Tuning the Hydrophobicity of a Hydrogel Using Self-Assembled Domains of Polymer Cross-Linkers



Abstract
:1. Introduction
2. Materials and Methods
2.1. Fluorescent Analysis of Self-Assembling of Polymer Cross-Linkers in a Solution
2.2. Hydrogel Preparation
2.3. Hydrogel Characterization
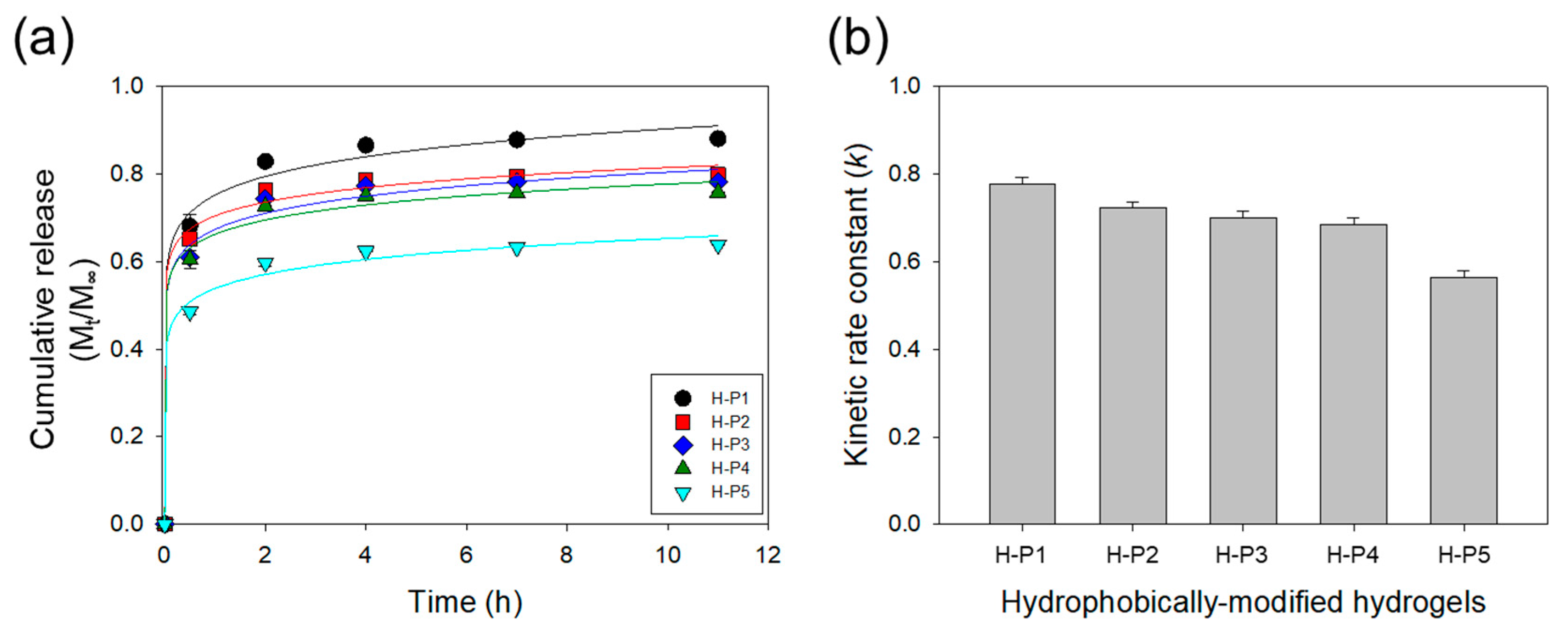
3. Results and Discussion
3.1. Analysis of Self-Assembling of Polymer Cross-Linkers in a Solution
3.2. Effects of the Hydrophobic Domains Incorporated into a Hydrogel
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Drury, J.L.; Mooney, D.J. Hydrogels for tissue engineering: scaffold design variables and applications. Biomaterials 2003, 24, 4337–4351. [Google Scholar] [CrossRef]
- Peppas, N.A.; Hilt, J.Z.; Khademhosseini, A.; Langer, R. Hydrogels in biology and medicine: from molecular principles to bionanotechnology. Adv. Mater. 2006, 18, 1345–1360. [Google Scholar] [CrossRef]
- Chan, V.; Park, K.; Collens, M.B.; Kong, H.; Saif, T.A.; Bashir, R. Development of Miniaturized Walking Biological Machines. Sci. Rep. 2012, 2, 857. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Arotzena, O.; Meier, J.G.; Del Amo, C.; García-Aznar, J.M. Characterization of Fibrin and Collagen Gels for Engineering Wound Healing Models. Materials 2015, 8, 1636–1651. [Google Scholar] [CrossRef] [Green Version]
- Boffito, M.; Sartori, S.; Ciardelli, G. Polymeric scaffolds for cardiac tissue engineering: requirements and fabrication technologies. Polym. Int. 2014, 63, 2–11. [Google Scholar] [CrossRef]
- Jeong, J.H.; Chan, V.; Cha, C.; Zorlutuna, P.; Dyck, C.; Hsia, K.J.; Bashir, R.; Kong, H. “Living” microvascular stamp for patterning of functional neovessels; orchestrated control of matrix property and geometry. Adv. Mater. 2012, 24, 58–63. [Google Scholar] [CrossRef] [PubMed]
- Thakur, V.K.; Thakur, M.K. Recent advances in green hydrogels from lignin: a review. Int. J. Biol. Macromol. 2015, 72, 834–847. [Google Scholar] [CrossRef]
- Vedadghavami, A.; Minooei, F.; Mohammadi, M.H.; Khetani, S.; Rezaei Kolahchi, A.; Mashayekhan, S.; Sanati-Nezhad, A. Manufacturing of hydrogel biomaterials with controlled mechanical properties for tissue engineering applications. Acta Biomater. 2017, 62, 42–63. [Google Scholar] [CrossRef]
- Wang, Y.; Ma, M.; Wang, J.; Zhang, W.; Lu, W.; Gao, Y.; Zhang, B.; Guo, Y. Development of a Photo-Crosslinking, Biodegradable GelMA/PEGDA Hydrogel for Guided Bone Regeneration Materials. Materials 2018, 11, 1345. [Google Scholar] [CrossRef]
- Cha, C.; Kim, E.S.; Kim, I.W.; Kong, H. Integrative design of a poly(ethylene glycol)-poly(propylene glycol)-alginate hydrogel to control three dimensional biomineralization. Biomaterials 2011, 32, 2695–2703. [Google Scholar] [CrossRef]
- Vo, T.N.; Ekenseair, A.K.; Spicer, P.P.; Watson, B.M.; Tzouanas, S.N.; Roh, T.T.; Mikos, A.G. In vitro and in vivo evaluation of self-mineralization and biocompatibility of injectable, dual-gelling hydrogels for bone tissue engineering. J. Control. Release 2015, 205, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Weiner, S.; Addadi, L. Design strategies in mineralized biological materials. J. Mater. Chem. 1997, 7, 689–702. [Google Scholar] [CrossRef]
- Gao, Y.; Koumoto, K. Bioinspired Ceramic Thin Film Processing: Present Status and Future Perspectives. Crystal Growth Des. 2005, 5, 1983–2017. [Google Scholar] [CrossRef]
- Tuncaboylu, D.C.; Argun, A.; Sahin, M.; Sari, M.; Okay, O. Structure optimization of self-healing hydrogels formed via hydrophobic interactions. Polymer 2012, 53, 5513. [Google Scholar] [CrossRef]
- Gao, Y.; Duan, L.; Guan, S.; Cheng, T.; Ren, X.; Wang, Y. The effect of hydrophobic alkyl chain length on the mechanical properties of latex particle hydrogels. RSC Adv. 2017, 7, 44673–44679. [Google Scholar] [CrossRef] [Green Version]
- Bogdanowicz, K.A.; Pirone, D.; Judit, P.R.; Ambrogi, V.; Reina, J.A.; Giamberini, M. In situ raman spectroscopy as a tool for structural insight into cation non-ionomeric polymer interactions during ion transport. Polymers 2018, 10, 416. [Google Scholar] [CrossRef]
- Chan, V.; Zorlutuna, P.; Jeong, J.H.; Kong, H.; Bashir, R. Three-dimensional photopatterning of hydrogels using stereolithography for long-term cell encapsulation. Lab Chip. 2010, 10, 2062–2070. [Google Scholar] [CrossRef] [PubMed]
- Bogdanowicz, K.A.; Rapsilber, G.A.; Reina, J.A.; Giamberini, M. Liquid crystalline polymeric wires for elective proton transport, part 1: Wires preparation. Polymer 2016, 92, 50–57. [Google Scholar] [CrossRef]
- Bogdanowicz, K.A.; Tylkowski, B.; Giamberini, M. Preparation and characterization of light-sensitive microcapsules based on a liquid crystalline polyester. Langmuir 2013, 29, 1601–1608. [Google Scholar] [CrossRef] [PubMed]
- Ritger, P.L.; Peppas, N.A. A simple equation for description of solute release I. Fickian and non-fickian release from non-swellable devices in the form of slabs, spheres, cylinders or discs. J. Control. Release 1987, 5, 23–36. [Google Scholar] [CrossRef]
- Kalyanasundaram, K.; Tomas, J.K. Environmental effects on vibronic band intensities in pyrene monomer fluorescence and their application in studies of micellar systems. J. Am. Chem. Soc. 1977, 99, 2039–2044. [Google Scholar] [CrossRef]
- Jeong, J.H.; Schmidt, J.J.; Cha, C.; Kong, H. Tuning responsiveness and structural integrity of pH responsive hydrogel using a poly(ethylene glycol) cross-linker. Soft Matter 2010, 6, 3930–3938. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Composition | Theoretical Pore-Size (Å) | Experimental Degree of Swelling | Contact Angle (°) | |
|---|---|---|---|---|---|
| 2PEGDA | 3PPGMA | ||||
| 1HMH-1 | 10.0 | 0.0 | 23.16 | 9.53 ± 0.21 | 44.30 ± 4.00 |
| HMH-2 | 9.5 | 0.5 | 23.47 | 8.85 ± 0.15 | 53.40 ± 0.96 |
| HMH-3 | 9.0 | 1.0 | 23.89 | 9.56 ± 0.26 | 54.84 ± 0.30 |
| HMH-4 | 8.0 | 2.0 | 24.85 | 9.55 ± 0.19 | 56.87 ± 1.05 |
| Sample | 3H-P1 | H-P2 | H-P3 | H-P4 | H-P5 |
|---|---|---|---|---|---|
| 1PEGDA | 20.0 | 19.85 | 19.7 | 19.5 | 18.0 |
| 2PPGMA | - | 0.15 | 0.3 | 0.5 | 2.0 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, H.-J.; Cho, S.; Oh, S.J.; Shin, S.G.; Ryu, H.W.; Jeong, J.H. Tuning the Hydrophobicity of a Hydrogel Using Self-Assembled Domains of Polymer Cross-Linkers. Materials 2019, 12, 1635. https://doi.org/10.3390/ma12101635
Kim H-J, Cho S, Oh SJ, Shin SG, Ryu HW, Jeong JH. Tuning the Hydrophobicity of a Hydrogel Using Self-Assembled Domains of Polymer Cross-Linkers. Materials. 2019; 12(10):1635. https://doi.org/10.3390/ma12101635
Chicago/Turabian StyleKim, Hee-Jin, Sungwoo Cho, Seung Joo Oh, Sung Gyu Shin, Hee Wook Ryu, and Jae Hyun Jeong. 2019. "Tuning the Hydrophobicity of a Hydrogel Using Self-Assembled Domains of Polymer Cross-Linkers" Materials 12, no. 10: 1635. https://doi.org/10.3390/ma12101635
APA StyleKim, H. -J., Cho, S., Oh, S. J., Shin, S. G., Ryu, H. W., & Jeong, J. H. (2019). Tuning the Hydrophobicity of a Hydrogel Using Self-Assembled Domains of Polymer Cross-Linkers. Materials, 12(10), 1635. https://doi.org/10.3390/ma12101635