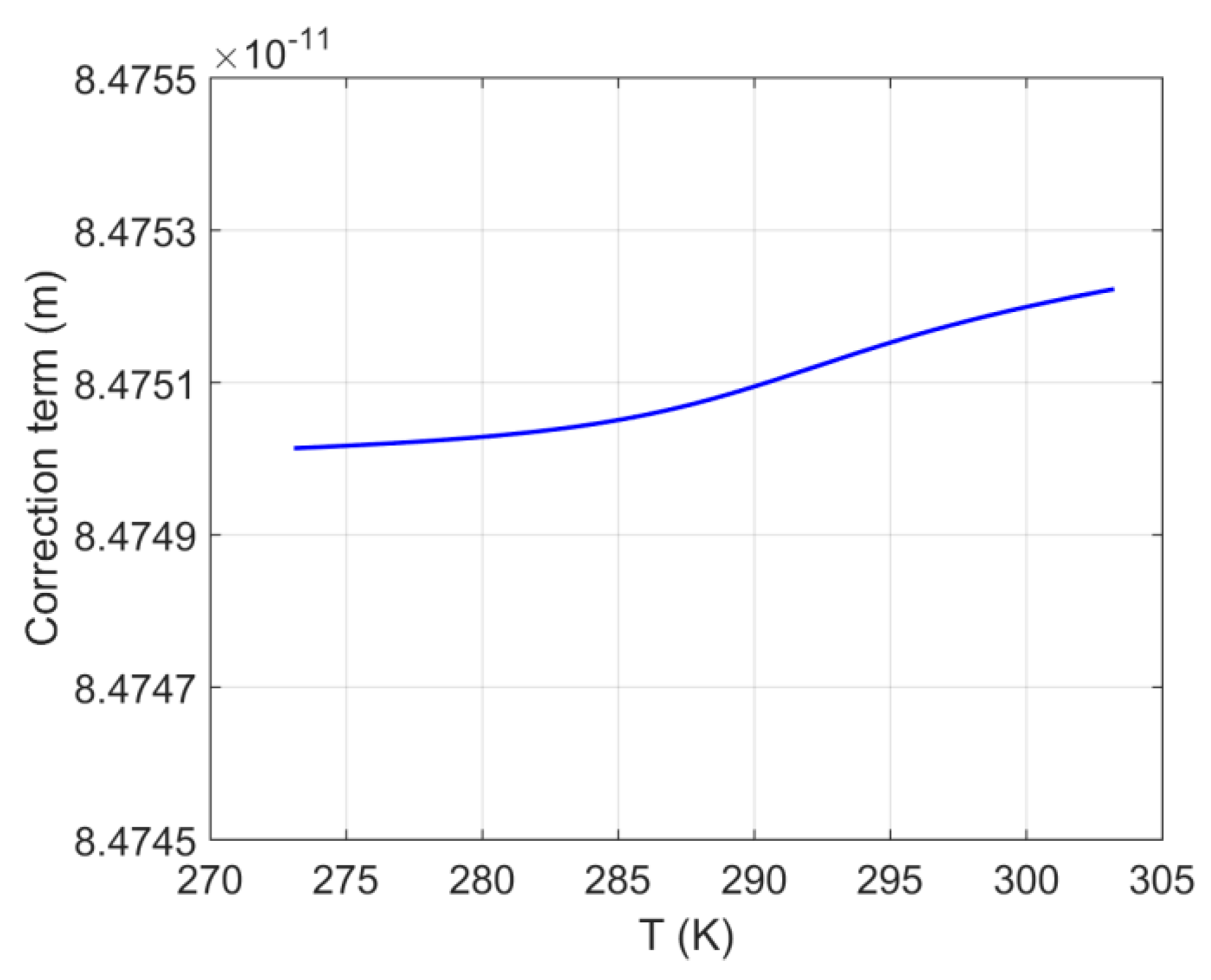
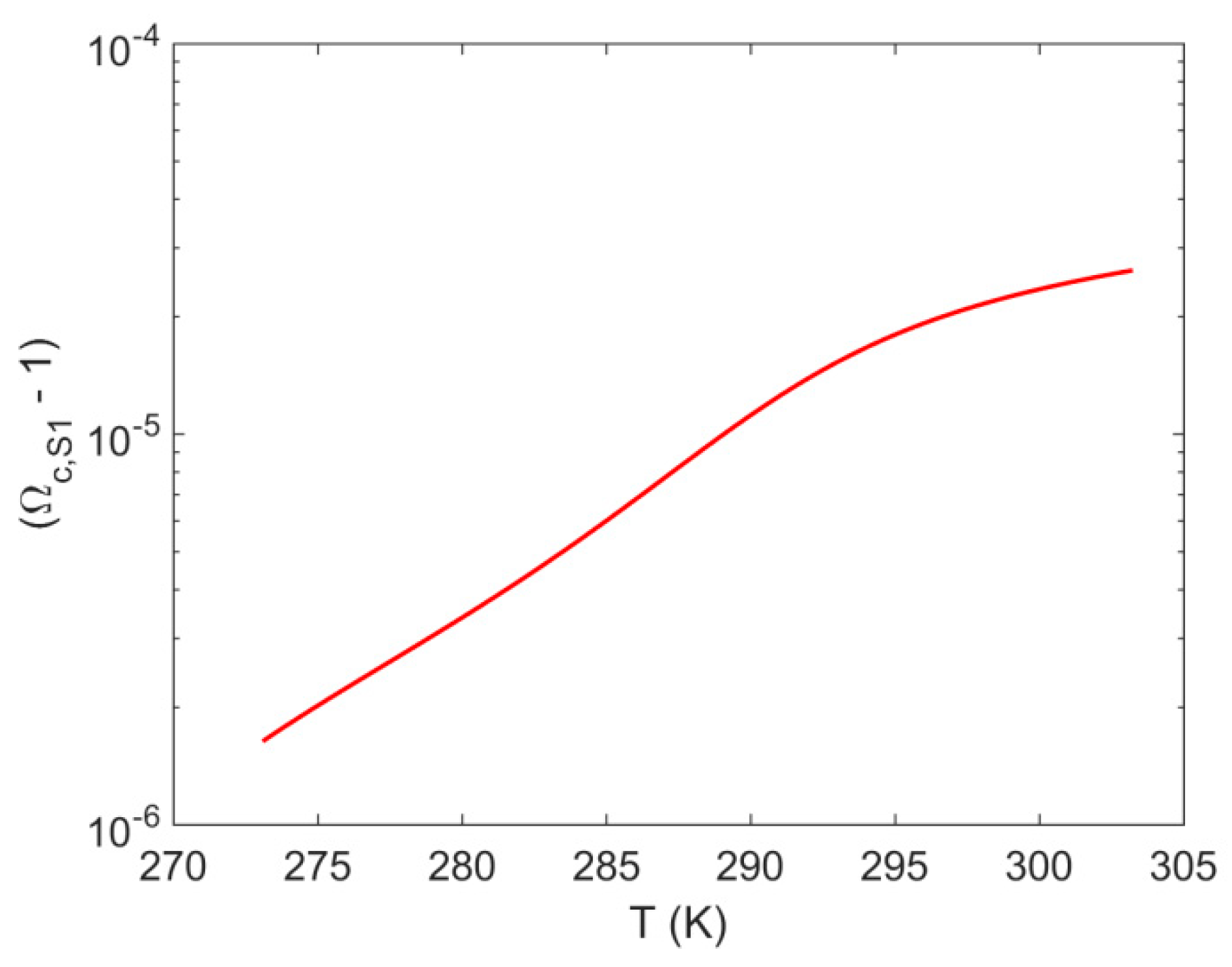
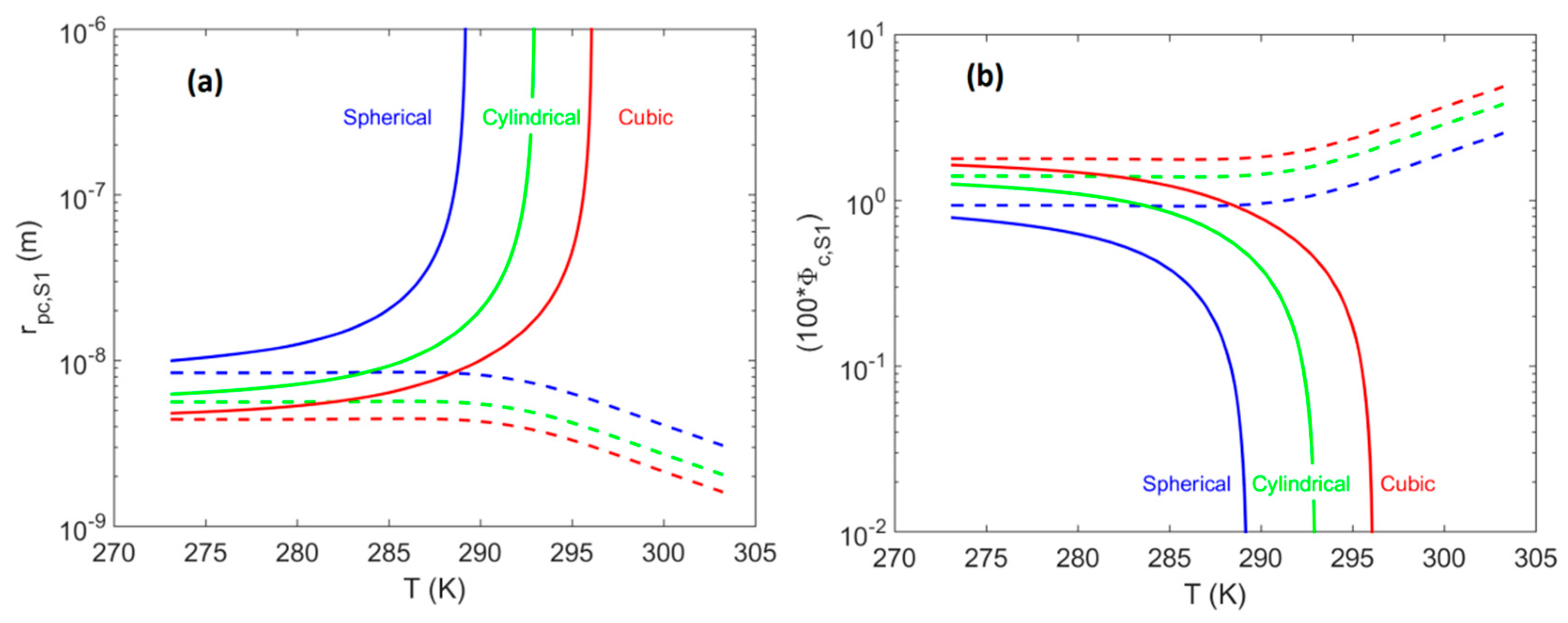
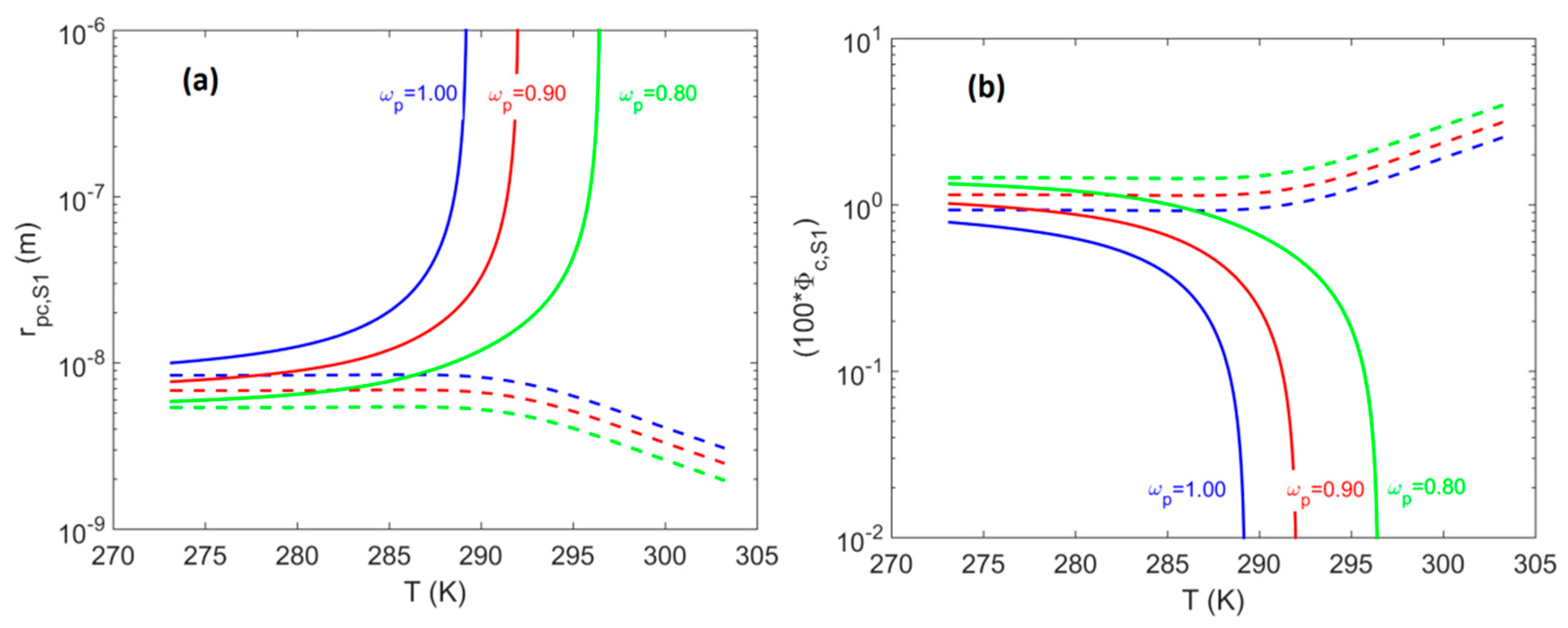
4.1. System S1: Single Pore-Body
- At pores located on the hydrate-dissociation front (i.e., where ), a shift of less than 2% is observed, with the shift magnitude having values in the following order: cubic > cylindrical > spherical.
- At pores located at points with the minimum pore pressure (i.e., where ) for well production via a pressure depletion scheme, a shift of less than 5% was observed, with the shift magnitude having values in the following order: cubic > cylindrical > spherical.
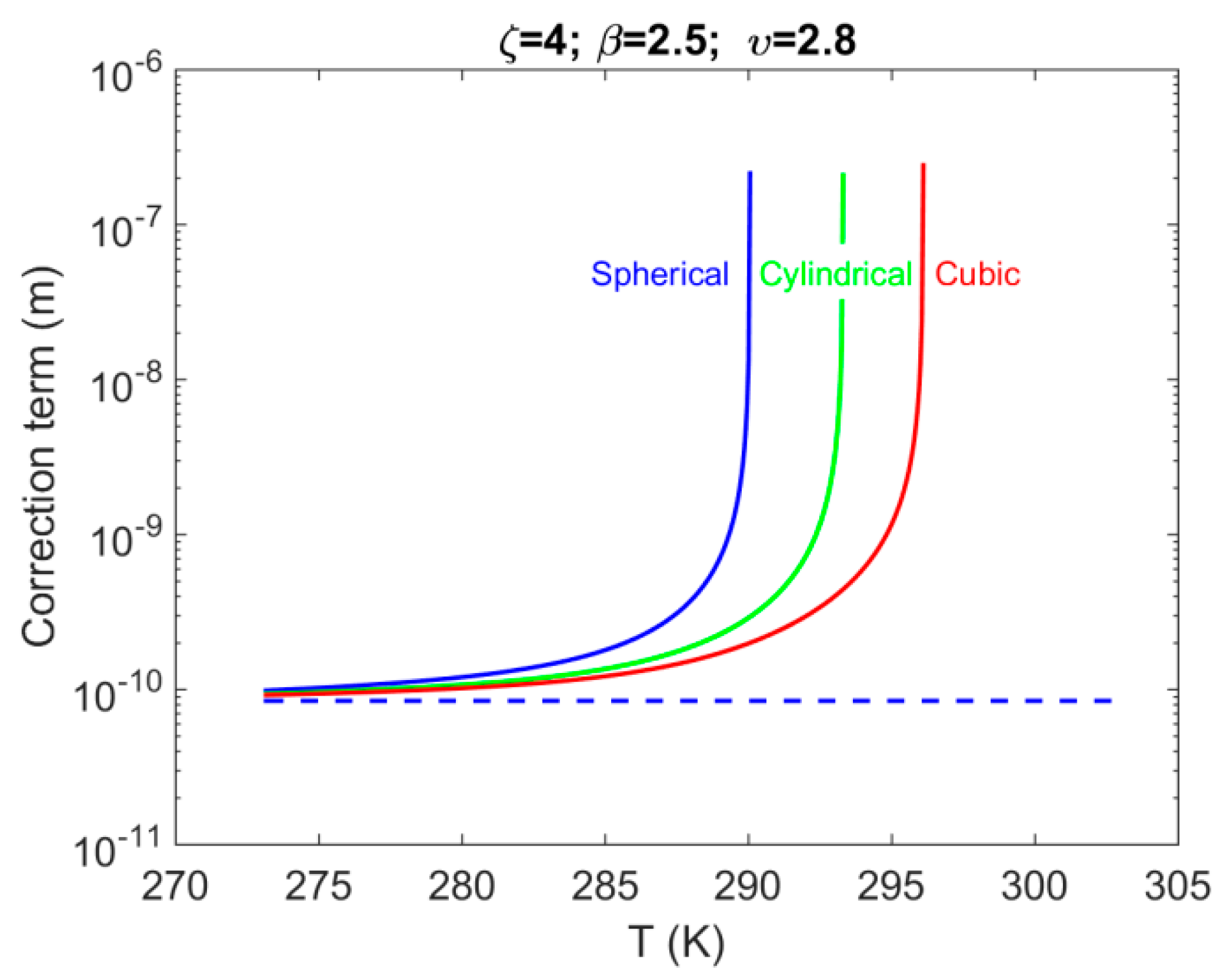
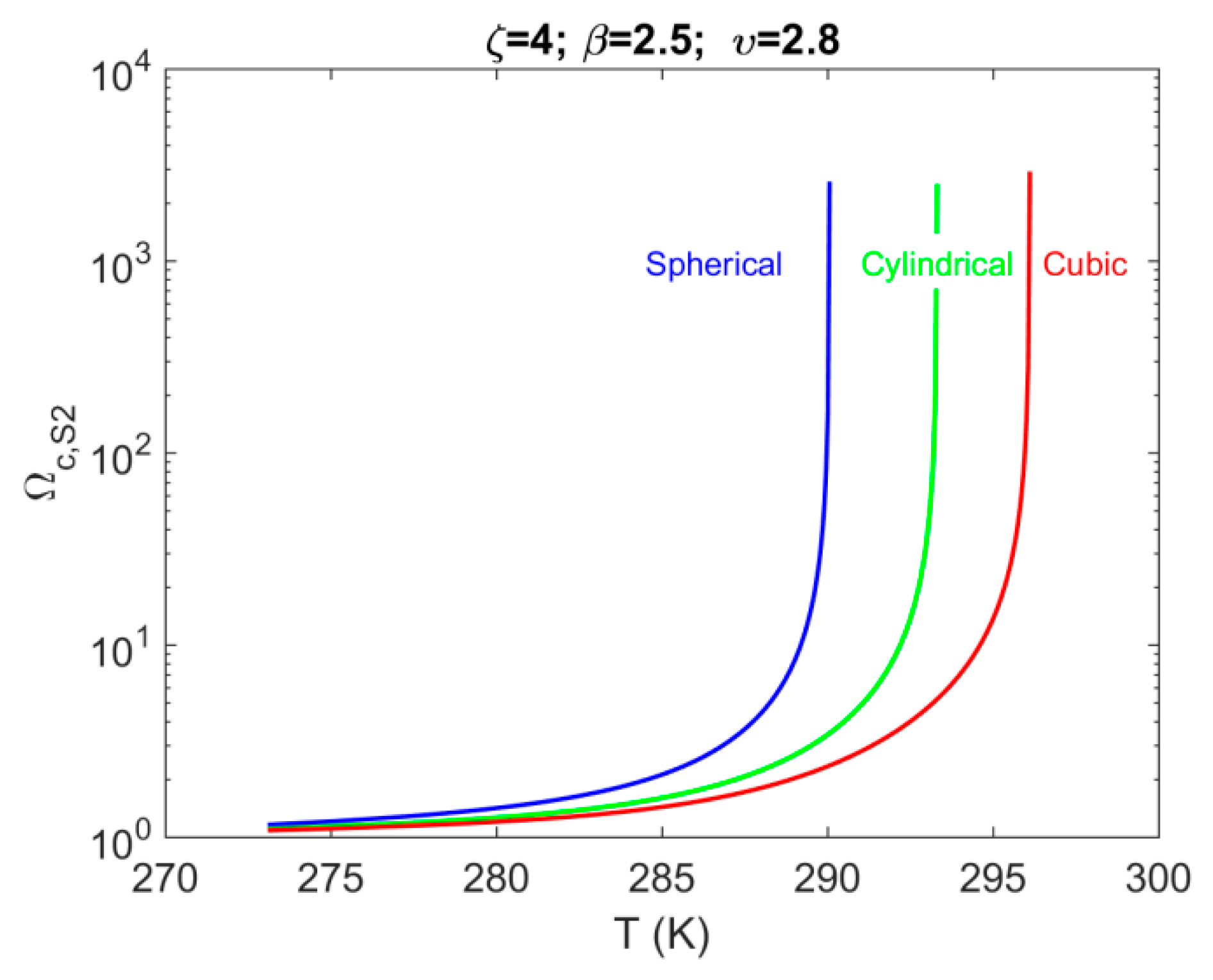
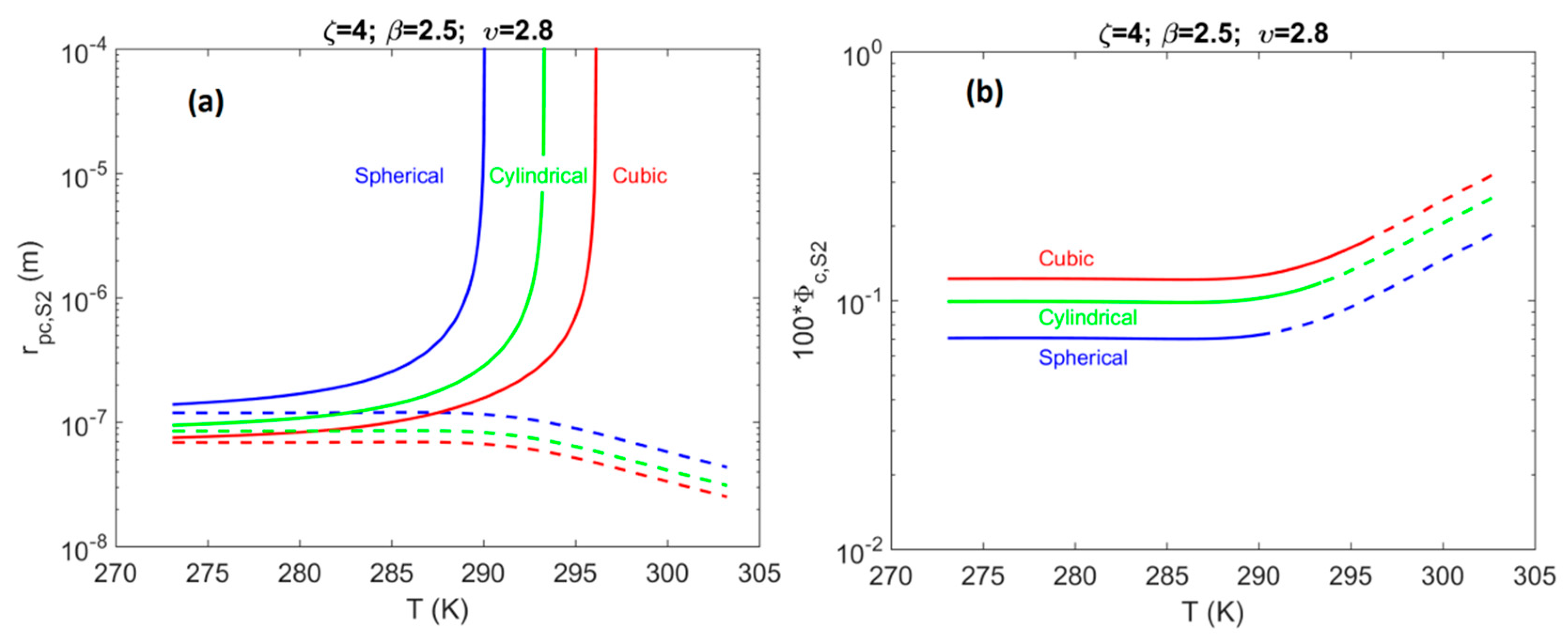
4.2. System S2: Single Pore-Body Connected with a Number of Pore-Throats
5. Conclusions
- As pore sizes decrease, the shift in the three-phase (H–Lw–V) equilibrium temperature becomes more important.
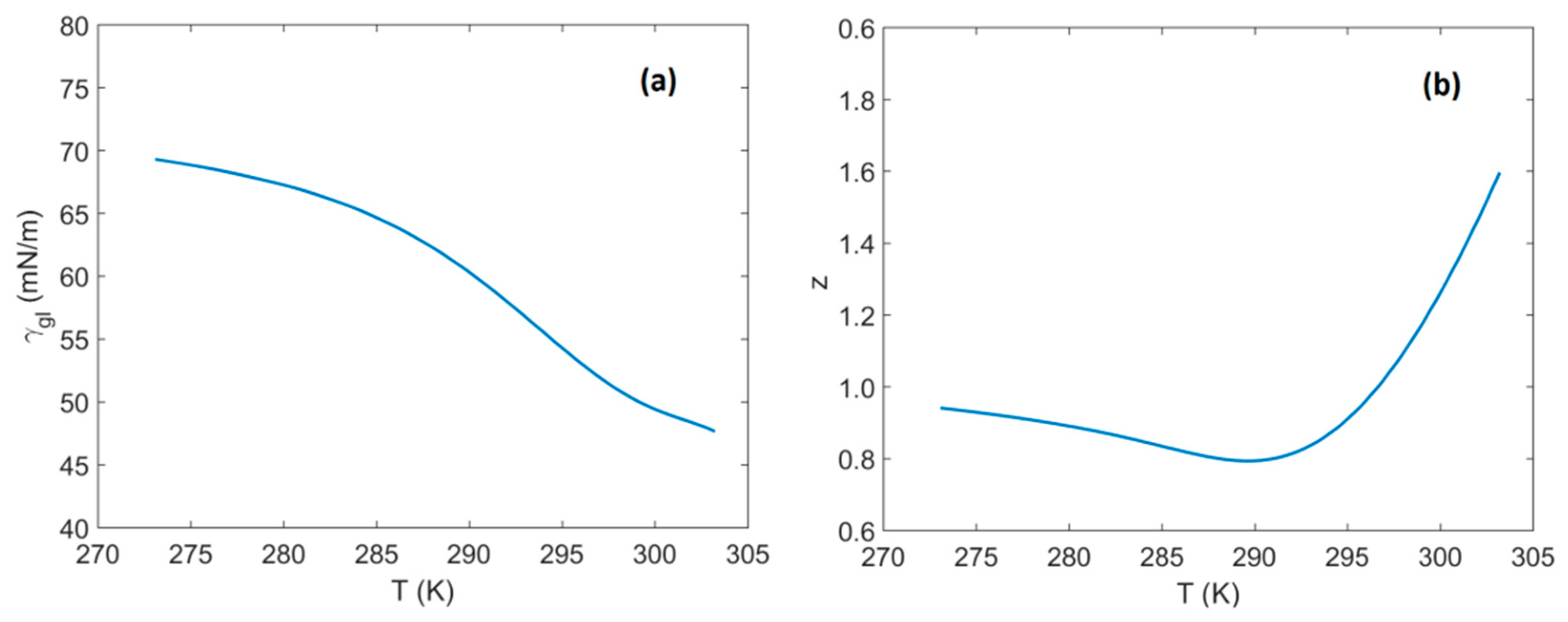
- In the current study the parameters , and are considered to be functions of temperature and their dependence is such that the parameter, , is constant (i.e., ) and thus, independent of temperature. The temperature dependence has been obtained from Monte Carlo simulations (), molecular dynamics simulations (), and hybrid methods that combine MD/MC simulations with experimental measurements ().
- For system S1, when accounting for the effect of confinement on the H–Lw–V equilibrium shift, results in a contribution up to 8% of the total value of . The shift magnitude was found to have values in the following order: cubic > cylindrical > spherical.
- For system S2, when accounting for the effect of confinement on the H–Lw–V equilibrium shift, results in a contribution up to 1% of the total value of . The shift magnitude was found to have values in the following order: cubic > cylindrical > spherical.
- The current analysis indicated that the contribution of the effect of the hydrate equilibrium shift (due to confinement in small pores) on the critical value of the pores, , in order to be able to produce the gas phase was less than 10%. The contribution to overcome the capillary thresholds of neighboring pore-throats accounted for the remaining 90%.
- Based on the previous observations we can conclude that for hydrate deposits within coarse-grained sediments or fractured systems, there is not going to be a significant negative effect, resulting from accounting for the confinement in small pores.
- Due to the lack of any experimental measurements (to the best of our knowledge) the provided discussion is limited only to the analytical solutions.
- The current analysis does not take into consideration the possible mobilization of the gas phase due to buoyancy or viscous gradients. While the contribution of buoyancy gradients is expected to be insignificant for such small gas clusters, the contribution of viscous gradients can be important, particularly in the vicinity of production wells. When accounting for such gradients, the gas production from hydrate deposits within formations with smaller pore sizes can become possible, due to gas mobilization.
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Nomenclature
| Parameter defined through: (–) | |
| Pore-body diameter (m) | |
| Constant with values: 1 (cylindrical pore-body) or 2 (spherical or cubic pore body) | |
| Geometric function defined through Equation (30) of Tsimpanogiannis [60] (–) | |
| Geometric function defined through Equation (32) of Tsimpanogiannis [60] (–) | |
| Length of unit porous block (UPB) (m) | |
| Component-specific constants for hydrate equilibrium: | |
| Capillary pressure (Pa) | |
| Pressure in the gas (g) phase (Pa) | |
| Pressure in the liquid (l) phase (Pa) | |
| Hydrate equilibrium pressure (Pa) | |
| Universal gas constant (J/mol K) | |
| Principle radii of curvature (m) | |
| Radius of pore-body (m) | |
| Radius of pore-throat (m) | |
| Critical pore-body radius of pore system S1 (m) | |
| Critical pore-body radius of pore system S2 (m) | |
| Correction term defined through Equation (12) for pore system S1 (m) | |
| Correction term defined through Equation (12) for pore system S2 (m) | |
| Gas saturation of pore system S1 (–) | |
| Gas saturation of pore system S2 (–) | |
| Critical gas saturation of pore system S1 (–) | |
| Critical gas saturation of pore system S2 (–) | |
| Hydrate saturation (–) | |
| Temperature (K) | |
| Hydrate equilibrium temperature (K) | |
| Volume of pore space that is occupied by hydrate (m3) | |
| Total volume of pore space (m3) | |
| Compressibility factor of methane gas (–) | |
| Parameter defined as: | |
| Constant defined through: (–) | |
| Constant defined through Equation (10) (–) | |
| Gas (Methane)/Liquid (Water) interfacial tension (N/m) | |
| Solid (Hydrate)/Liquid (Water) interfacial tension (N/m) | |
| Latent heat of hydrate dissociation/melting (kJ/mol methane) | |
| Coordination number of pore network (–) | |
| Contact angle (o) | |
| Cavity fractional occupancy of the small (S) cavities (–) | |
| Cavity fractional occupancy of the large (L) cavities (–) | |
| solid–liquid interface curvature defined through Equation (5) | |
| Constant defined through Equation (28) of Tsimpanogiannis [60] (mol/m3) | |
| Density of hydrate (kg/m3) | |
| Constant defined through: (–) | |
| Function defined through Equation (15) (–) | |
| Function defined through Equation (13) (–) | |
| Constant indicating amount of wetting liquid attached to pore-body wall (–) |
References
- Sloan, E.D.; Koh, C.A. Clathrate Hydrates of Natural Gases, 3rd ed.; CRC Press: Boca Raton, FL, USA, 2008. [Google Scholar]
- Li, X.-S.; Xu, C.-G.; Zhang, Y.; Ruan, X.-K.; Li, G.; Wang, Y. Investigation into gas production from natural gas hydrate: A review. Appl. Energy 2016, 172, 286–322. [Google Scholar] [CrossRef] [Green Version]
- Rossi, F.; Gambelli, A.M. Thermodynamic phase equilibrium of single-guest hydrate and formation data of hydrate in presence of chemical additives: A review. Fluid Phase Equilib. 2021, 536, 112958. [Google Scholar] [CrossRef]
- Tsimpanogiannis, I.N.; Costandy, J.; Kastanidis, P.; El Meragawi, S.; Michalis, V.K.; Papadimitriou, N.I.; Karozis, S.N.; Diamantonis, N.I.; Moultos, O.A.; Romanos, G.E.; et al. Using clathrate hydrates for gas storage and gas-mixture separations: Experimental and computational studies at multiple length scales. Mol. Phys. 2018, 116, 2041–2060. [Google Scholar] [CrossRef]
- Kelland, M.A. History of the Development of Low Dosage Hydrate Inhibitors. Energy Fuels 2006, 20, 825–847. [Google Scholar] [CrossRef]
- Kelland, M.A. A review of kinetic hydrate inhibitors from an environmental perspective. Energy Fuels 2019, 32, 12001–12012. [Google Scholar] [CrossRef]
- Collett, T.; Bahk, J.J.; Baker, R.; Boswell, R.; Divins, D.; Frye, M.; Goldberg, D.; Husebø, J.; Koh, C.; Malone, M.; et al. Methane Hydrates in Nature-Current Knowledge and Challenges. J. Chem. Eng. Data 2015, 60, 319–329. [Google Scholar] [CrossRef]
- Chong, Z.R.; Yang, S.H.B.; Babu, P.; Linga, P.; Li, X.-S. Review of Natural Gas Hydrates as an Energy Resource: Prospects and Challenges. Appl. Energy 2016, 162, 1633–1652. [Google Scholar] [CrossRef]
- Kvenvolden, K.A. A Review of the Geochemistry of Methane in Natural Gas Hydrate. Org. Geochem. 1995, 23, 997–1008. [Google Scholar] [CrossRef]
- Buffett, B.A.; Archer, D. Global inventory of methane clathrate: Sensitivity to changes in the deep ocean. Earth Planet. Sci. Lett. 2004, 227, 185–199. [Google Scholar] [CrossRef]
- Milkov, A.V. Global Estimates of Hydrate-Bound Gas in Marine Sediments: How Much is Really out There? Earth Sci. Rev. 2004, 66, 183–197. [Google Scholar] [CrossRef]
- Klauda, J.B.; Sandler, S.I. Predictions of gas hydrate phase equilibria and amounts in natural sediment porous media. Mar. Petrol. Geol. 2003, 20, 459–470. [Google Scholar] [CrossRef]
- Boswell, R.; Collett, T.S. Current perspectives on gas hydrate resources. Energy Environ. Sci. 2011, 4, 1206–1215. [Google Scholar]
- Wallmann, K.; Pinero, E.; Burwicz, E.; Haeckel, M.; Hensen, C.; Dale, A.; Ruepke, L. The global inventory of methane hydrate in marine sediments: A theoretical approach. Energies 2012, 5, 2449. [Google Scholar] [CrossRef]
- Pinero, E.; Marquardt, M.; Hensen, C.; Haeckel, M.; Wallmann, K. Estimation of the global inventory of methane hydrates in marine sediments using transfer functions. Biogeosciences 2013, 10, 959–975. [Google Scholar] [CrossRef] [Green Version]
- You, K.; Flemings, P.B.; Malinverno, A.; Collett, T.S.; Darnell, K. Mechanisms of methane hydrate formation in geological systems. Rev. Geophys. 2019, 57, 1146–1196. [Google Scholar] [CrossRef]
- Buffett, B.A. Clathrate hydrates. Annu. Rev. Earth Planet. Sci. 2000, 28, 477–507. [Google Scholar] [CrossRef]
- Walsh, M.R.; Hancock, S.H.; Wilson, S.; Patil, S.L.; Moridis, G.J.; Boswell, R.; Collett, T.S.; Koh, C.A.; Sloan, E.D. Preliminary report on the commercial viability of gas production from natural gas hydrates. Energy Econ. 2009, 31, 815–823. [Google Scholar] [CrossRef]
- Ruppel, C.D.; Kessler, J.D. The interaction of climate change and methane hydrates. Rev. Geophys. 2017, 55, 126–168. [Google Scholar] [CrossRef]
- Nisbet, E.G.; Fisher, R.E.; Lowry, D.; France, J.L.; Allen, G.; Bakkaloglu, S.; Broderick, T.J.; Cain, M.; Coleman, M.; Fernandez, J.; et al. Methane mitigation: Methods to reduce emissions, on the path to the Paris agreement. Rev. Geophys. 2020, 58, e2019RG000675. [Google Scholar] [CrossRef]
- EPA (USA). Available online: https://www.epa.gov/ghgemissions/understanding-global-warming-potentials (accessed on 8 September 2021).
- IPCC. Climate Change 2014: Synthesis Report. Contribution of Working Groups I, II and III to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change (IPCC); IPCC: Geneva, Switzerland, 2014; Available online: https://www.ipcc.ch/assessment-report/ar5/ (accessed on 5 August 2021).
- Vanneste, M.; Sultan, N.; Garziglia, S.; Forsberg, C.F.; L’Heureux, J.-S. Seafloor instabilities and sediment deformation processes: The need for integrated multidisciplinary investigations. Mar. Geol. 2014, 352, 183–214. [Google Scholar] [CrossRef] [Green Version]
- McConnell, D.R.; Zhang, Z.; Boswell, R. Review of progress in evaluating gas hydrate drilling hazards. Mar. Petrol. Geol. 2012, 34, 209–223. [Google Scholar] [CrossRef]
- Handa, Y.P.; Stupin, D. Thermodynamic Properties and Dissociation Characteristics of Methane and Propane Hydrates in 70--Radius Silica Gel Pores. J. Phys. Chem. 1992, 96, 8599–8603. [Google Scholar] [CrossRef]
- Uchida, T.; Ebinuma, T.; Ishizaki, T. Dissociation condition measurements of methane hydrate in confined small pores of porous glass. J. Phys. Chem. B 1999, 103, 3659–3662. [Google Scholar] [CrossRef]
- Uchida, T.; Ebinuma, T.; Takeya, S.; Nagao, J.; Narita, H. Effects of pore sizes on dissociation temperatures and pressures of methane, carbon dioxide, and propane hydrates in porous media. J. Phys. Chem. B 2002, 106, 820–826. [Google Scholar] [CrossRef]
- Wilder, J.W.; Sheshardi, K.; Smith, D.H. Resolving apparent contradictions in equilibrium measurements for clathrate hydrates in porous media. J. Phys. Chem. B 2001, 105, 9970–9972. [Google Scholar] [CrossRef]
- Sheshardi, K.; Wilder, J.W.; Smith, D.H. Measurements of Equilibrium Pressures and Temperatures for Propane Hydrate in Silica Gels with Different Pore-Size Distributions. J. Phys. Chem. B 2001, 105, 2627–2631. [Google Scholar]
- Smith, D.H.; Wilder, J.W.; Sheshardi, K. Methane hydrate equilibria in silica gels with broad pore-size distribution. AICHE J. 2002, 48, 393–400. [Google Scholar] [CrossRef]
- Anderson, R.; Llamendo, M.; Tohidi, B.; Burgass, R.W. Experimental measurement of methane and carbon dioxide clathrate hydrate equilibria in mesoporous silica. J. Phys. Chem. B 2003, 107, 3507–3514. [Google Scholar] [CrossRef]
- Kang, S.-P.; Lee, J.-W.; Ryu, H.-J. Phase behavior of methane and carbon dioxide hydrates in meso- and macro-sized porous media. Fluid Phase Equilib. 2008, 274, 68–72. [Google Scholar] [CrossRef]
- Kang, S.-P.; Lee, J.-W. Formation characteristics of synthesized natural gas hydrates in meso- and macroporous silica gels. J. Phys. Chem. B 2010, 114, 6973–6978. [Google Scholar] [CrossRef] [PubMed]
- Aladko, E.Y.; Dyadin, Y.A.; Fenelonov, V.B.; Larionov, E.G.; Manakov, A.Y.; Mel’gunov, M.S.; Zhurko, F.V. Formation and decomposition of ethane, propane, and carbon dioxide hydrates in silica gel mesopores under high pressure. J. Phys. Chem. B 2006, 110, 19717–19725. [Google Scholar] [CrossRef]
- Seo, Y.; Lee, S.; Cha, I.; Lee, J.D.; Lee, H. Phase equilibria and thermodynamic modeling of ethane and propane hydrates in porous silica gels. J. Phys. Chem. B 2009, 113, 5487–5492. [Google Scholar] [CrossRef]
- Lee, S.; Seo, Y. Experimental measurement and thermodynamic modeling of the mixed CH4 + C3H8 clathrate hydrate equilibria in silica gel pores: Effects of pore size and salinity. Langmuir 2010, 26, 9742–9748. [Google Scholar] [CrossRef] [PubMed]
- Zarifi, M.; Javanmardi, J.; Hashemi, H.; Eslamimanesh, A.; Mohammadi, A.H. Experimental study and thermodynamic modelling of methane and mixed C1 + C2 + C3 clathrate hydrates in the presence of mesoporous silica gel. Fluid Phase Equilib. 2016, 423, 17–24. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, X.-S.; Wang, Y.; Chen, Z.-Y.; Yan, K.-F. Decomposition conditions of methane hydrate in marine sediments from South China Sea. Fluid Phase Equilib. 2016, 413, 110–115. [Google Scholar] [CrossRef]
- Liu, H.; Zhan, S.; Guo, P.; Fan, S.; Zhang, S. Understanding the characteristic of methane hydrate equilibrium in materials and its potential application. Chem. Eng. J. 2018, 349, 775–781. [Google Scholar] [CrossRef]
- Tsimpanogiannis, I.N. A novel hybrid method for the calculation of methane hydrate—Water interfacial tension along the three-phase (hydrate–liquid water–vapor) equilibrium line. J. Chem. Phys. 2021, 155, 024702. [Google Scholar] [CrossRef] [PubMed]
- Gambelli, A.M. Analyses of CH4 and CO2 hydrate formation to define the optimal pressure for CO2 injection to maximize the replacement efficiency into natural gas hydrate in presence of a silica-based natural porous medium, via depressurization techniques. Chem. Eng. Process—Process Intens. 2021, 167, 108512. [Google Scholar] [CrossRef]
- Clennell, M.B.; Hovland, M.; Booth, J.S.; Henry, P.; Winters, W.J. Formation of natural gas hydrates in marine sediments: 1. Conceptual model of gas hydrate growth conditioned by host sediments. J. Geophys. Res. B 1999, 104, 22985–23003. [Google Scholar] [CrossRef]
- Clennell, M.B.; Henry, P.; Hovland, M.; Booth, J.S.; Winters, W.J.; Thomas, M. Formation of natural gas hydrates in marine sediments. Ann. N. Y. Acad. Sci. 2000, 912, 887–896. [Google Scholar] [CrossRef]
- Henry, P.; Thomas, M.; Clennell, M.B. Formation of natural gas hydrates in marine sediments 2. Thermodynamic calculations of stability conditions in porous sediments. J. Geophys. Res. B 1999, 104, 23005–23022. [Google Scholar] [CrossRef] [Green Version]
- Anderson, R.; Llamendo, M.; Tohidi, B.; Burgass, R.W. Characteristics of clathrate hydrate equilibria in mesopores and interpretation of experimental data. J. Phys. Chem. B 2003, 107, 3500–3506. [Google Scholar] [CrossRef]
- van der Waals, J.H.; Platteeuw, J.C. Clathrate Solutions. Adv. Chem. Phys. 1959, 2, 1–57. [Google Scholar]
- Parrish, W.R.; Prausnitz, J.M. Dissociation pressures of gas hydrates formed by gas mixtures. Ind. Eng. Chem. Proc. Des. Dev. 1972, 11, 26–35. [Google Scholar] [CrossRef]
- Holder, G.D.; Corbin, G.; Papadopoulos, K.D. Thermodynamic and molecular properties of gas hydrates from mixtures containing methane, argon, and krypton. Ind. Eng. Chem. Fund. 1980, 19, 282–286. [Google Scholar] [CrossRef]
- Holder, G.D.; Zetts, S.P.; Pradhan, N. Phase behavior in systems containing clathrate hydrates. Rev. Chem. Eng. 1988, 5, 1–70. [Google Scholar] [CrossRef]
- Clarke, M.A.; Pooladi-Darvish, M.; Bishnoi, P.R. A method to predict equilibrium conditions of gas formation in porous media. Ind. Eng. Chem. Res. 1999, 38, 2485–2490. [Google Scholar] [CrossRef]
- Klauda, J.B.; Sandler, S.I. Modeling gas hydrate equilibria in laboratory and natural porous media. Ind. Eng. Chem. Res. 2001, 40, 4197–4208. [Google Scholar] [CrossRef]
- Peddireddy, S.; Lee, S.-Y.; Lee, J.W. Variable contact angle model for gas hydrate equilibrium in porous media. AIChE J. 2006, 52, 1228–1234. [Google Scholar] [CrossRef]
- Sun, R.; Duan, Z. An accurate model to predict the thermodynamic stability of methane hydrate and methane solubility in marine environments. Chem. Geol. 2007, 244, 248–262. [Google Scholar] [CrossRef]
- Song, Y.; Yang, M.; Chen, Y.; Li, Q. An improved model for predicting hydrate phase equilibrium in marine sediment environment. J. Natural Gas Chem. 2010, 19, 241–245. [Google Scholar] [CrossRef]
- Duan, Z.; Li, D.; Chen, Y.; Sun, R. The influence of temperature, pressure, salinity and capillary force on the formation of methane hydrate. Geosci. Front. 2011, 2, 125–135. [Google Scholar] [CrossRef] [Green Version]
- Li, S.-L.; Ma, Q.-L.; Sun, C.-Y.; Chen, L.-T.; Liu, B.; Feng, X.-J.; Wang, X.-Q.; Chen, G.-J. A fractal approach on modeling gas hydrate phase equilibria in porous media. Fluid Phase Equilib. 2013, 356, 277–283. [Google Scholar] [CrossRef]
- Zhou, J.; Liang, W.; Wei, C. Phase Equilibrium Condition for Pore Hydrate: Theoretical Formulation and Experimental Validation. J. Geophys. Res. Solid Earth 2019, 124, 12703–12721. [Google Scholar] [CrossRef]
- Azimi, A.; Javanmardi, J.; Mohammadi, A.H. Development of thermodynamic frameworks for modeling of clathrate hydrates stability conditions in porous media. J. Mol. Liquids 2021, 329, 115463. [Google Scholar] [CrossRef]
- De La Fuente, M.; Vaunat, J.; Marin-Moreno, H. Modelling Methane Hydrate Saturation in Pores: Capillary Inhibition Effects. Energies 2021, 14, 5627. [Google Scholar] [CrossRef]
- Tsimpanogiannis, I.N. Study of the critical gas saturation during methane hydrate dissociation at the single-pore scale: Analytical solutions for large pores. J. Natural Gas Sci. Eng. 2020, 83, 103577. [Google Scholar] [CrossRef]
- Davie, M.K.; Buffett, B.A. Sources of methane for marine gas hydrate: Inferences from a comparison of observations and numerical models. Earth Planet. Sci. Lett. 2003, 206, 51–63. [Google Scholar] [CrossRef]
- Xie, Y.; Li, R.; Wang, X.-H.; Zheng, T.; Cui, J.-L.; Yuan, Q.; Qin, H.-B.; Sun, C.-Y.; Chen, G.-J. Review on the accumulation behavior of natural gas hydrates in porous sediments. J. Natural Gas Sci. Eng. 2020, 83, 103520. [Google Scholar] [CrossRef]
- Minshull, T.A.; Marin-Moreno, H.; Betlem, P.; Bialas, J.; Bunz, S.; Burwitz, E.; Cameselle, A.L.; Çifçi, G.; Giustiniani, M.; Hillman, J.; et al. Hydrate occurrence in Europe: A review of available evidence. Mar. Petrol. Geol. 2020, 111, 735–764. [Google Scholar] [CrossRef]
- Waite, W.F.; Santamarina, J.C.; Cortes, D.D.; Dugan, B.; Espinoza, D.N.; Germaine, J.; Jang, J.; Jung, J.W.; Kneafsey, T.J.; Shin, H.; et al. Physical properties of hydrate-bearing sediments. Rev. Geophys. 2009, 47, RG4003. [Google Scholar] [CrossRef]
- Tohidi, B.; Anderson, R.; Clennell, M.B.; Burgass, R.W.; Biderkab, A.B. Visual Observation of Gas-Hydrate Formation and Dissociation in Synthetic Porous Media by Means of Glass Micromodels. Geology 2001, 29, 867–870. [Google Scholar] [CrossRef]
- Makogan, Y.F.; Holditch, S.A.; Makogan, T.Y. Russian field illustrates gas–hydrate production. Oil Gas J. 2005, 103, 43–47. [Google Scholar]
- Tsimpanogiannis, I.N.; Lichtner, P.C. Parametric study of methane hydrate dissociation in oceanic sediments driven by thermal stimulation. J. Petr. Sci. Eng. 2007, 56, 165–175. [Google Scholar] [CrossRef]
- Papadimitriou, N.I.; Tsimpanogiannis, I.N.; Stubos, A.K. Monte Carlo simulations of methane hydrate. In Proceedings of the 7th International Conference on Gas Hydrates, Edinburgh, UK, 17–21 July 2011. [Google Scholar]
- Papadimitriou, N.I.; Tsimpanogiannis, I.N.; Economou, I.G.; Stubos, A.K. Storage of Methane in Clathrate Hydrates: Monte Carlo Simulations of sI Hydrates and Comparison with Experimental Measurements. J. Chem. Eng. Data 2016, 61, 2886–2896. [Google Scholar] [CrossRef]
- Papadimitriou, N.I.; Tsimpanogiannis, I.N.; Economou, I.G.; Stubos, A.K. Identifications of conditions for increase methane storage capacity in sII and sH clathrate hydrates from Monte Carlo simulations. J. Chem. Thermodyn. 2018, 117, 128–137. [Google Scholar] [CrossRef]
- Lasich, M.; Mohammadi, A.H.; Bolton, K.; Vrabec, J.; Ramjugernath, D. Phase equilibria of methane clathrate hydrates from Grand Canonical Monte Carlo simulations. Fluid Phase Equilib. 2014, 369, 47–54. [Google Scholar] [CrossRef]
- Brumby, P.E.; Yuhara, D.; Wu, D.T.; Sum, A.K.; Yasuoka, K. Cage occupancy of methane hydrates from Gibbs ensemble Monte Carlo simulations. Fluid Phase Equilib. 2016, 413, 242–248. [Google Scholar] [CrossRef]
- Papadimitriou, N.I.; Tsimpanogiannis, I.N.; Economou, I.G.; Stubos, A.K. Influence of combining rules on the cavity occupancy of clathrate hydrates by Monte Carlo simulations. Mol. Phys. 2014, 112, 2258–2274. [Google Scholar] [CrossRef]
- Papadimitriou, N.I.; Tsimpanogiannis, I.N.; Stubos, A.K.; Martin, A.; Rovetto, L.J.; Florusse, L.J.; Peters, C.J. experimental and computational investigation of the sII binary He-THF hydrate. J. Phys. Chem. B 2011, 115, 1411–1415. [Google Scholar] [CrossRef]
- Tsimpanogiannis, I.N.; Economou, I.G. Monte Carlo simulation studies of clathrate hydrates: A review. J. Supercrit. Fluids 2018, 134, 51–61. [Google Scholar] [CrossRef]
- Duan, Z.; Moller, N.; Weare, J.H. An equation of state for the CH4-CO2-H2O system: I. Pure systems from 0 to 1000 °C and 0 to 8000 bar. Geochim. Cosm. Acta 1992, 56, 2605–2617. [Google Scholar] [CrossRef]
- Tsimpanogiannis, I.N.; Yortsos, Y.C.; Poulou, S.; Kanellopoulos, N.; Stubos, A.K. Scaling Theory of Drying in Porous Media. Phys. Rev. E. 1999, 59, 4353–4365. [Google Scholar] [CrossRef]
- Yiotis, A.G.; Boudouvis, A.G.; Stubos, A.K.; Tsimpanogiannis, I.N.; Yortsos, Y.C. Effect of Liquid Films on the Isothermal Drying of Porous Media. Phys. Rev. E. 2003, 68, 037303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yiotis, A.G.; Tsimpanogiannis, I.N.; Stubos, A.K. Fractal Characteristics and Scaling of the Drying Front in Porous Media: A Pore Network Study. Dry. Technol. 2010, 28, 981–990. [Google Scholar] [CrossRef]
- Liu, X.; Flemings, P.B. Capillary effects on hydrate stability in marine sediments. J. Geophys. Res. 2011, 116, B07102. [Google Scholar] [CrossRef]
- Moridis, G.M. Numerical Studies of Gas Production from Methane Hydrates. SPE J. 2003, 8, 359–370. [Google Scholar] [CrossRef]
- Dufal, S.; Galindo, A.; Jackson, G.; Haslam, A.J. Modelling the effect of methanol, glycol inhibitors and electrolytes on the equilibrium stability of hydrates with the SAFT-VR approach. Mol. Phys. 2012, 110, 1223–1240. [Google Scholar] [CrossRef]
- El Meragawi, S.; Diamantonis, N.I.; Tsimpanogiannis, I.N.; Economou, I.G. Hydrate – Fluid Phase Equilibria Modeling using PC-SAFT and Peng-Robinson Equations of State. Fluid Phase Equilib. 2016, 413, 209–219. [Google Scholar] [CrossRef]
- Tsimpanogiannis, I.N.; Lichtner, P.C. Pore-Network Study of Methane Hydrate Dissociation. Phys. Rev. E 2006, 74, 056303. [Google Scholar] [CrossRef] [Green Version]
- Tsimpanogiannis, I.N.; Lichtner, P.C. Gas Saturation Resulting from Methane Hydrate Dissociation in a Porous Medium: Comparison between Analytical and Pore-Network Results. J. Phys. Chem. C 2013, 113, 11104–11116. [Google Scholar] [CrossRef]
- Du, C.; Yortsos, Y.C. A numerical study of the critical gas saturation in a porous medium. Transp. Porous Media 1999, 35, 205–225. [Google Scholar] [CrossRef]
- Tsimpanogiannis, I.N.; Yortsos, Y.C. The Critical Gas Saturation in a Porous Medium in the Presence of Gravity. J. Colloid Interface Sci. 2004, 270, 388–395. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Tsuji, S.; Jia, J.; Tsuji, T.; Matsuoka, T. Modeling CO2–Water–Mineral Wettability and Mineralization for Carbon Geosequestration. Acc. Chem. Res. 2017, 50, 1530–1540. [Google Scholar] [CrossRef]
- Tsimpanogiannis, I.N.; Michalis, V.K.; Economou, I.G. Enthalpy of dissociation of methane hydrates at a wide pressure and temperature range. Fluid Phase Equilib. 2019, 489, 30–40. [Google Scholar] [CrossRef]
- Papadimitriou, N.I.; Tsimpanogiannis, I.N.; Economou, I.G.; Stubos, A.K. Monte Carlo simulations of the separation of the binary gas mixture (CH4 + CO2) using hydrates. Phys. Chem. Chem. Phys. 2018, 20, 28026–28038. [Google Scholar] [CrossRef] [PubMed]
- Sahoo, S.K.; Marín-Moreno, H.; North, L.J.; Falcon-Suarez, I.; Madhusudhan, B.N.; Best, A.I.; Minshull, T.A. Presence and consequences of coexisting methane gas with hydrate under two phase water-hydrate stability conditions. J. Geophys. Res. Solid Earth 2018, 123, 3377–3390. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Study | |||
|---|---|---|---|
| Uchida et al. [27] | 17 ± 3 | 914 | 54.2 |
| Uchida et al. [27] (a) | 34 ± 6 | 914 | 54.2 |
| Uchida et al. [26] | 39 | 914 | 45.92 |
| Uchida et al. [26] (b) | 78 | 914 | 45.92 |
| Uchida et al. [26] (c) | 32.72 | 914 | 45.92 |
| Anderson et al. [45] | 32 ± 1 to 35 ± 1 | 907–914 | 53.2–57.7 |
| Anderson et al. [45] (d) | 32 ± 3 | – | – |
| Zarifi et al. [37] | 31 ± 3 | 914 | 57.8 |
| Liu et al. [39] | 32 ± 2 | 914 | 54.2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tsimpanogiannis, I.N.; Stamatakis, E.; Stubos, A.K. Study of the Critical Pore Radius That Results in Critical Gas Saturation during Methane Hydrate Dissociation at the Single-Pore Scale: Analytical Solutions for Small Pores and Potential Implications to Methane Production from Geological Media. Energies 2022, 15, 210. https://doi.org/10.3390/en15010210
Tsimpanogiannis IN, Stamatakis E, Stubos AK. Study of the Critical Pore Radius That Results in Critical Gas Saturation during Methane Hydrate Dissociation at the Single-Pore Scale: Analytical Solutions for Small Pores and Potential Implications to Methane Production from Geological Media. Energies. 2022; 15(1):210. https://doi.org/10.3390/en15010210
Chicago/Turabian StyleTsimpanogiannis, Ioannis Nikolaos, Emmanuel Stamatakis, and Athanasios Konstantinos Stubos. 2022. "Study of the Critical Pore Radius That Results in Critical Gas Saturation during Methane Hydrate Dissociation at the Single-Pore Scale: Analytical Solutions for Small Pores and Potential Implications to Methane Production from Geological Media" Energies 15, no. 1: 210. https://doi.org/10.3390/en15010210
APA StyleTsimpanogiannis, I. N., Stamatakis, E., & Stubos, A. K. (2022). Study of the Critical Pore Radius That Results in Critical Gas Saturation during Methane Hydrate Dissociation at the Single-Pore Scale: Analytical Solutions for Small Pores and Potential Implications to Methane Production from Geological Media. Energies, 15(1), 210. https://doi.org/10.3390/en15010210