Mechanical, Thermodynamic and Electronic Properties of Wurtzite and Zinc-Blende GaN Crystals


Abstract
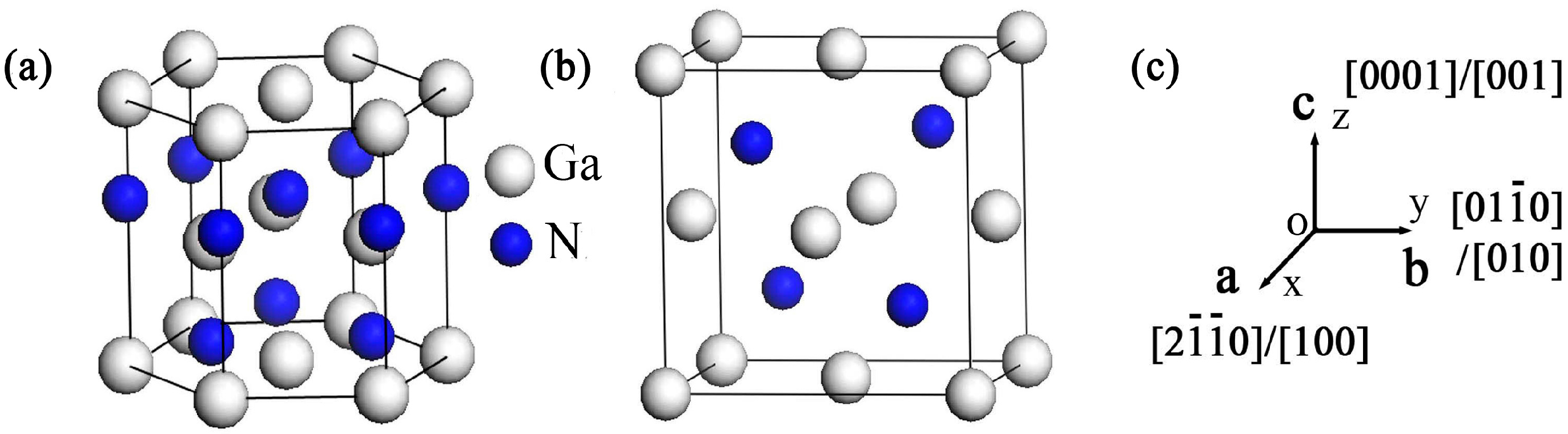
:1. Introduction
2. Computational Method and Details
3. Results and Discussion
3.1. Elastic Constants
3.2. Elastic Properties
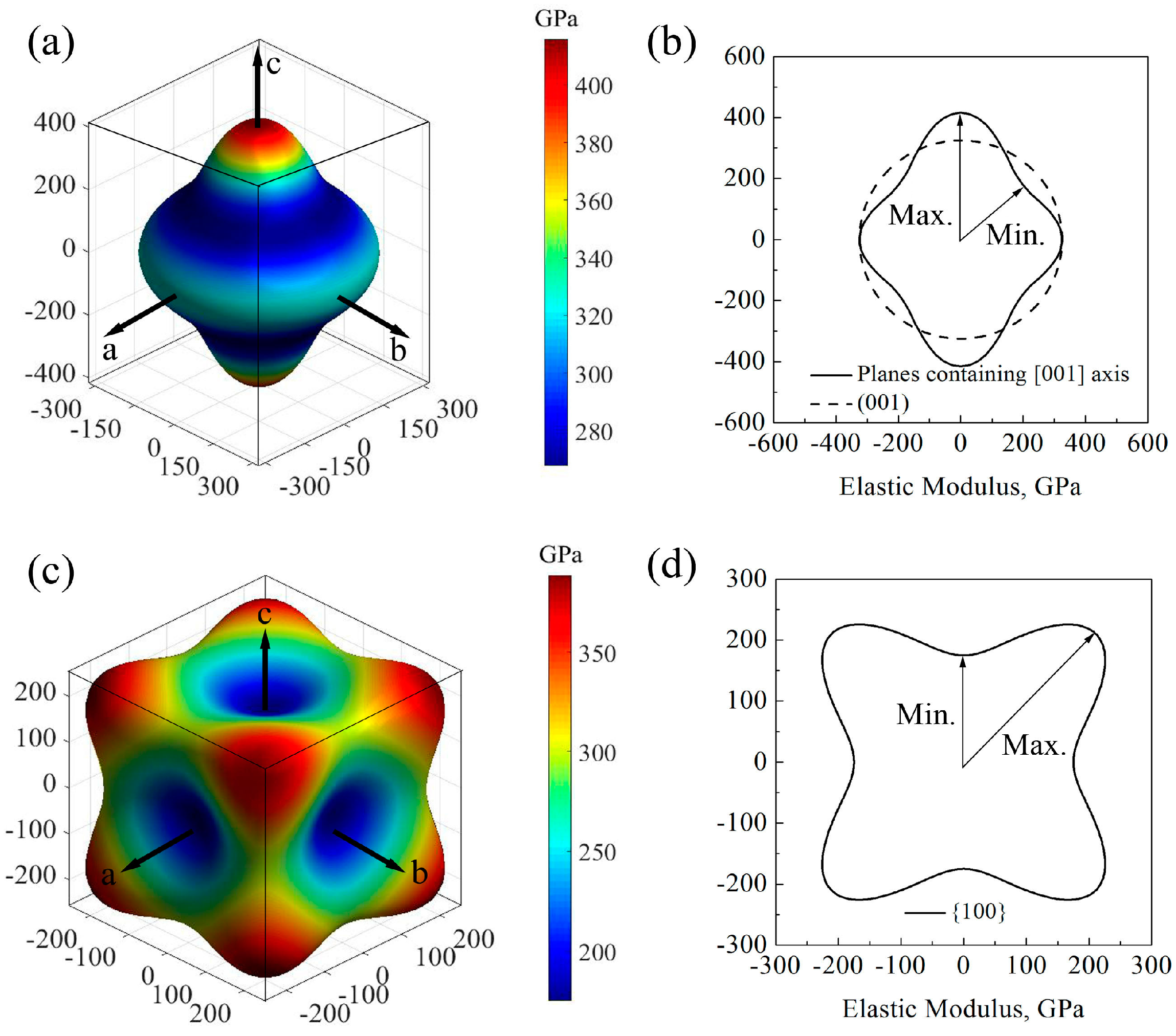
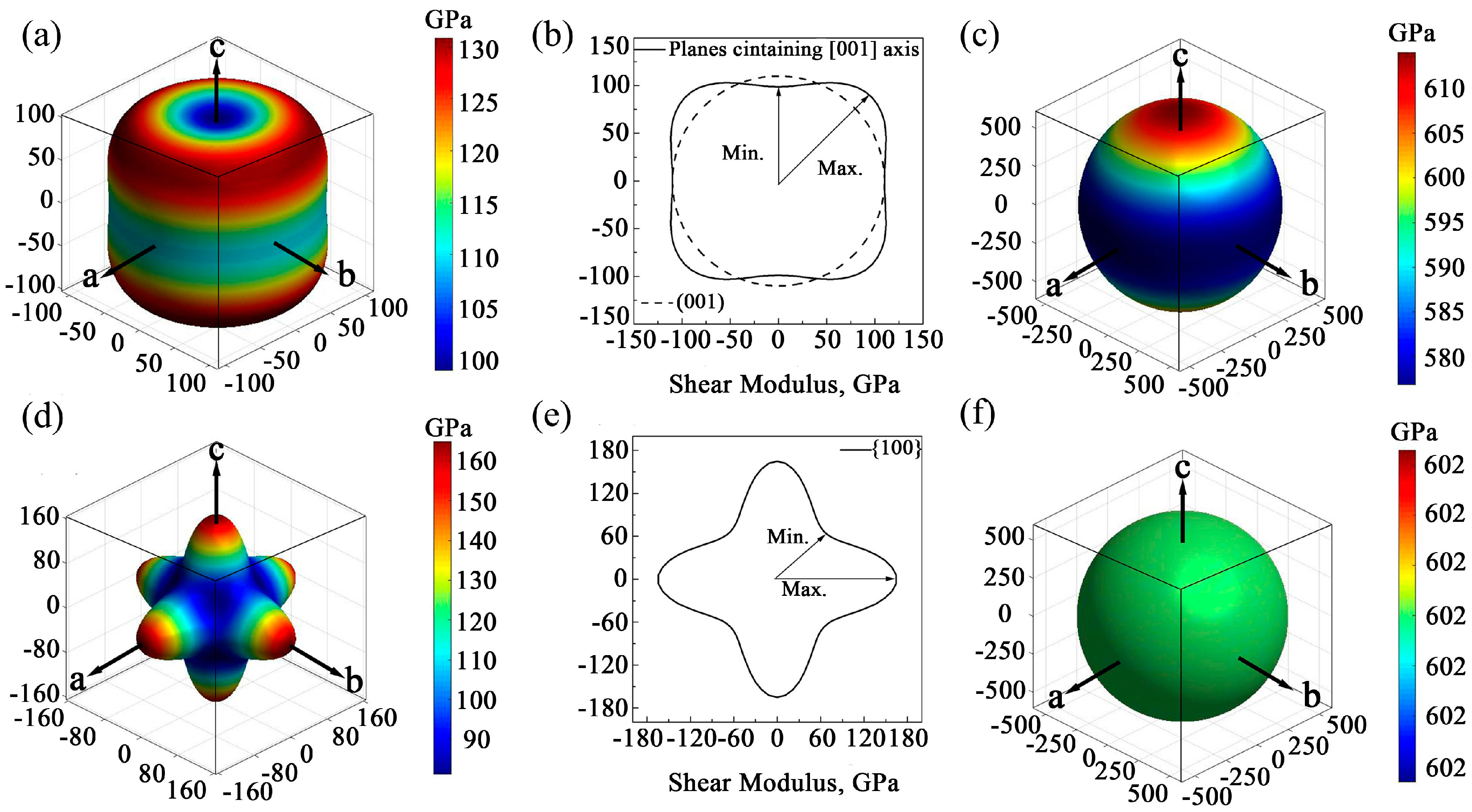
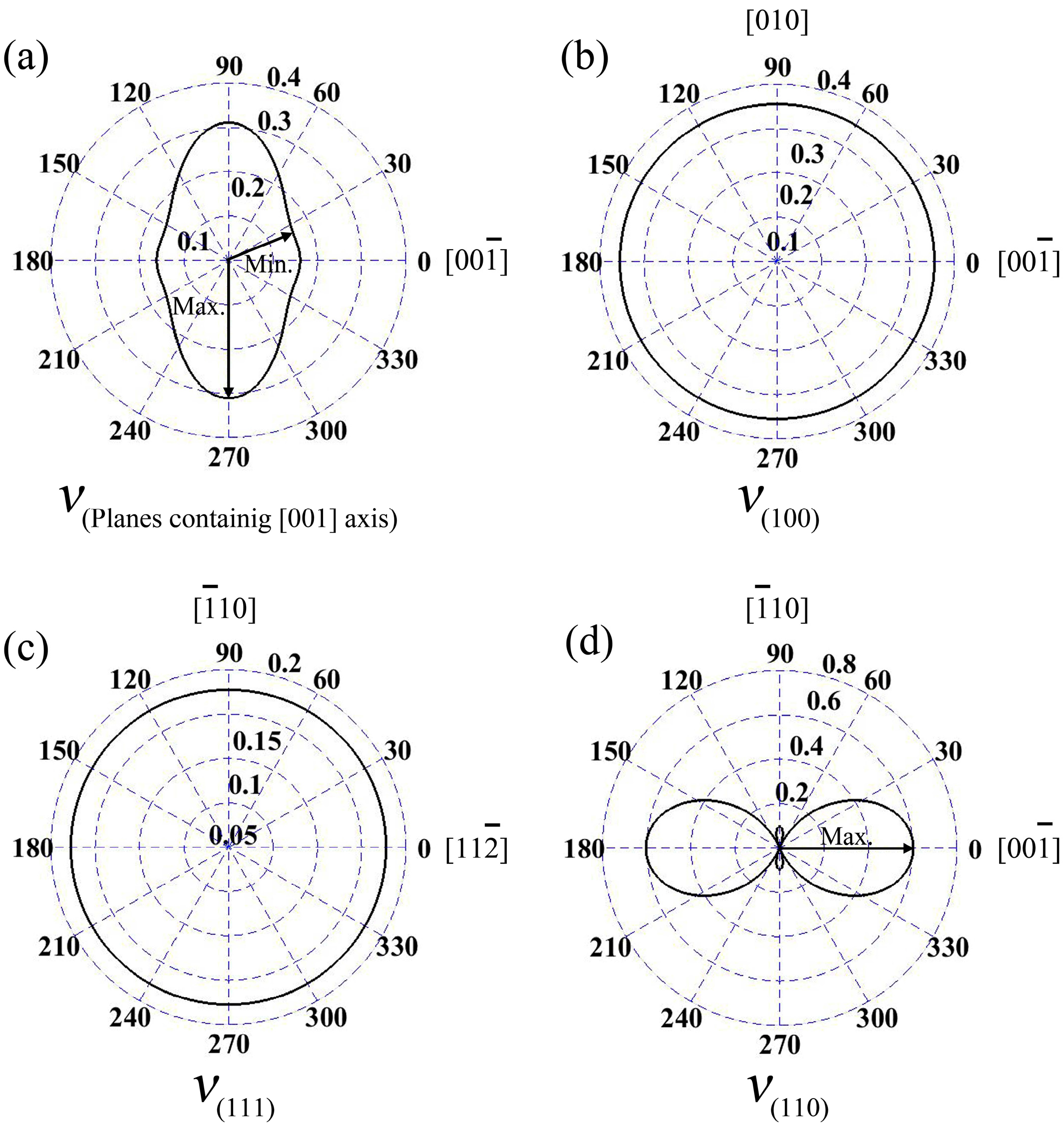
3.3. Elastic Anisotropy
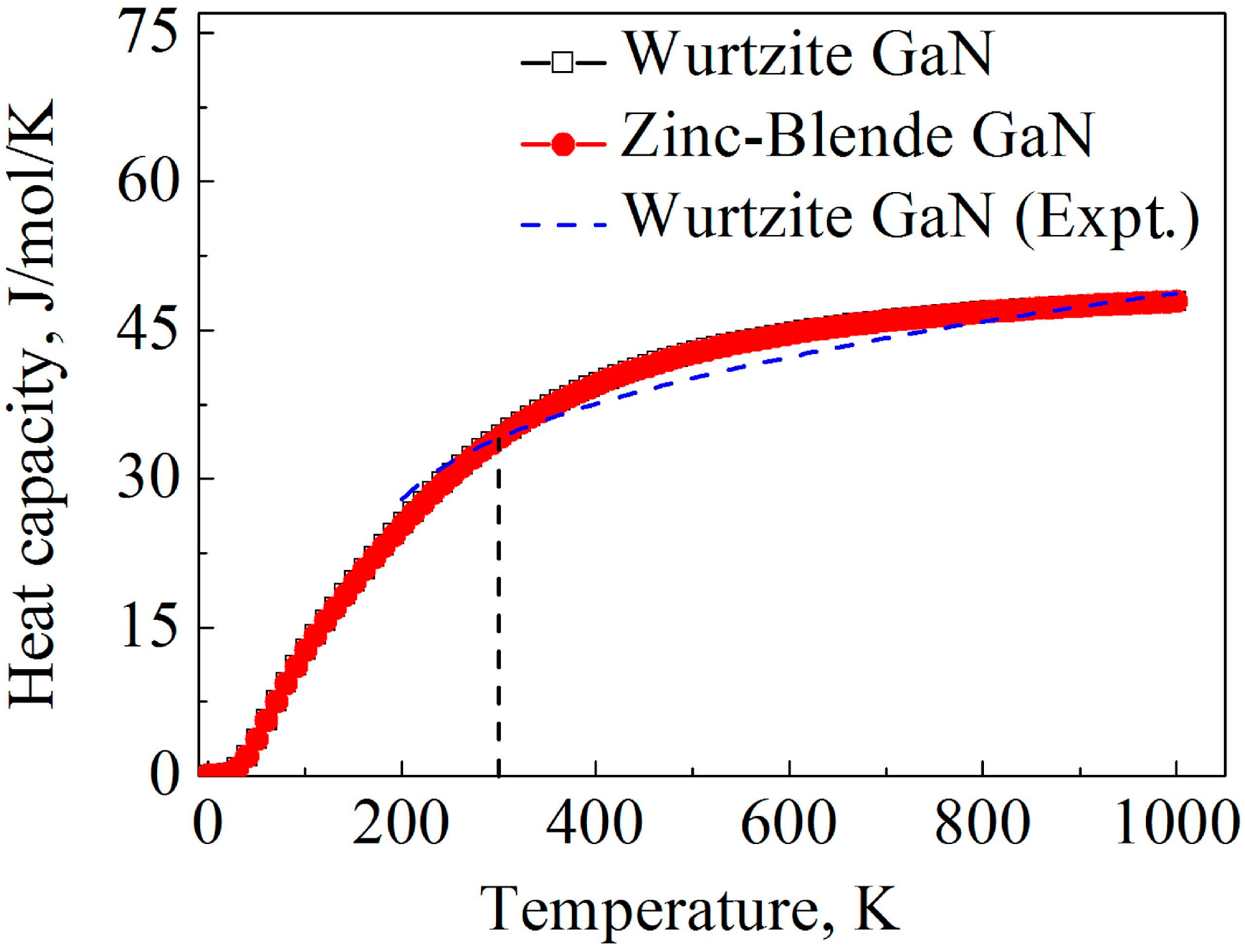
3.4.Thermodynamic Properties
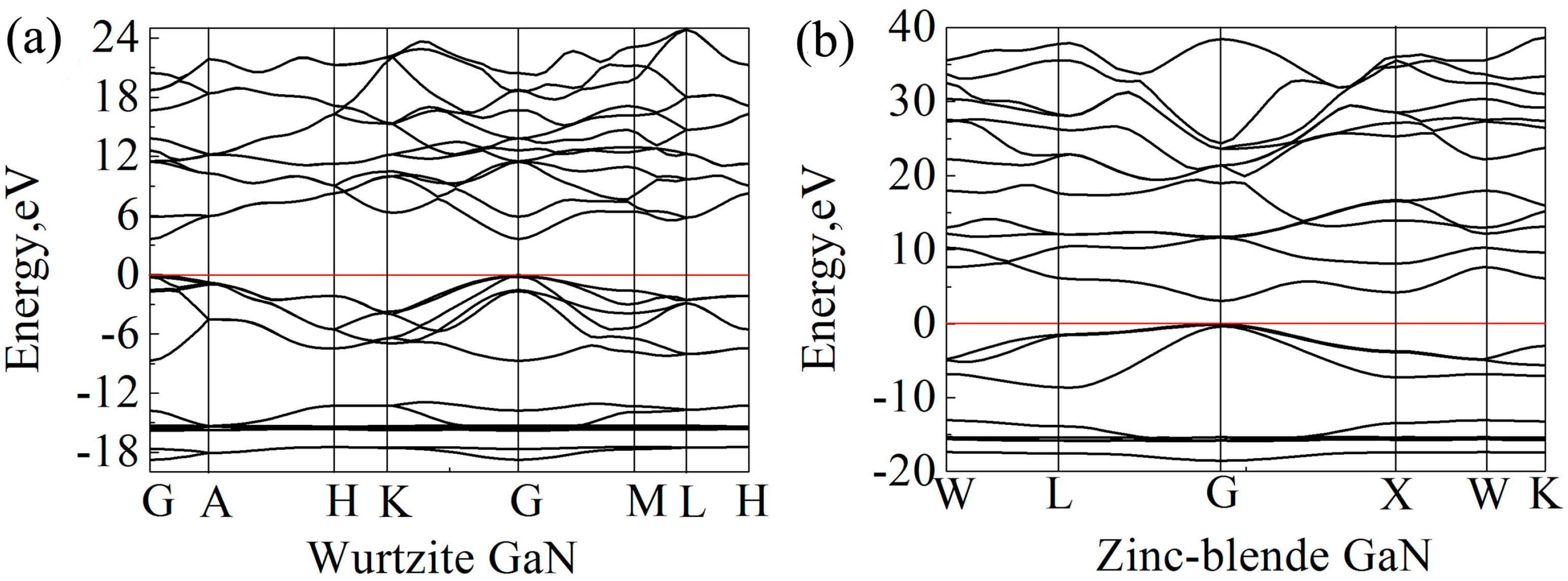
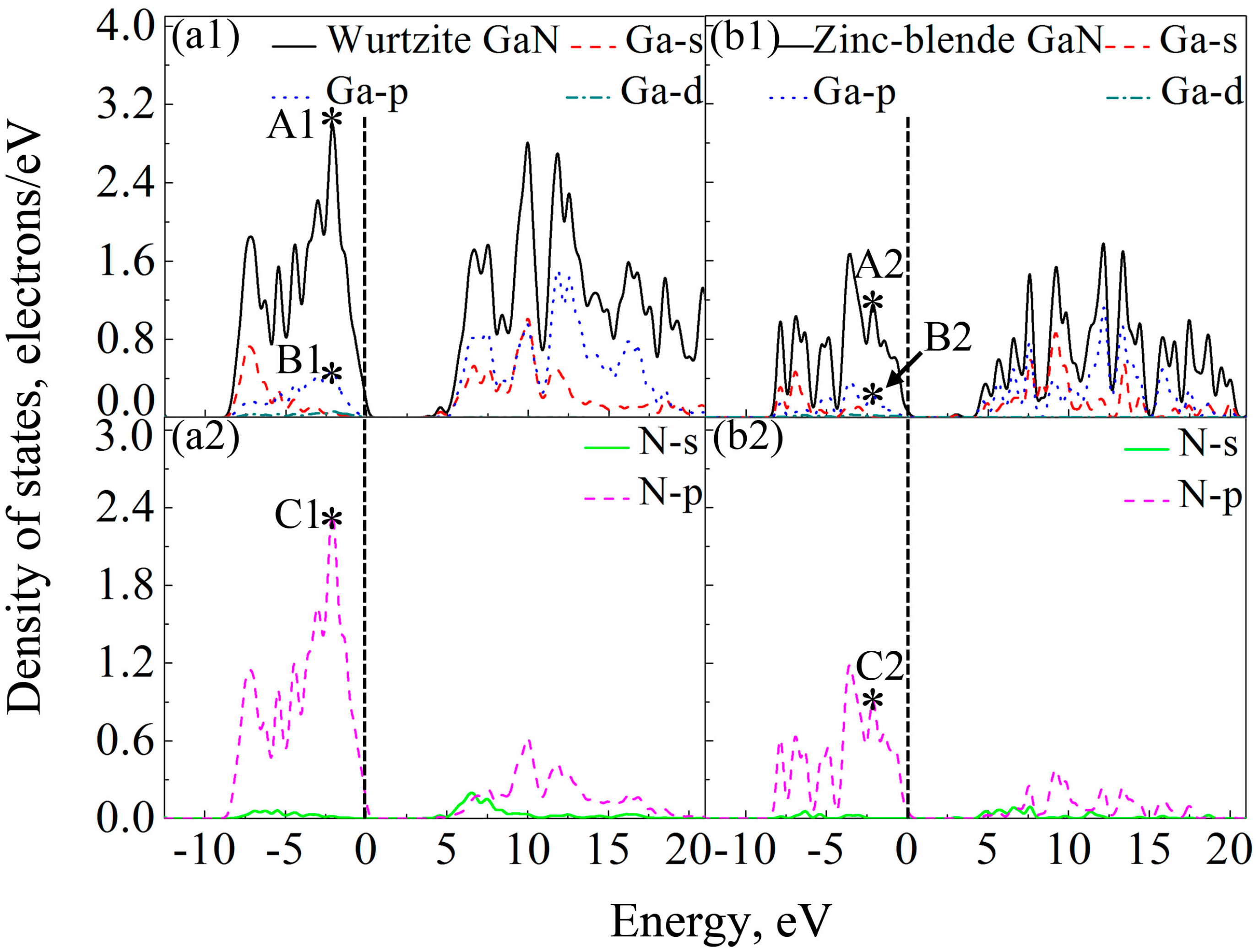
3.5. Electronic Properties
4. Conclusions
- Generally, the two GaN polycrystals have common or similar elastic properties. The bulk moduli of the wurtzite and zinc-blende GaN polycrystals are very close, and polycrystalline wurtzite GaN has slightly larger shear modulus and elastic modulus but smaller Poisson’s ratio and shows more evident characteristic of brittleness.
- Except for the bulk modulus, the anisotropic behavior of the two GaN monocrystal is quite different, or even opposite. For wurtzite GaN monocrystal, the maximum and minimum elastic moduli are located at orientations [001] and <111>, respectively, while they are in the orientations <111> and <100> in the monocrystal zinc-blende GaN, respectively. Compared to their respective elastic moduli, the shear moduli of the two GaN monocrystals have completely opposite direction dependences. Thus, the differences in their anisotropic behavior should draw enough attention.
- For wurtzite GaN, Poisson’s ratios at the planes containing [001] axis are anisotropic. The maximum value is 0.31 and it is located at the directions vertical to [001] axis. For zinc-blende GaN, Poisson’s ratios at planes (100) and (111) are isotropic, while the Poisson’s ratio at plane (110) exhibits dramatically anisotropic phenomenon.
- The Debye temperatures calculated based on elastic constants and average sound velocities of wurtzite and zinc-blende GaN are 641.8 and 620.2 K, respectively, and wurtzite GaN exhibits slightly higher stiffness and melt point for the higher Debye temperature. At the same temperature, the two GaN crystals have the same heat capacity. Zinc-blende GaN is hoped to be more amenable to doping than the wurtzite GaN [15,16], thus it can be a first decent approach of wurtzite GaN due to many common or similar properties.
- The exchange-correlation functional theory HSE06 is recommended to calculate the band gaps. Band gaps are located at the G point for the two crystals, and the band gaps of wurtzite and zinc-blende GaN are 3.62 and 3.06 eV, respectively. At the G point, the lowest energy of conduction band in the wurtzite GaN is larger than that in the zinc-blende GaN, resulting in a wider band gap.
- Densities of states in the orbital hybridization between Ga and N atoms of wurtzite GaN are much higher, indicating more electrons in corresponding atomic orbitals participate in forming Ga-N ionic bonds, which is why the wurtzite GaN has larger elastic modulus, higher Debye temperature, smaller Poisson’s ratio and shows more evident characteristic of brittleness.
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Van de Walle, C.G. Wide-Band-Gap Semiconductors; Elsevier: Amsterdam, The Netherlands, 2012. [Google Scholar]
- Kanoun, M.B.; Goumri-Said, S.; Merad, A.E.; Merad, G.; Cibert, J.; Aourag, H. Zinc-blende AlN and GaN under pressure: Structural, electronic, elastic and piezoelectric properties. Semicond. Sci. Technol. 2004, 19, 1220–1231. [Google Scholar] [CrossRef]
- Soltani, A.; Cordier, Y.; Gerbedoen, J.C.; Joblot, S.; Okada, E.; Chmielowska, M.; Ramdani, M.R.; De Jaeger, J.C. Assessment of transistors based on GaN on silicon substrate in view of integration with silicon technology. Semicond. Sci. Technol. 2013, 28, 094003. [Google Scholar] [CrossRef]
- Zhang, Y.; Wu, Z.F.; Gao, P.F.; Fang, D.-Q.; Zhang, S.-L. Enhanced visible light absorption in ZnO/GaN heterostructured nanofilms. J. Alloys Compd. 2017, 704, 478–483. [Google Scholar] [CrossRef]
- Nakamura, S. The roles of structural imperfections in InGaN-based blue light-emitting diodes and laser diodes. Science 1998, 281, 956–961. [Google Scholar] [CrossRef]
- Wang, X.; Chang, B.; Ren, L.; Gao, P. Influence of the p-type doping concentration on reflection-mode GaN photocathode. Appl. Phys. Lett. 2011, 98, 082109. [Google Scholar] [CrossRef]
- Chen, J.; Xue, C.; Zhuang, H.; Yang, Z.; Qin, L.; Li, H.; Huang, Y. Catalytic synthesis and optical properties of large-scale GaN nanorods. J. Alloys Compd. 2009, 468, L1–L4. [Google Scholar] [CrossRef]
- Paskova, T.; Hanser, D.A.; Evans, K.R. GaN Substrates for III-Nitride Devices. Proc. IEEE 2010, 98, 1324–1338. [Google Scholar] [CrossRef]
- Zhuang, D.; Edgar, J.H. Wet etching of GaN, AlN, and SiC: A review. Mater. Sci. Eng. R Rep. 2005, 48, 1–46. [Google Scholar] [CrossRef]
- Gao, X.; Man, B.; Zhang, C.; Leng, J.; Xu, Y.; Wang, Q.; Zhang, M.; Meng, Y. The important role of Ga vacancies in the ferromagnetic GaN thin films. J. Alloys Compd. 2017, 699, 596–600. [Google Scholar] [CrossRef]
- Sawicki, M.; Devillers, T.; Gałęski, S.; Simserides, C.; Dobkowska, S.; Faina, B.; Grois, A.; Navarro-Quezada, A.; Trohidou, K.N.; Majewski, J.A.; et al. Origin of low-temperature magnetic ordering in Ga1−xMnxN. Phys. Rev. B 2012, 85, 205204. [Google Scholar] [CrossRef]
- Stefanowicz, S.; Kunert, G.; Simserides, C.; Majewski, J.A.; Stefanowicz, W.; Kruse, C.; Figge, S.; Li, T.; Jakieła, R.; Trohidou, K.N.; et al. Phase diagram and critical behavior of the random ferromagnet Ga1−xMnxN. Phys. Rev. B 2013, 88, 081201. [Google Scholar] [CrossRef]
- Simserides, C.; Majewski, J.A.; Trohidou, K.N.; Dietl, T. Theory of ferromagnetism driven by superexchange in dilute magnetic semi-conductors. EPJ Web Conf. 2014, 75, 01003. [Google Scholar] [CrossRef]
- Paisley, M.J.; Sitar, Z.; Posthill, J.B.; Davis, R.F. Growth of cubic phase gallium nitride by modified molecular-beam epitaxy. J. Vac. Sci. Technol. A Vac. Surf. Films 1989, 7, 701–705. [Google Scholar] [CrossRef]
- Lei, T.; Fanciulli, M.; Molnar, R.J.; Moustakas, T.D.; Graham, R.J.; Scanlon, J. Epitaxial growth of zinc blende and wurtzitic gallium nitride thin films on (001) silicon. Appl. Phys. Lett. 1991, 59, 944–946. [Google Scholar] [CrossRef]
- Pankove, J.I. Perspective on gallium nitride. MRS Online Proc. Library Arch. 1989, 162. [Google Scholar] [CrossRef]
- Zywietz, T.K.; Neugebauer, J.; Scheffler, M. The adsorption of oxygen at GaN surfaces. Appl. Phys. Lett. 1999, 74, 1695–1697. [Google Scholar] [CrossRef]
- Adelmann, C.; Brault, J.; Mula, G.; Daudin, B.; Lymperakis, L.; Neugebauer, J. Gallium adsorption on (0001) GaN surfaces. Phys. Rev. B 2003, 67, 165419. [Google Scholar] [CrossRef]
- Rosa, A.L.; Neugebauer, J.; Northrup, J.E.; Lee, C.D.; Feenstra, R.M. Adsorption and incorporation of silicon at GaN (0001) surfaces. Appl. Phys. Lett. 2002, 80, 2008–2010. [Google Scholar] [CrossRef]
- Lee, D.S.; Lee, J.H.; Lee, Y.H.; Lee, D.D. GaN thin films as gas sensors. Sens. Actuators B Chem. 2003, 89, 305–310. [Google Scholar] [CrossRef]
- Pearton, S.; Zolper, J.; Shul, R.; Ren, F. GaN: Processing, defects, and devices. J. Appl. Phys. 1999, 86, 1–78. [Google Scholar] [CrossRef]
- Reshchikov, M.A.; Usikov, D.O.D.A.; Helava, H.; Makarov, Yu. Carbon defects as sources of the green and yellow luminescence bands in undoped GaN. Phys. Rev. B 2014, 90, 235203. [Google Scholar] [CrossRef]
- Li, L.; Yu, J.; Hao, Z.; Wang, L.; Wang, J.; Han, Y.; Li, H.; Xiong, B.; Sun, C.; Luo, Y. Influence of point defects on optical properties of GaN-based materials by first principle study. Comput. Mater. Sci. 2017, 129, 49–54. [Google Scholar] [CrossRef]
- Li, E.; Wu, B.; Lv, S.; Cui, Z.; Ma, D.; Shi, W. Field emission properties of Ge-doped GaN nanowires. J. Alloys Compd. 2016, 681, 324–329. [Google Scholar] [CrossRef]
- Kamimura, J.; Bogdanoff, P.; Ramsteiner, M.; Corfdir, P.; Feix, F.; Geelhaar, L.; Riechert, H. P-Type Doping of GaN Nanowires Characterized by Photoelectrochemical Measurements. Nano Lett. 2017, 17, 1529–1537. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.P.; Zhang, Z.H.; Tan, S.T.; Hernandezmartinez, P.L.; Zhu, B.B.; Lu, S.P.; Kang, X.J.; Sun, X.W.; Demir, H.V. Investigation of p-type depletion doping for InGaN/GaN-based light-emitting diodes. Appl. Phys. Lett. 2017, 110, 033506. [Google Scholar] [CrossRef]
- Yang, M.; Chang, B.; Hao, G.; Wang, H.; Wang, M. Optoelectronic properties of GaN, AlN, and GaAlN alloys. Opt. Int. J. Light Electron Opt. 2015, 126, 3357–3361. [Google Scholar] [CrossRef]
- Li, S.; Ouyang, C. First principles study of wurtzite and zinc blende GaN: A comparison of the electronic and optical properties. Phys. Lett. A 2005, 336, 145–151. [Google Scholar] [CrossRef]
- Luo, B.; Wu, X.; Li, G. Electronic structure, elastic and thermal properties of semiconductor GaX (X = N, P, As, Sb) with zinc blende from first-principles calculation. Int. J. Mod. Phys. B 2014, 28, 1450183. [Google Scholar] [CrossRef]
- Chen, W.H.; Cheng, H.C.; Yu, C.F. The mechanical, thermodynamic, and electronic properties of cubic Au4Al crystal via first-principles calculations. J. Alloys Compd. 2016, 689, 857–864. [Google Scholar] [CrossRef]
- Guechi, A.; Merabet, A.; Chegaar, M.; Bouhemadou, A.; Guechi, N. Pressure effect on the structural, elastic, electronic and optical properties of the Zintl phase KAsSn, first principles study. J. Alloys Compd. 2015, 623, 219–228. [Google Scholar] [CrossRef]
- Liu, X.K.; Zhou, W.; Zheng, Z.; Peng, S.M. The elastic and thermodynamic properties of ZrMo2 from first principles calculations. J. Alloys Compd. 2014, 615, 975–982. [Google Scholar] [CrossRef]
- Piskorska-Hommel, E.; Winiarski, M.J.; Kunert, G.; Hommel, D. Polarization-dependent XAFS and density functional theory investigations of the quality of the epitaxial GaMnN structure. J. Alloys Compd. 2017, 725, 632–638. [Google Scholar] [CrossRef]
- Segall, M.D.; Lindan, P.J.D.; Probert, M.J.; Pickard, C.J.; Hasnip, P.J.; Clark, S.J.; Payne, M.C. First-principles simulation: Ideas, illustrations and the CASTEP code. J. Phys. Condens. Matter 2002, 14, 2717–2744. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Hammer, B.; Hansen, L.B.; Nørskov, J.K. Improved adsorption energetics within density-functional theory using revised Perdew-Burke-Ernzerhof functionals. Phys. Rev. B 1999, 59, 7413–7421. [Google Scholar] [CrossRef] [Green Version]
- Vanderbilt, D. Soft self-consistent pseudopotentials in a generalized eigenvalue formalism. Phys. Rev. B 1990, 41, 7892–7895. [Google Scholar] [CrossRef]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Heyd, J.; Scuseria, G.E.; Ernzerhof, M. Erratum: “Hybrid functionals based on a screened Coulomb potential”. [J. Chem. Phys. 2003, 118, 8207 (2003)]. J. Chem. Phys. 2006, 124, 219906. [Google Scholar] [CrossRef]
- Chen, W.H.; Yu, C.F.; Cheng, H.C.; Lu, S.T. Crystal size and direction dependence of the elastic properties of Cu3Sn through molecular dynamics simulation and nanoindentation testing. Microelectron. Reliab. 2012, 52, 1699–1710. [Google Scholar] [CrossRef]
- Yoshikawa, A.; Ohshima, E.; Fukuda, T.; Tsuji, H.; Oshima, K. Crystal growth of GaN by ammonothermal method. J. Cryst. Growth 2004, 260, 67–72. [Google Scholar] [CrossRef]
- Nye, J.F. Lindsay, R.B. Physical Properties of Crystals: Their Representation by Tensors and Matrices; Clarendon Press: Oxford, UK, 1985. [Google Scholar]
- Polian, A.; Grimsditch, M.; Grzegory, I. Elastic constants of gallium nitride. J. Appl. Phys. 1996, 79, 3343–3344. [Google Scholar] [CrossRef]
- Shimada, K.; Sota, T.; Suzuki, K. First-principles study on electronic and elastic properties of BN, AlN, and GaN. J. Appl. Phys. 1998, 84, 4951–4958. [Google Scholar] [CrossRef]
- Wright, A.F. Elastic properties of zinc-blende and wurtzite AlN, GaN, and InN. J. Appl. Phys. 1997, 82, 2833–2839. [Google Scholar] [CrossRef]
- Hill, R. The Elastic Behaviour of a Crystalline Aggregate. Proc. Phys. Soc. Sect. A 1952, 65, 349–354. [Google Scholar] [CrossRef]
- Kanoun, M.B.; Goumri-Said, S.; Reshak, A.H. Theoretical study of mechanical, electronic, chemical bonding and optical properties of Ti2SnC, Zr2SnC, Hf2SnC and Nb2SnC. Comput. Mater. Sci. 2009, 47, 491–500. [Google Scholar] [CrossRef]
- Pugh, S. XCII. Relations between the elastic moduli and the plastic properties of polycrystalline pure metals. Lond. Rdinb. Dublin Philos. Mag. J. Sci. 1954, 45, 823–843. [Google Scholar] [CrossRef]
- Bannikov, V.V.; Shein, I.R.; Ivanovskii, A.L. Electronic structure, chemical bonding and elastic properties of the first thorium-containing nitride perovskite TaThN3. Phys. Status Solidi RRL 2007, 1, 89–91. [Google Scholar] [CrossRef]
- Chen, X.; Liang, J.; Xu, Y.; Xu, T.; Jiang, P.; Yu, Y.; Lu, K. Structure and Debye temperature of wurtzite GaN. Mod. Phys. Lett. B 1999, 13, 285–290. [Google Scholar] [CrossRef]
- Xia, H.; Xia, Q.; Ruoff, A.L. High-pressure structure of gallium nitride: Wurtzite-to-rocksalt phase transition. Phys. Rev. B 1993, 47, 12925–12928. [Google Scholar] [CrossRef]
- Yonenaga, I. Hardness, yield strength, and dislocation velocity in elemental and compound semiconductors. Mater. Trans. 2005, 46, 1979–1985. [Google Scholar] [CrossRef]
- Nowak, R.; Pessa, M.; Suganuma, M.; Leszczynski, M.; Grzegory, I.; Porowski, S.; Yoshida, F. Elastic and plastic properties of GaN determined by nano-indentation of bulk crystal. Appl. Phys. Lett. 1999, 75, 2070–2072. [Google Scholar] [CrossRef]
- Kisielowski, C.; Krüger, J.; Ruvimov, S.; Suski, T.; Ager, J., III; Jones, E.; Liliental-Weber, Z.; Rubin, M.; Weber, E.; Bremser, M. Strain-related phenomena in GaN thin films. Phys. Rev. B 1996, 54, 17745–17753. [Google Scholar] [CrossRef]
- Vurgaftman, I.; Meyer, J.R.; Ram-Mohan, L.R. Band parameters for III–V compound semiconductors and their alloys. J. Appl. Phys. 2001, 89, 5815–5875. [Google Scholar] [CrossRef]
- Kei Lau, A.K.M. Elastic anisotropy factors for orthorhombic, tetragonal, and hexagonal crystals. Phys. Rev. B 1998, 58, 8980. [Google Scholar]
- Panda, K.B.; Ravi Chandran, K.S. Determination of elastic constants of titanium diboride (TiB2) from first principles using FLAPW implementation of the density functional theory. Comput. Mater. Sci. 2006, 35, 134–150. [Google Scholar] [CrossRef]
- Zhang, J.M.; Zhang, Y.; Xu, K.W.; Ji, V. Anisotropic elasticity in hexagonal crystals. Thin Solid Films 2007, 515, 7020–7024. [Google Scholar] [CrossRef]
- Zhang, J.M.; Zhang, Y.; Xu, K.W.; Ji, V. Young’s modulus surface and Poisson’s ratio curve for cubic metals. J. Phys. Chem. Solids 2007, 68, 503–510. [Google Scholar] [CrossRef]
- Baroni, S.; De Gironcoli, S.; Dal Corso, A.; Giannozzi, P. Phonons and related crystal properties from density-functional perturbation theory. Rev. Mod. Phys. 2001, 73, 515–562. [Google Scholar] [CrossRef]
- Chen, X.L.; Lan, Y.C.; Liang, J.K.; Cheng, X.R.; Xu, Y.P.; Xu, T.; Jiang, P.Z.; Lu, K.Q. Structure and Heat Capacity of Wurtzite GaN from 113 to 1073 K. Chin. Phys. Lett. 1999, 16, 107–108. [Google Scholar] [CrossRef]
- Kushwaha, A.K. Vibrational, elastic properties and sound velocities of ZnGa2O4 spinel. Comput. Mater.Sci. 2014, 85, 259–263. [Google Scholar] [CrossRef]
- Roder, C.; Einfeldt, S.; Figge, S.; Hommel, D. Temperature dependence of the thermal expansion of GaN. Phys. Rev. B 2005, 72, 085218. [Google Scholar] [CrossRef]
- Rubio, A.; Corkill, J.L.; Cohen, M.L.; Shirley, E.L.; Louie, S.G. Quasiparticle band structure of AlN and GaN. Phys. Rev. B Condens. Matter 1993, 48, 11810. [Google Scholar] [CrossRef] [PubMed]
- Dingle, R.; Sell, D.D.; Stokowski, S E.; Ilegems, M. Absorption, Reflectance, and Luminescence of GaN Epitaxial Layers. Phys. Rev. B Condens. Matter 1971, 3, 1211–1218. [Google Scholar] [CrossRef]
- Ilegems, M.; Dingle, R.; Logan, R.A. Luminescence of Zn- and Cd-doped GaN. J. Appl. Phys. 1972, 43, 3797–3800. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Structure | Method | Lattice Constants | |||
|---|---|---|---|---|---|
| a0 = b0 (Å) | c0 (Å) | c0/a0 | V0 | ||
| Wurtzite GaN | GGA (present) | 3.242 | 5.280 | 1.629 | 48.075 |
| LDA (present) | 3.156 | 5.145 | 1.631 | 44.373 | |
| Expt. [41] | 3.189 | 5.185 | 1.626 | 45.671 | |
| Zinc-blende GaN | GGA (present) | 4.582 | 4.582 | 1.000 | 96.221 |
| LDA (present) | 4.461 | 4.461 | 1.000 | 88.800 | |
| Expt. [15] | 4.490 | 4.490 | 1.000 | 90.519 | |
| Structure | C11 = C22 | C12 | C13 = C23 | C33 | C44 = C55 | C66 | RMSE * | Note |
|---|---|---|---|---|---|---|---|---|
| Wurtzite GaN | 353.93 | 82.59 | 51.79 | 380.44 | 98.49 | 135.67 | 37.90 | GGA |
| 374.35 | 126.56 | 80.85 | 441.94 | 98.86 | 123.89 | 21.35 | LDA | |
| 390 ± 15 | 145 ± 20 | 106 ± 20 | 398 ± 20 | 105 ± 10 | 123 ± 10 | - | Expt. [43] | |
| Zinc-blende GaN | 242.41 | 122.05 | 122.05 | 242.41 | 146.82 | 146.82 | - | GGA |
| 286.93 | 152.77 | 152.77 | 286.93 | 164.97 | 164.97 | - | LDA | |
| 285 | 161 | 161 | 285 | 149 | 149 | - | LDA [44] | |
| 293 | 159 | 159 | 293 | 155 | 155 | - | LDA [45] |
| Structure | S11=S22 | S12 | S13= S23 | S33 | S44 | S55 | S66 | Note |
|---|---|---|---|---|---|---|---|---|
| Wurtzite GaN | 3.026 | −0.659 | −0.322 | 2.716 | 10.153 | 10.153 | 7.371 | GGA |
| 3.079 | −0.957 | −0.388 | 2.405 | 10.115 | 10.115 | 8.071 | LDA | |
| Zinc-blende GaN | 6.224 | −2.084 | −2.084 | 6.224 | 6.811 | 6.811 | 6.811 | GGA |
| 5.712 | −2.027 | −2.027 | 5.712 | 6.061 | 6.061 | 6.061 | LDA |
| Structure | B, GPa | G, GPa | E, GPa | ν | B/G | Note |
|---|---|---|---|---|---|---|
| Wurtzite GaN | 162.3 | 124.1 | 296.7 | 0.20 | 1.31 | GGA |
| 196.3 | 121.7 | 302.6 | 0.24 | 1.61 | LDA | |
| 170 [50] 188 [51] | 116 [52] | 295 ± 3 [53] | 0.23 ± 0.06 [54] | - | Expt. | |
| Zinc-blende GaN | 162.2 | 102.2 | 253.4 | 0.24 | 1.59 | GGA |
| 199.1 | 113.1 | 285.3 | 0.26 | 1.76 | LDA | |
| 203.7 | 110.71 | 281.18 | 0.27 | 1.84 | Expt. [55] |
| Structure | Symmetry | Anisotropy Factor A | Calculation Results | |
|---|---|---|---|---|
| GGA | LDA | |||
| Wurtzite GaN | Planes containing the [001] axis | 0.63 | 0.61 | |
| Zinc-blende GaN | {100} | 2.44 | 2.46 | |
| {110} | * | 1.85 | 1.85 | |
| Structure | ρ, g/cm | νt, m/s | νl, m/s | νm, m/s | θD, K |
|---|---|---|---|---|---|
| Wurtzite GaN | 6.268 | 4406.386 | 7563.581 | 4888.006 | 641.8 |
| Zinc-blende GaN | 6.264 | 4249.685 | 7474.802 | 4724.200 | 620.2 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qin, H.; Luan, X.; Feng, C.; Yang, D.; Zhang, G. Mechanical, Thermodynamic and Electronic Properties of Wurtzite and Zinc-Blende GaN Crystals. Materials 2017, 10, 1419. https://doi.org/10.3390/ma10121419
Qin H, Luan X, Feng C, Yang D, Zhang G. Mechanical, Thermodynamic and Electronic Properties of Wurtzite and Zinc-Blende GaN Crystals. Materials. 2017; 10(12):1419. https://doi.org/10.3390/ma10121419
Chicago/Turabian StyleQin, Hongbo, Xinghe Luan, Chuang Feng, Daoguo Yang, and Guoqi Zhang. 2017. "Mechanical, Thermodynamic and Electronic Properties of Wurtzite and Zinc-Blende GaN Crystals" Materials 10, no. 12: 1419. https://doi.org/10.3390/ma10121419
APA StyleQin, H., Luan, X., Feng, C., Yang, D., & Zhang, G. (2017). Mechanical, Thermodynamic and Electronic Properties of Wurtzite and Zinc-Blende GaN Crystals. Materials, 10(12), 1419. https://doi.org/10.3390/ma10121419