3.1. Definition of the Curing Schedules
As said, we need curing schedules composed of a 1st pre-curing step until the gelation of the systems, followed by a post-curing step at 180 °C for 2 h in order to get completion of epoxy-amine reactions. Our aim was to determine three different pre-curing steps for determining the effects on morphologies and properties. First, it is important to assess the influence of PES on polymerization kinetics. Evolution of the viscosity versus reaction time for the neat and the two modified epoxy systems under isothermal conditions, 150 °C has been measured (see
Supporting Information, Figure S1). Dissolving the PES into the epoxy-amine system considerably increases its viscosity by more than one order of magnitude. It is noteworthy that PES is usually added as viscosity enhancing agent in semi-products like prepregs. To evaluate the effect of PES on the cure kinetics of the epoxy-amine network, epoxy conversion was measured by DSC for the neat and the two PES-modified systems. The systems were heated at 10 °C·min
−1 from room temperature up to 300 °C. The exothermic peak corresponding to the heat of reaction appears above 100 °C and ends at about 280 °C (see
Supporting Information, Figure S2). Assuming that a total conversion is reached at the end of the enthalpy peak, it is possible to plot the conversion of reaction as a function of temperature and subsequently as function of time (
Figure 3).
Differences between the cure kinetics of the three systems are very slight. A dilution effect slowing down the initial reaction rate was observed for a TP-modified TS system [
25] but in our case, the PES concentration is too low to induce this phenomenon. It is interesting to notice that the functionality of the phenoxy-end-modified PES does not lead to any change in the overall cure kinetics, even if –OH groups could have catalyzed the epoxy-amine reaction [
1]. From these results, the same curing schedule can be considered for the neat and the PES-modified epoxy-amine systems.
Then the Conversion Temperature Transformation (CTT) phase diagram for the neat epoxy system has been determined [
1]. As mentioned previously, gelation and/or vitrification determine the extent of phase separation and as a consequence final morphologies. It is therefore essential to determine at which conversion and time these physical transitions occur depending on the selected curing conditions.
The dependence of the glass temperature, T
g, and the conversion, x, of a TS system could be fitted by the Di Benedetto model:
where T
g0 and T
g∞ are the glass transition temperatures of the non-reacted system and of the fully cured one, respectively. Pascault and Williams reported later that λ parameter corresponds to the ratio ΔC
p∞/ΔC
p0, with ΔC
p∞ and ΔC
p0 being the associated variations of isobaric calorific capacity from initial state to fully cured state, respectively [
26].
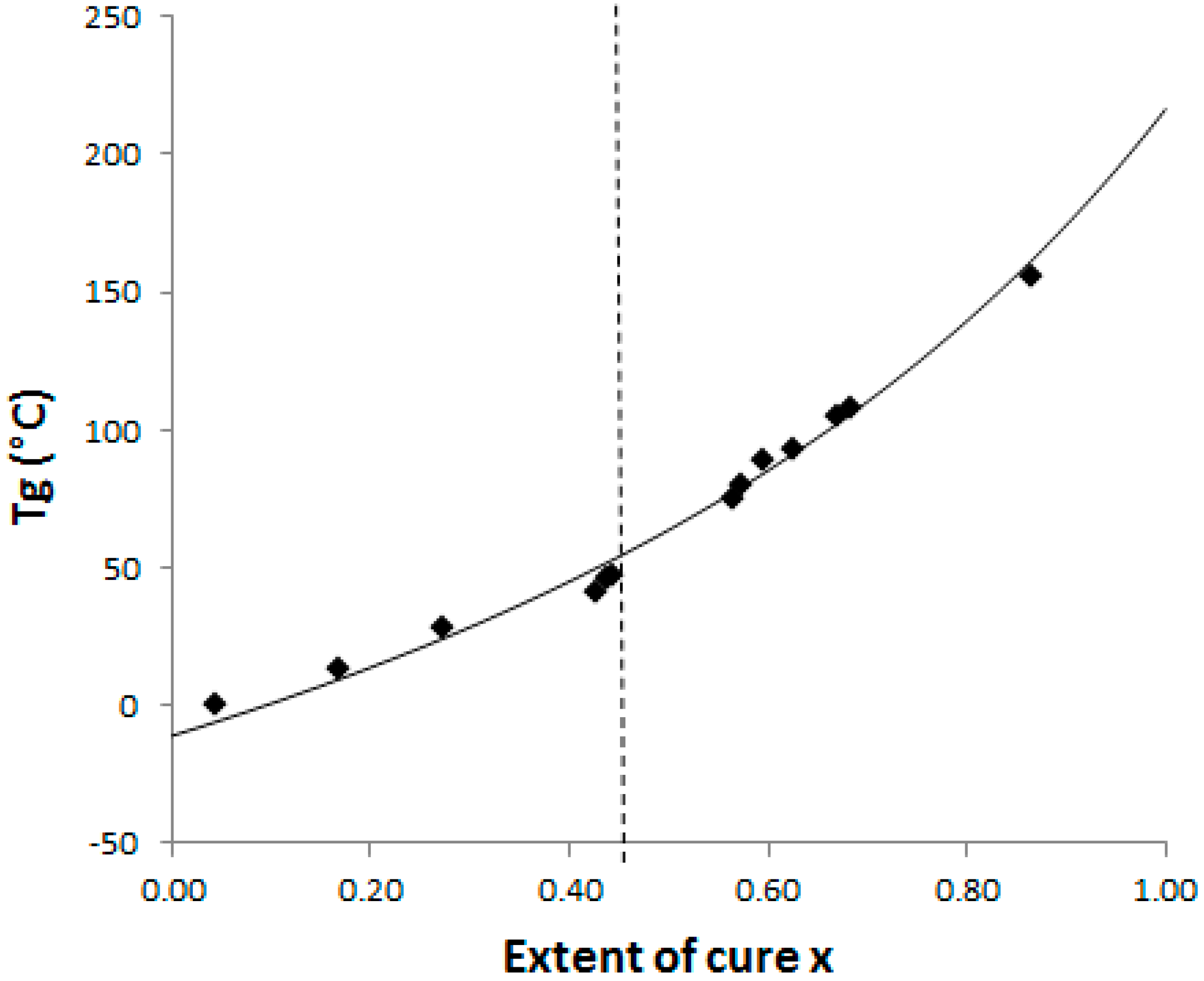
The Di Benedetto dependence is plotted for the neat system (
Figure 4) from experiments done in isothermal conditions at 130 °C. In such a complex (initially off-stoichiometric and excess of epoxy) reactive system, it is quite difficult to estimate when a total conversion is achieved. After 2 h at 180 °C, the system displays some residual heat of reaction, meaning that residual epoxies can react on themselves. The Di Benedetto dependence was then built thanks to a point corresponding to a not completely built epoxy network, as done by Jordan and al.: the point (x = 1; T
g∞) is replaced by an intermediate (x
max < 1; T
gmax) [
27]. For the same reasons, it was impossible to experimentally determine a value of ΔC
p∞. Thus the parameter λ was determined by fitting the Di Benedetto curve to the experimental points.
The values of T
g and ΔC
p for x = 0 (measured) and x = 1 (calculated) are listed in
Table 1. Very close values for ΔC
p0 were already found by Girard-Reydet and al. [
28] and Eloundou and al. [
29] (ΔC
p0 = 0.54 J·g
−1·K
−1). Nevertheless, ΔC
p∞ values differ (as the authors reported 0.19 J·g
−1·K
−1). Such discrepancies could be attributed to the stoichiometric ratio, r = 0.9 considered in this work, introducing some dangling chain ends.
To complete the CTT phase diagram, the conversion of the system at gelation, x
gel has to be known. It can be calculated thanks to the ‘Flory–Stockmayer’ equation [
30]:
where f
e and f
a are the epoxy and amine functionalities respectively, r is the initial stoichiometric ratio. This calculation assumes that all the functions own the same reactivity and that no intermolecular reaction occurs. The nominal values of 2 and 3 were considered for the functionalities of the epoxy monomers (DGEBF and TGmAP respectively), and average epoxy functionality was calculated considering the mass distribution. Keeping in mind that the system is amine-deficient, x
gel was found equal to 0.45. This value was experimentally confirmed since the lower conversion for which insoluble species were evidenced in tetrahydrofurane (THF) was found to be 0.45 (complete solubility was observed for a conversion of 0.43). It will be considered that the conversion at gelation is not significantly affected by the presence of PES, as observed by Yu and al for anhydride-cured epoxy/PES blends with high PES contents (up to 20 wt %) if compared to our study [
31].
As our aim is to know when phase changes (gelation and vitrification) occur during the curing schedules, it is now important to estimate the T
g value at the gel point, so called
gelT
g. With the help of this CTT phase diagram (
Figure 4),
gelT
g was found to be 55 °C. Based on
gelT
g value and to ensure that the systems can reach gelation in a reasonable delay, the following curing temperatures have been considered: 90 °C, 130 °C and 150 °C. According to these temperatures, no vitrification effect will be suffered and morphologies will be definitively defined at gelation. Gel times have also been measured in these isothermal conditions by rheology under a multi-frequency mode using the tanδ cross-over (see
Supporting Information, Figure S3) [
29]. The apparent activation energy of gelation E
a could be determined since the gel time follows an Arrhenius’ law in respect to the temperature. A value of E
a = 64 kJ·mol
−1 is found and is in agreement with results previously reported in the literature [
32]. The gelation time and T
gx for the three different pre-curing temperatures are listed in
Table 2.
For all the resulting networks, the glass transition temperature, T
gx is higher than
gelT
g, ensuring that gelation occurs during the pre-curing step. It could also be noted that for a pre-curing at 150 °C, vitrification is also reached but at longer times than gelation. These curing conditions will be applied both to neat epoxy-amine system and PES-based blends, followed by a post-curing at 180 °C during 2 h (see
Supporting Information, Figure S4). It is then expected that these different curing schedules will bring important modifications in the developed morphologies for the PES-based systems.
3.2. Morphologies of the Epoxy/PES Blends
After curing, all the blends are transparent and brown-colored, i.e., the neat and the PES-modified epoxy-amine networks cannot be visually differentiated (see
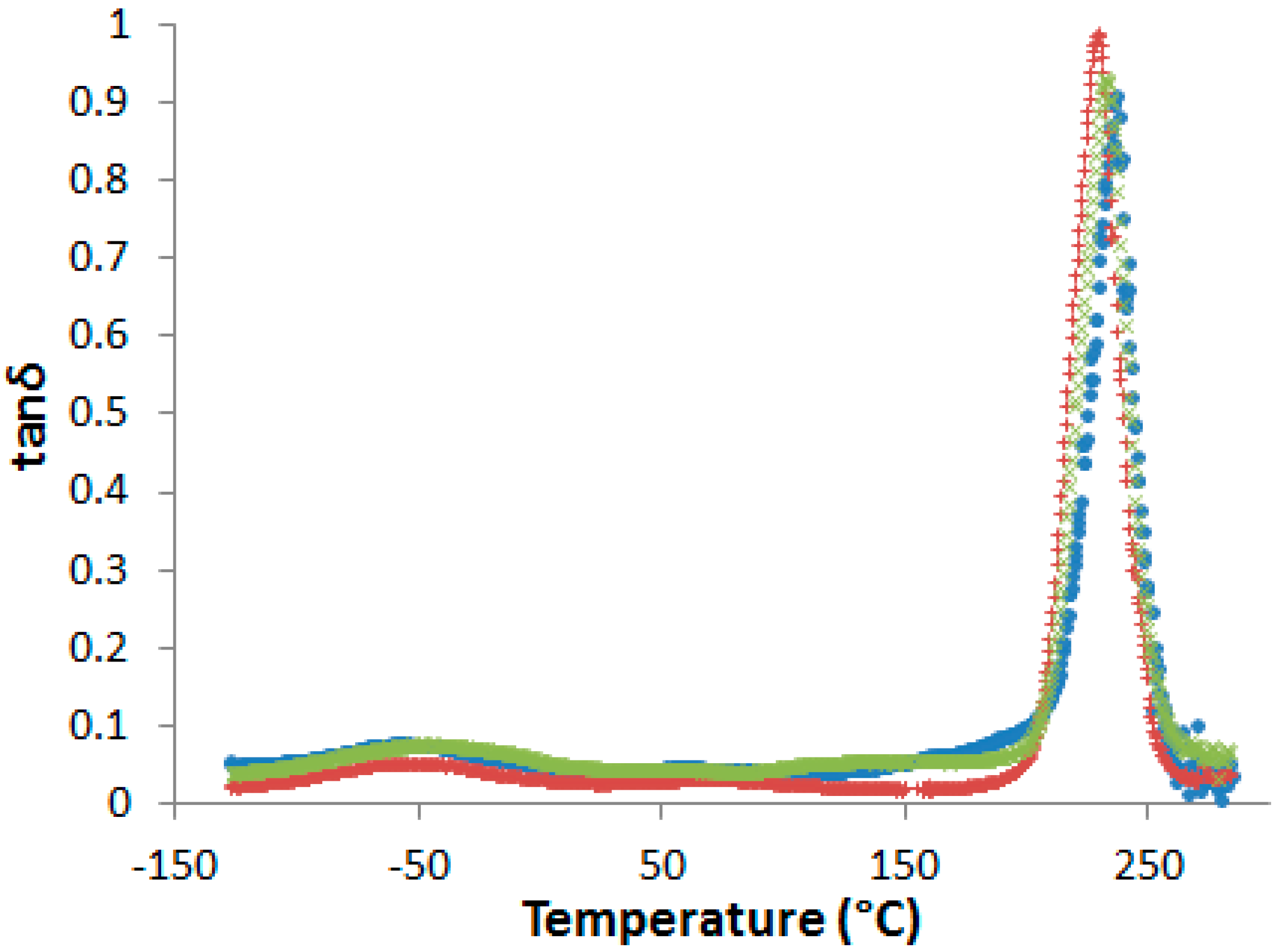
Supporting Information, Figure S5). No evidence of phase separation in cured samples could be evidenced by DSC or by dynamic mechanical analyses (DMA) (
Figure 5).
In fact, even if a slight decrease of the α-peak position, T
α, is observed in presence of PES, no other relaxation related to rich-PES separated phase can be detected on dynamic mechanical spectra. However, some authors observed a unique tanδ peak in phase-separated epoxy-PES blends due to the close values of the glass transition temperatures of the cured epoxy network and PES [
33,
34]. As a consequence, dynamic mechanical spectroscopy is not sufficient to state on phase separation.
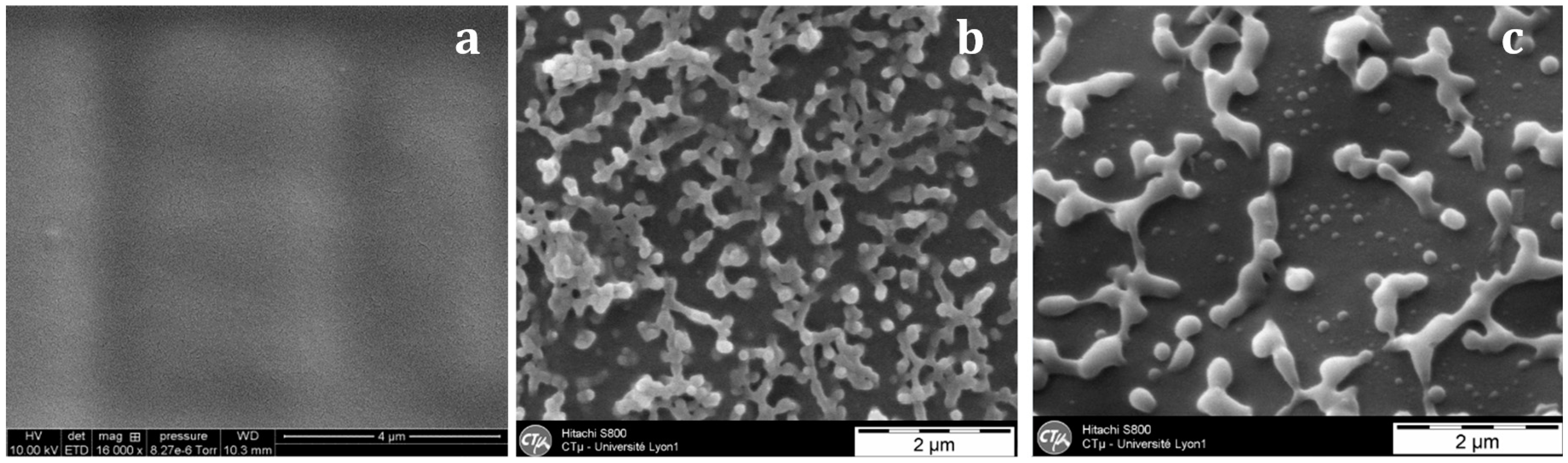
With the nf-PES-based blend, the pre-curing temperature strongly influences the final morphology (
Figure 6). By Scanning Electron Microscopy (SEM), it is not possible to evidence separated domains for blends cured at 90 °C. Besides, vermicular morphologies are observed for blends cured at 130 °C and 150 °C. For these vermicular morphologies, the average width of the PES-rich domains is about 250 nm for curing at 130 °C against 400 nm for curing at 150 °C. It has to be noticed that the samples were expected to be opaque or translucent and not transparent since the separated PES-rich domains having such dimensions use to scatter light. However, Inoue reported in a similar system that the PES and the epoxy/amine matrix own close refractive index, explaining why even microphase-separated materials can be transparent [
20]. Transmission Electron Microscopy (TEM) clearly evidences the PES-rich phases (
Figure 7). Furthermore, it can be seen that the vermicular morphologies developed in the nf-PES-based blends pre-cured at 130 °C and 150 °C do not lead to a PES-continuous phase, i.e., that PES nanostructures do not percolate. Vermicular morphology was also obtained by Akay and Cracknell for an epoxy network modified with a similar PES; they found continuity of the PES-rich phase at higher PES contents, i.e., 30 wt % [
35].
For a better understanding of the mechanisms leading to a given morphology, it is important to know the value of the critical composition, Φ
TP,crit, for phase-inversion. The ‘Flory-Huggins’ equation allows to estimate this composition [
3,
4].
Assuming a Schulz-Flory’ distribution of molar masses of the TP:
where V
TP and V
TS are the molar volumes of the thermoplastic polymer and the thermoset precursors respectively. h is the degree of polymerization of the TP.
This critical composition is calculated for the uncured epoxy-amine system. However, it has been showed that it does not suffer a significant change during curing of the TS system and as a consequence, it will be assumed to stay constant during all the curing process [
36]. The phase diagram is then shifted only along the temperature axis during curing, due to the loss of miscibility between PES and the growing epoxy oligomers (
Figure 1). Volumic fraction φ
TP,crit is found to be equal to 9%, i.e., when converted in weight fraction, φ
TP,crit = 11 wt %. Keeping in mind that PES has been initially introduced at ω
0 = 13 wt %, it implies that the modified system is expected to go through a mechanism of spinodal decomposition [
2,
4,
37].
As already mentioned, for nf-PES based blends, the higher T
cure, the bigger the PES-rich separated domains are. As the phase separation proceeds via spinodal decomposition, co-continuous phases are formed in the early stages of the phase separation. The final morphology will depend on the competition between the thermodynamics of phase separation and the polymerization reaction rate. It has been shown that this competition usually leads to nodular morphologies for sufficiently long times before gelation, in order to decrease interfacial energy between phases [
5,
19]. In this study, the PES content is too low to induce co-continuous morphologies. The increase of the PES-rich phase size with T
precure can be explained in terms of molecular mobility when phase separation occurs. It can be argued that the conversion at which the phase separation takes place decreases when T
precure is increased since the LCST will be reached at a lower conversion (
Figure 1). Thus the nodules which are formed in a less viscous matrix for high T
precure (since both conversion and viscosity are lower for higher T
precure) have a higher tendency to coalesce and so get larger.
Another explanation concerning the vermicular structure could arise from the vitrification of the PES-separated phase during curing. In fact, after the phase separation, the two phases get purified, the matrix becoming richer and richer in epoxy-amine and as a consequence, the PES-rich separated domains richer and richer in PES. As a consequence, the glass transition temperature of the PES-rich domain increases since the PES has a much higher T
g than the epoxy-system before gelation [
25]. It can be reasonably anticipated that the PES-rich phase vitrifies more easily for low T
precure temperatures, freezing the vermicular morphology. Unfortunately, no experimental evidence was found to validate this hypothesis even by using DSC. In fact, the glass transition of the PES-rich phase is always hidden by the residual enthalpy peak of the reaction of the remaining reactive species. Vitrification of the epoxy-rich matrix does not play a role on the morphology development as it occurs only for curing at 150 °C and a long time after gelation (
Table 2). By the way, PES-rich phase owning a higher T
g than the epoxy-rich phase, would in all cases see its morphology fixed by its own vitrification. It is also known that the surface tension of an epoxy-amine system (prior to gelation) which is varying with conversion during curing and temperature can also interfere on the interfacial interactions between TS prepolymers and PES [
38].
The –OH end groups on the f-PES induce huge differences on the resulting morphologies of f-PES-based blends. As for the nf-PES, no phase separation could be evidenced by SEM for a blend cured at 90 °C (
Figure 8a). For higher T
precure, occurrence of phase separation is clearly evidenced, leading to nanometer-scale PES-rich domains. For curing temperatures from 130 °C to 180 °C, the higher T
precure, the bigger the nodular domains: average diameter is about 40–50 nm for 130 °C (
Figure 8b), and about 150 nm for 150 °C (
Figure 8c) and even bigger than 200 nm for 180 °C (no pre-cure,
Figure 8d).
Comparing resulting morphologies in the two PES-based blends, it can be seen that for both PES-based blends, the size of the dispersed domains increases with the pre-curing temperature, but more important is the fact that for a same T
precure, the average size of the PES-separated domains is quite lower with f-PES than with nf-PES. As the two considered PES have quite similar molar masses, it can be considered that the two modified systems display the same φ
TP,crit value. Thus, such a huge difference between morphologies can only be explained by the presence of the phenoxy chain-ends of the f-PES. It is well known that phenoxy and oxirane are able to react to form an ether linkage [
1,
35]. Such a chemical reaction between f-PES and epoxy prepolymers can be expected in our system. Some phenoxy functions brought by the f-PES are then able to react with epoxy prepolymers to form an epoxy-PES block copolymer at the interface. For instance, Macosko and al. showed that compatibilizing a blend of two immiscible polymers inhibits the coalescence of the dispersed phase in the matrix, leading to quite finer morphologies [
39]. Some authors also observed noticeable differences between non-reactive and reactive TP modifiers in a TS system. Yoon and al. managed to keep a NH
2-terminated polysulfone modified epoxy-amine system homogeneous after curing whereas the non-reactive one leads to phase separation [
12]. Finer morphologies were also obtained with the phase-separated reactive polysulfone if compared with the non-reactive one. A NH
2-terminated PES was also successfully used by Kim and al. to greatly reduce the size of the separated domains of epoxy/PES blend compared with a non-reactive PES [
8]. In this work, a similar phenomenon could result from the in-situ formation of an epoxy-PES block copolymer. One can wonder if the epoxy-PES copolymer formation takes place during the curing of the blends or if it already happened during the PES-dissolution step at 130 °C (See “Samples Preparation” part). Epoxy-PES mixtures obtained after PES-dissolution were analyzed via
1H Nuclear Magnetic Resonance (
1H NMR) to evidence some differences between nf-PES and f-PES based blends. No concluding evidence was obtained due to the low amount of in-situ formed species.
Coming back to the behavior of the samples pre-cured at 90 °C. TEM analyses were carried out on these blends (see
Supporting Information, Figure S6). Some PES-rich domains having an average diameter of about 20 nm are evidenced in both nf-PES and f-PES based blends, whereas a homogeneous structure is evidenced at nanoscale for the neat network. It proves that the PES-modified systems undergo phase-separation even when cured at 90 °C. It means that the two modified systems exhibit phase separation for all the curing temperatures considered in this study. Additionally, it can be stated that before gelation of the epoxy-amine system, the LCST is lower than 90 °C.
3.3. Fracture Properties
It is now of great interest to investigate the effect of the developed morphologies exposed above on the matrix crack resistance of the PES modified systems. The possible toughening effects cannot be related to a PES-induced network plasticization (
Figure 5). In fact, as reported before, only one relaxation peak associated to the glass transition of the epoxy-amine network is observed for the PES-based blends whereas it is established that the morphologies are biphasic. As two different α relaxation peaks corresponding to two phases in presence should be observed, it can be assumed that in our case the proximity of the T
α of PES and neat epoxy-amine network does not allow to clearly state on the existence of two phases.
Pure PES exhibiting a slightly higher Tα (239 °C) than the one of the neat epoxy-amine network (236 °C), it should then be expected that the Tα of PES-based blends is higher than the one of the neat epoxy-amine network. The lower Tα of the epoxy-amine matrix in the presence of PES could be related to a lower TS conversion. Taking into account the structures of the chemicals, it would not be surprising that the amine has more affinity than the epoxy prepolymers with the PES and is then retained longer in the PES-rich phase during purification even at high TS conversions: stoichiometry ratio would then be altered and the final properties modified. However, such a small difference in Tα after post-curing demonstrates that the epoxy-rich phase is only slightly modified, i.e., the intrinsic properties of the matrix such as crack resistance will not be different from neat epoxy-amine networks.
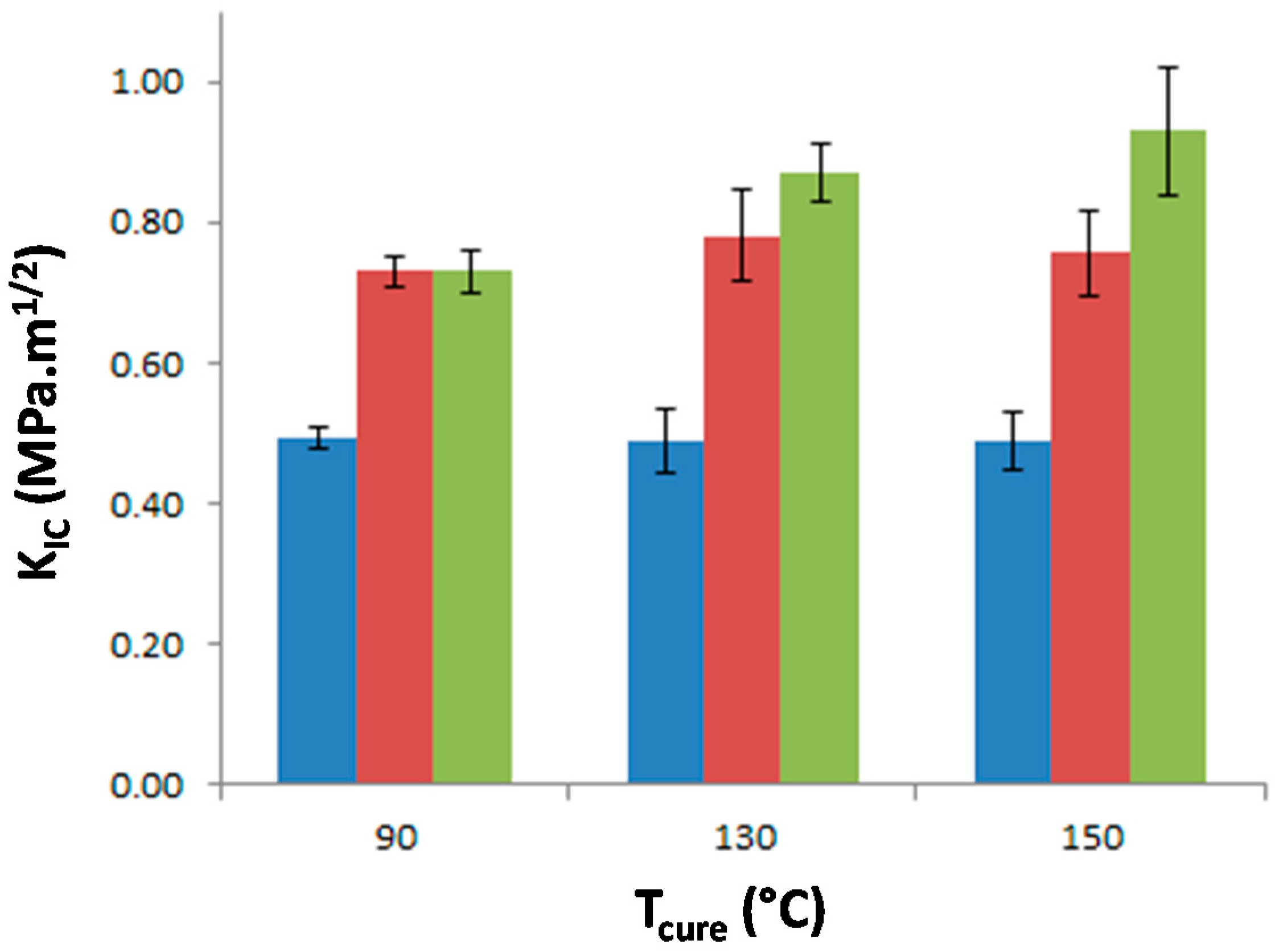
The stress intensity factor, K
IC, of the materials was measured as a function of T
precure to investigate the effect of the morphology on the matrix crack propagation resistance (
Figure 9). The neat epoxy-amine network appears very brittle as K
IC is equal to 0.49 MPa·m
1/2 whatever T
precure which confirms that no significant effect on the network architecture is undergone. The incorporation of either f-PES or nf-PES dramatically modifies the fracture toughness for all T
precure. Thus the PES-induced toughness improvement can solely be attributed to the generated morphologies since all epoxy networks exhibit similar architecture. The introduction of f-PES leads to an increase of K
IC up to 0.73 MPa·m
1/2, i.e., 50% higher than for neat epoxy-amine network. For these f-PES-based blends, it is worth noting that the improvement in toughness does not depend on T
precure whereas the curing conditions have an impact on the final PES-rich domains size. In fact, considering spherical particles with an average diameter of 20 nm for the 90 °C-cured blend and 150 nm for the 150 °C-cured blend, the matrix-particles interface is about 50 times larger as T
precure is increased from 90 °C to 150 °C.
Very different results are obtained for blends based on nf-PES: the higher Tprecure, the higher the resulting KIC.
SEM micrographs of the non-chemically treated fractured samples allow identification of the toughening mechanisms. Neat epoxy-amine network presents a mirror-like surface (
Figure 10a) whereas crack deviation is observed on PES-based blends (
Figure 10b). Yee and al. proposed a list of potential TP-based toughening mechanisms, in particular for epoxies [
5]. In our case the PES-separated domains are not ductile at room temperature since their glass transition temperature is close to 230 °C.
Some toughening mechanisms are not possible in our systems: for instance TP particles bridging is only related to rubbery particles, and TP particles transformation implies semi-crystalline TP whereas PES is amorphous. Shear yielding could contribute to a large extent of the toughening effect. In fact, since epoxy-amine network and PS own different moduli (compression modulus for neat epoxy-amine network: 2.8 GPa, and PES: 1.6 GPa), stress concentration field around PES-rich domains superimpose which can induce yielding in a large volume of matter ahead from the crack tip. Kishi and al found that incorporation of PES in a DGEBA-DDS network allows a large increase of its ability to shear-yielding, evidenced by optical microscopic observations of compression samples under cross-polarized light [
33]. An enhanced interfacial adhesion could have been expected with f-PES thanks to reaction between –OH chain-ends and epoxy-prepolymers [
38]. However SEM micrographs of surface fracture of PES-based blends under a high magnification reveal that matrix/particle debonding occurred with both PES (
Figure 11), i.e., interfacial adhesion is low. As a consequence crack pinning and bowing which both require high levels of interfacial adhesion are not expected to occur. Oppositely, multiple crack generation in the matrix, often observed in highly crosslinked matrices reinforced by rigid particles, should take place. Eventually, crack-path deflection is also one of the most probable toughening mechanisms.
For f-PES-based blends, the fact that toughness is independent of the PES-rich particle size could be explained by a “plateau effect”. In fact, for such nano-size morphologies, the particles are so numerous that their size does not influence any more the reinforcement extent. The two types of PES lead to a similar toughening effect for blends cured at 90 °C, which seems obvious according to the fact that average particles sizes are similar, as well as the matrix/PES interfacial adhesion. Higher values of K
IC obtained with vermicular morphologies in nf-PES-based blends can be explained by the fact that crack-path is much more altered than in the case of spherical-like domains-based morphologies. This phenomenon is highlighted by the large difference in roughness between the fractured surfaces (
Figure 11a,b). With such elongated PES-rich domains, it can also be possible that a part of these domains are broken during crack propagation. PES being quite tougher (K
IC = 2.1 MPa·m
1/2) than the epoxy-rich phase, toughening of the blend is again enhanced.
It is quite interesting to notice that these vermicular morphologies lead to a high toughening extent compared to other results on PES-reinforced epoxy networks reported in literature. In this study, an increase of 85% is obtained from neat epoxy network (K
IC = 0.49 MPa·m
1/2) to nf-PES-based blends exhibiting vermicular morphologies (K
IC = 0.90 MPa·m
1/2). To our knowledge, for similar PES contents, i.e., below 15–20 wt %, such an improvement of highly crosslinked epoxies was not already reported. For instance, Bejoy and al. managed to improve by 50% K
IC of DGEBA-DDS network thanks to a PES “P3500” from Gharda Chemicals [
40]. Bucknall and Partridge also reached 50% improvement rate on networks based on tri and tetra-functional epoxy pre-polymers cured either by dicyanodiamide or DDS wih a PES “100P” from Victrex [
41]. It is also surprising that some authors who worked with the same phenoxy-terminated PES used in this study (5003P from Sumitomo) did not observe any toughening enhancement on epoxy networks. For example, Kishi and al. considered DGEBA-DDS networks containing up to 15 wt % PES [
34], as well as McKinnon and al. who worked on a triglycidyl para-amino phenol-DDS network which was not significantly toughen for PES contents ≤15 wt % [
10]. Of course it has to be kept in mind that toughening improvement depends on several parameters which make direct comparison quite difficult: TP molar mass, curing schedule, final crosslinking density of the TS matrix, etc.
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}