Corrosion Resistance and Apatite-Forming Ability of Composite Coatings formed on Mg–Al–Zn–Ca Alloys


Abstract
:
1. Introduction
2. Materials and Methods
2.1. Specimen Preparation
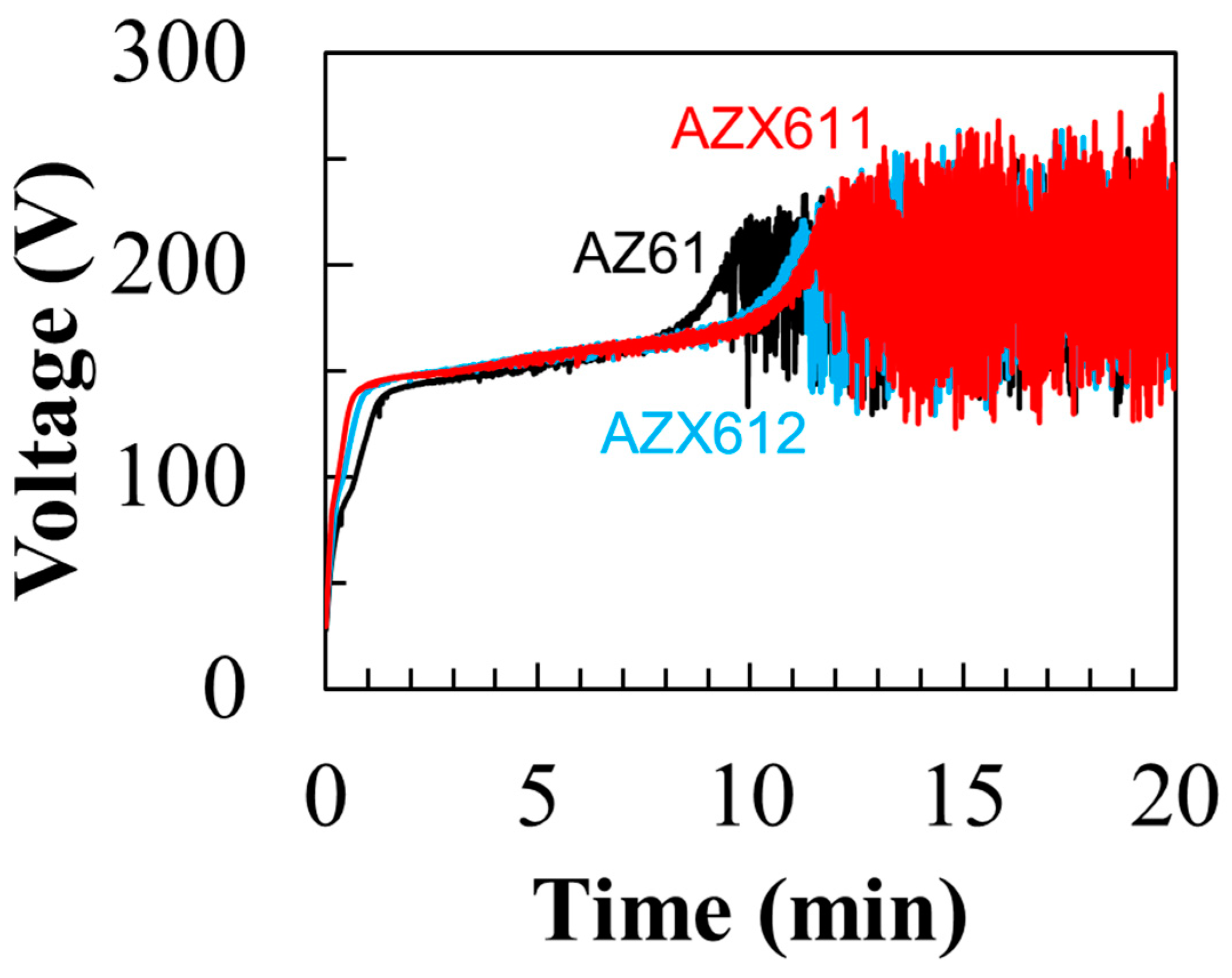
2.2. Plasma Electrolytic Oxidation
2.3. Electrochemical Corrosion
2.4. Apatite-Forming Ability Test
2.5. Surface Analyses
3. Results
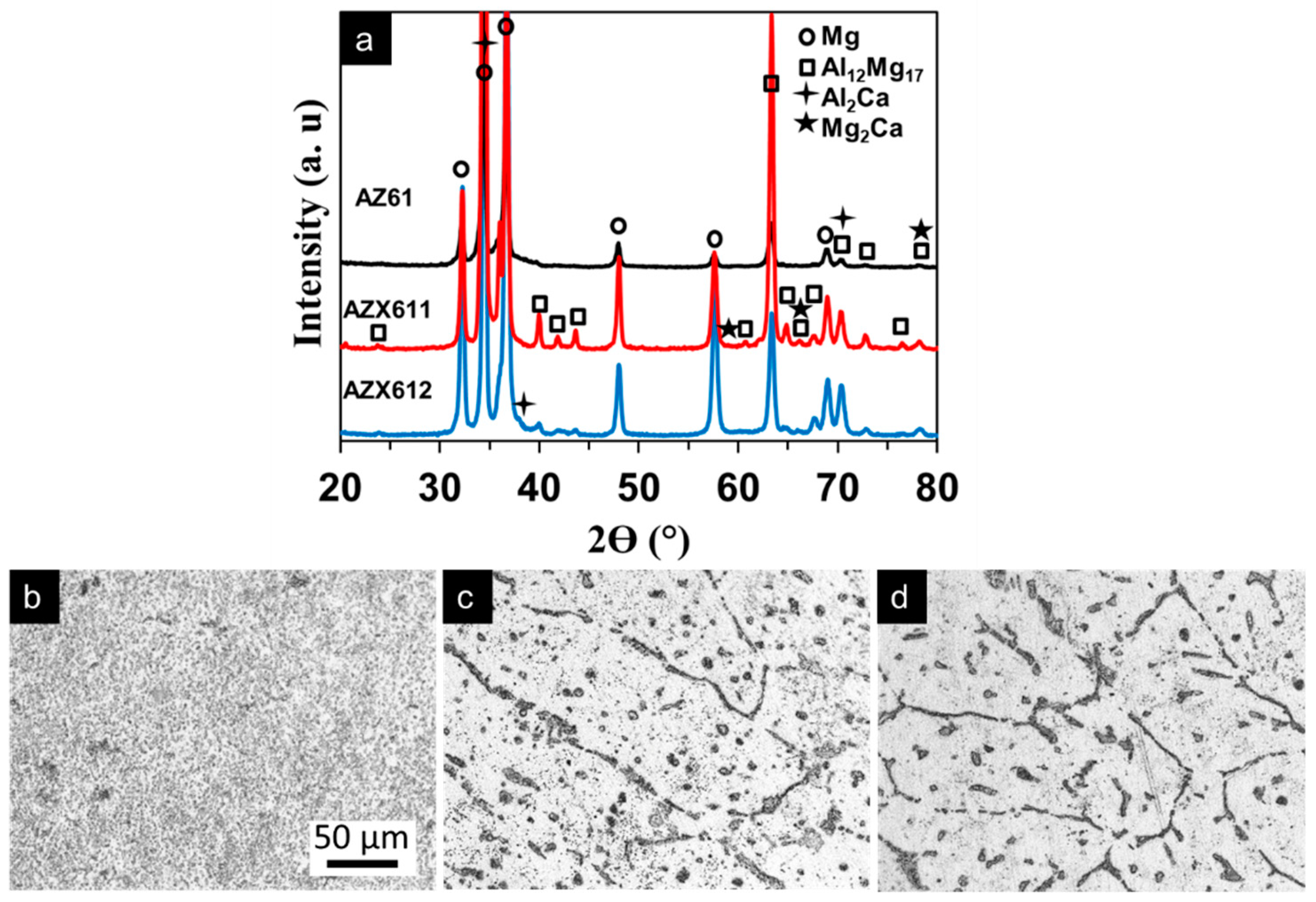
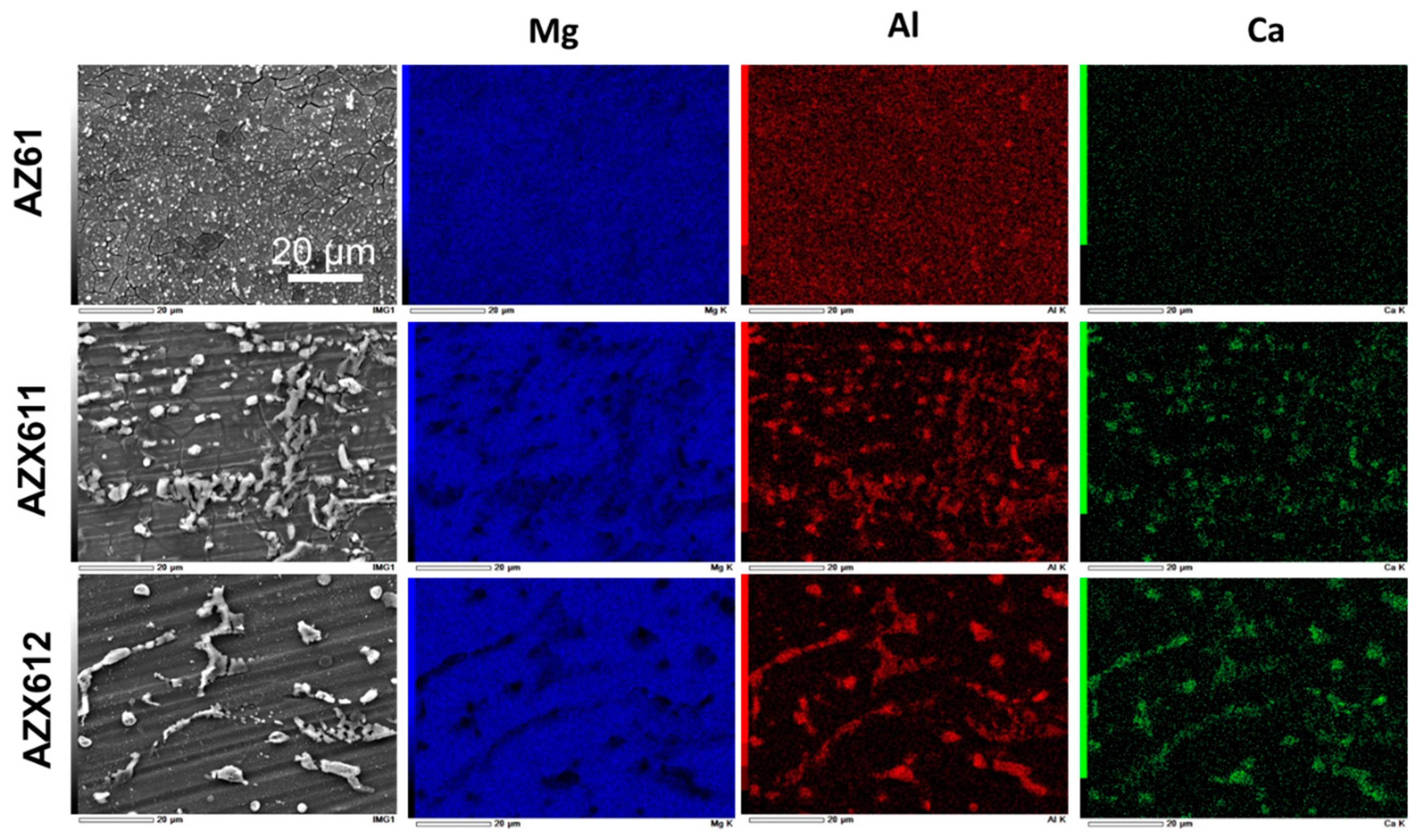
3.1. Alloy Microstructure
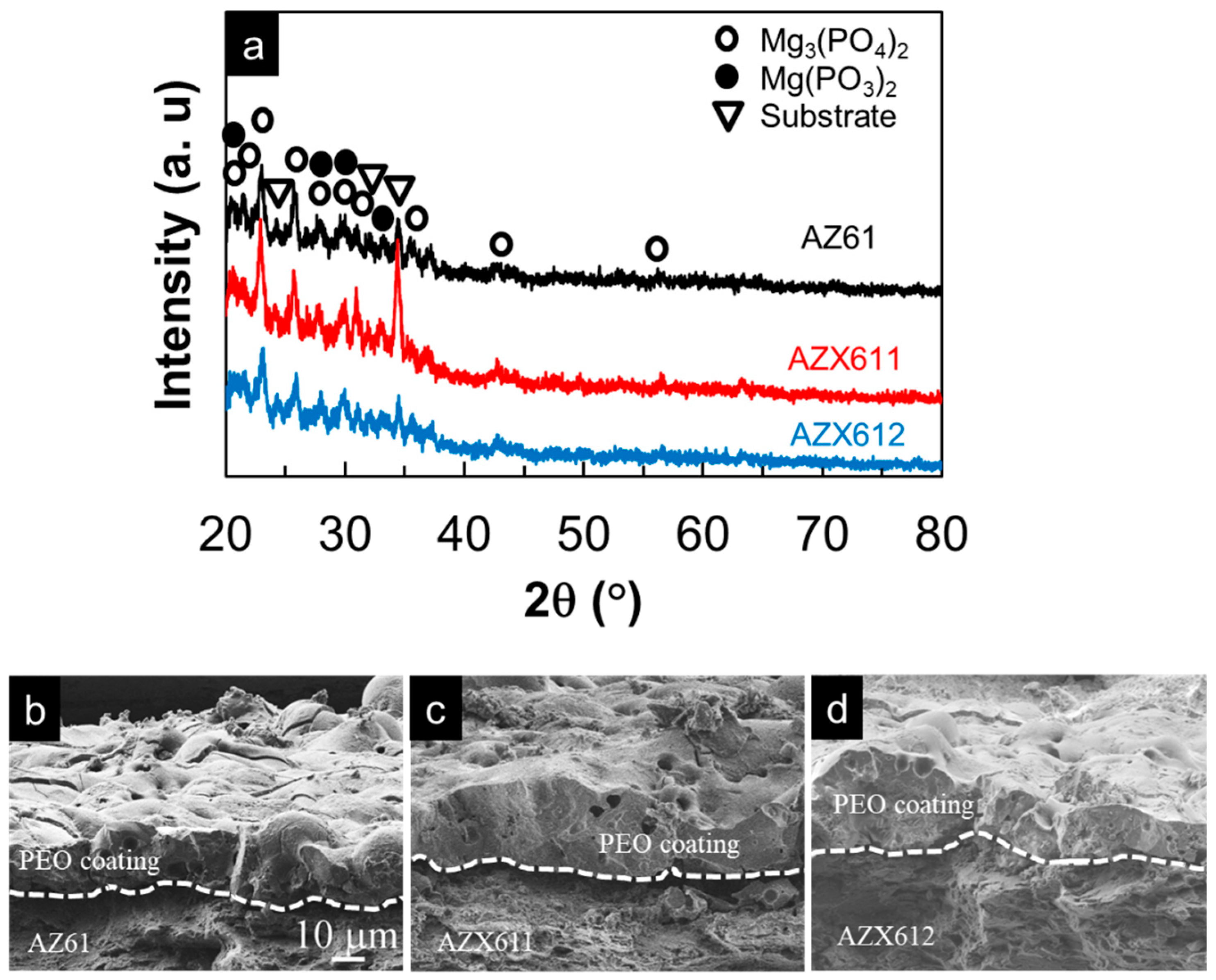
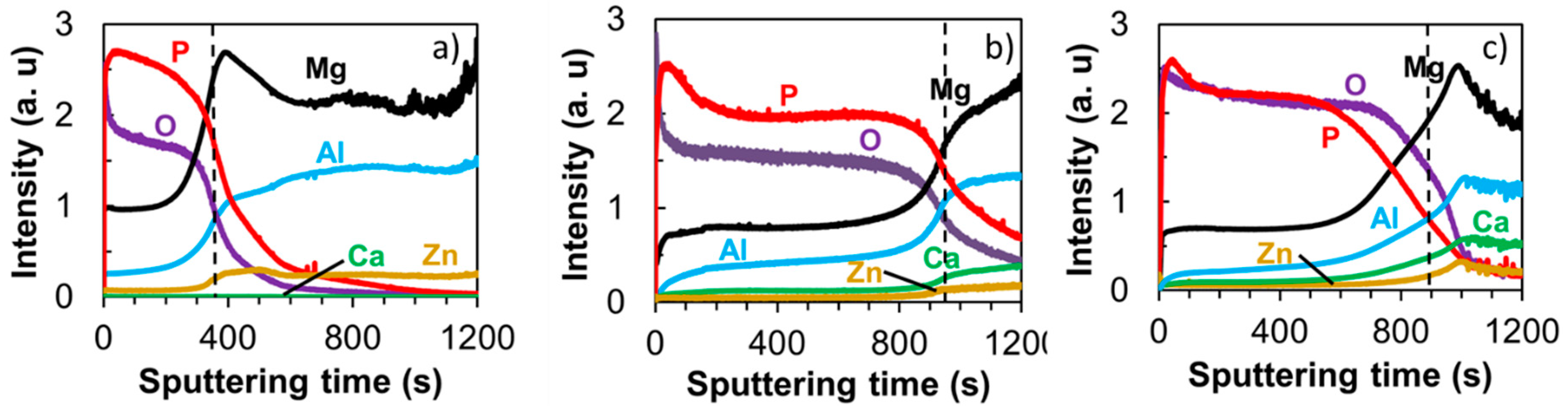
3.2. PEO-Coating Structure and Composition
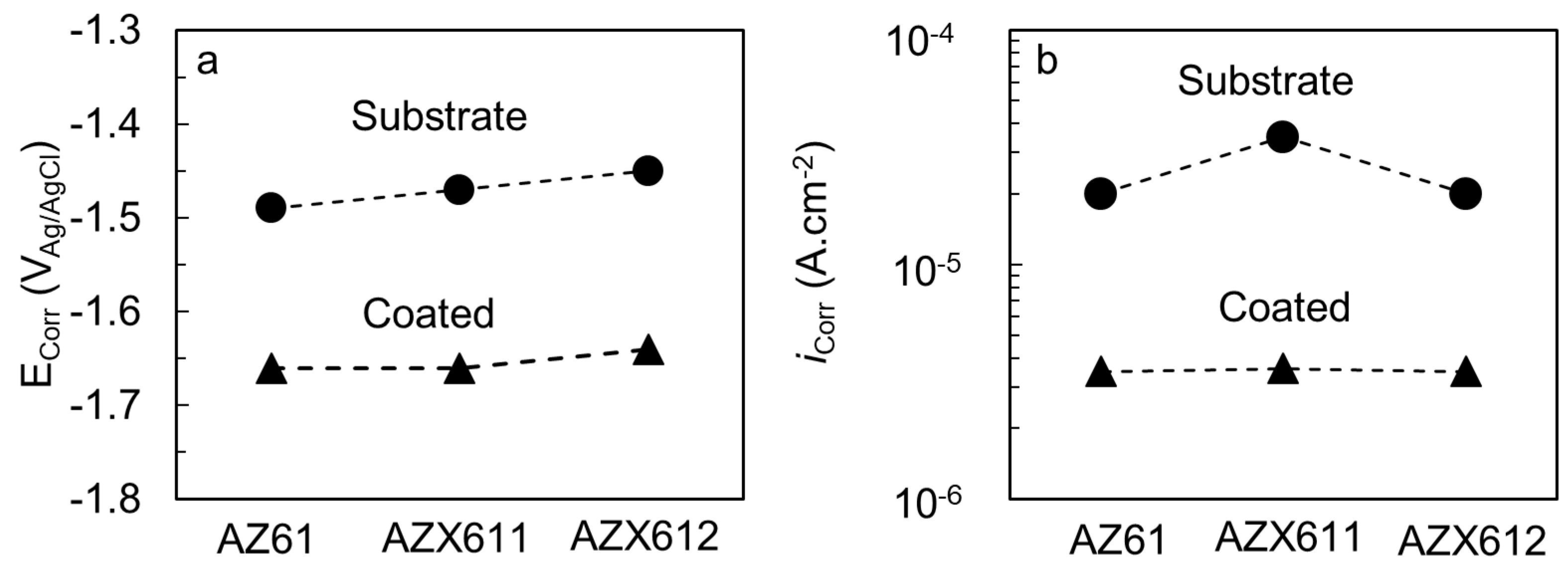
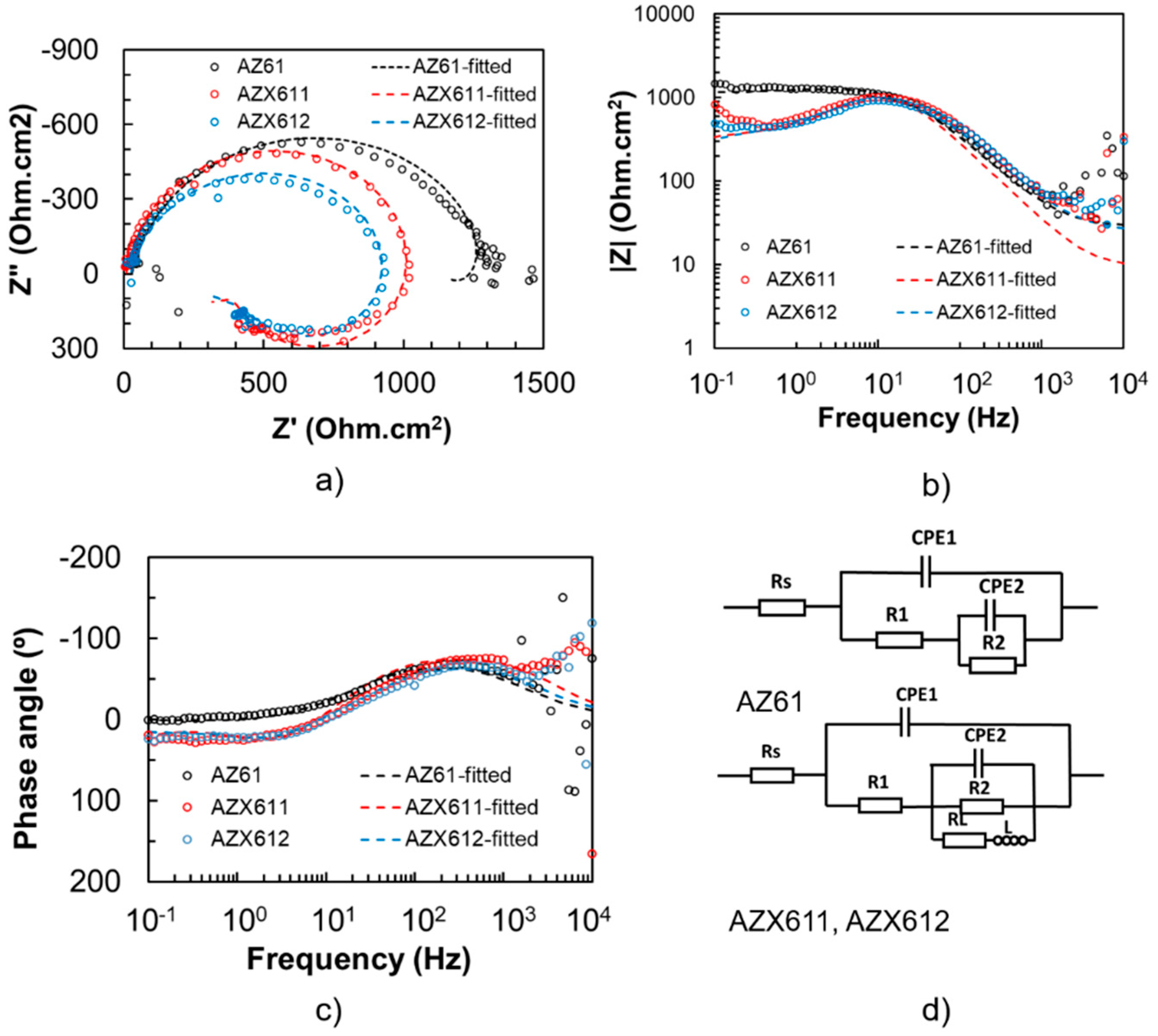
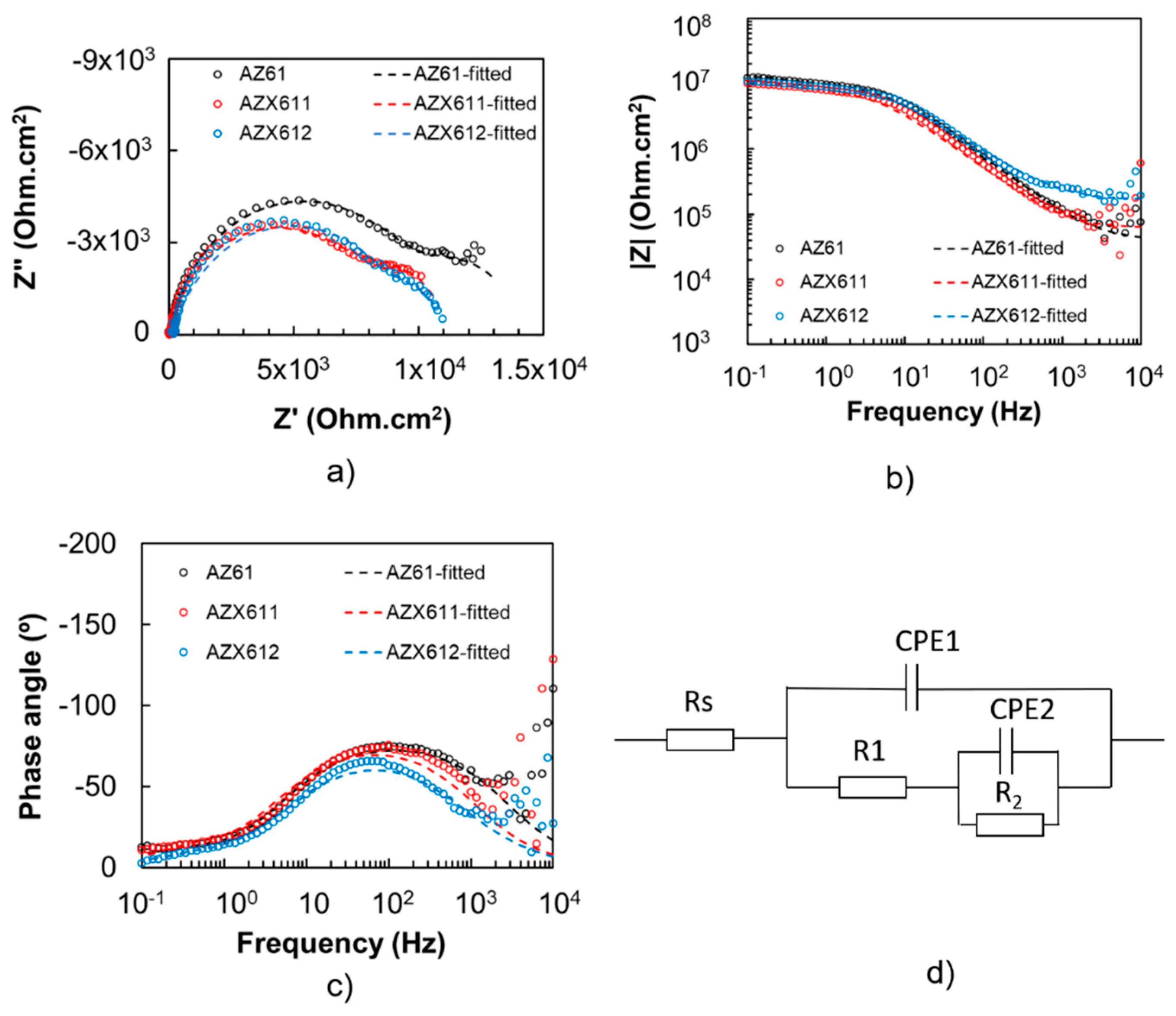
3.3. Electrochemical Corrosion
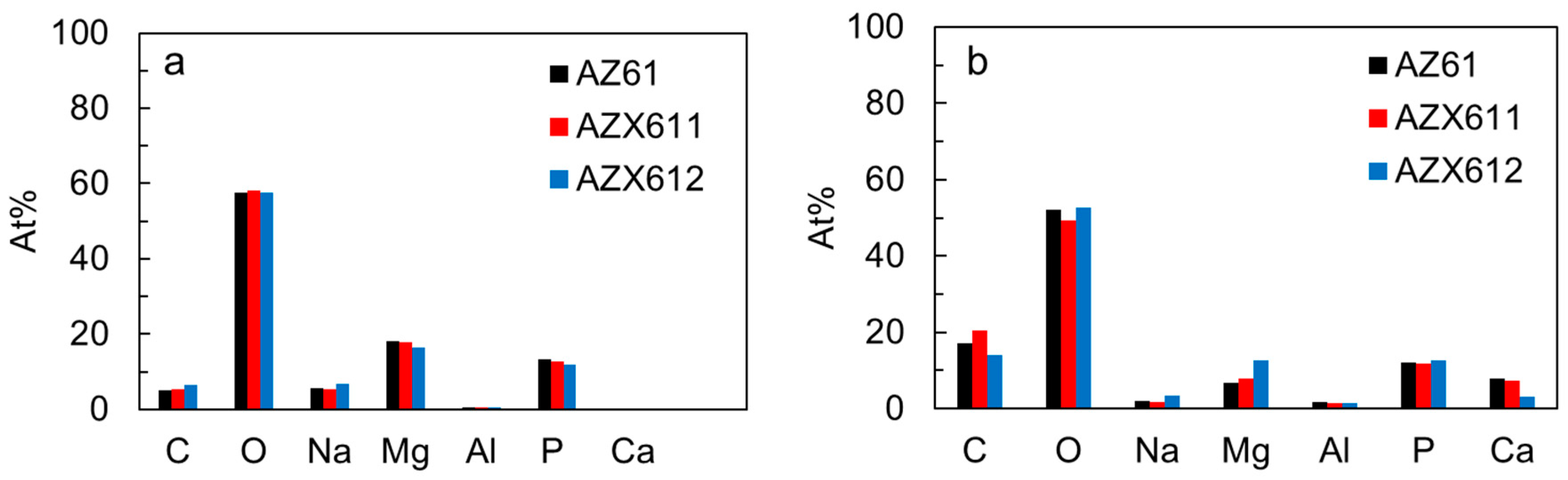
3.4. Apatite-Forming Ability
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Song, G. Control of biodegradation of biocompatable magnesium alloys. Corros. Sci. 2007, 49, 1696–1701. [Google Scholar] [CrossRef]
- Kirkland, N.; Lespagnol, J.; Birbilis, N.; Staiger, M.; Kirkland, N. A survey of bio-corrosion rates of magnesium alloys. Corros. Sci. 2010, 52, 287–291. [Google Scholar] [CrossRef]
- Witte, F.; Hort, N.; Vogt, C.; Cohen, S.; Ulrich, K.; Willumeit, R.; Feyerabend, F. Current Opinion in Solid State and Materials Science Degradable biomaterials based on magnesium corrosion. Curr. Opin. Solid State Mater. Sci. 2008, 12, 63–72. [Google Scholar] [CrossRef]
- Patel, J.L.; Saka, N. Microplasmic ceramic coating. Int. Ceram. Rev. 2001, 50, 398–401. [Google Scholar]
- White, L.; Koo, Y.; Neralla, S.; Sankar, J.; Yun, Y. Enhanced mechanical properties and increased corrosion resistance of a biodegradable magnesium alloy by plasma electrolytic oxidation (PEO). Mater. Sci. Eng. B 2016, 208, 39–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Echeverry-Rendon, M.; Duque, V.; Quintero, D.; Robledo, S.M.; Harmsen, M.C.; Echeverria, F. Improved corrosion resistance of commercially pure magnesium after its modification by plasma electrolytic oxidation with organic additives. J. Biomater. Appl. 2018, 33, 725–740. [Google Scholar] [CrossRef] [PubMed]
- Matykina, E.; Garcia, I.; Arrabal, R.; Mohedano, M.; Mingo, B.; Sancho, J.; Merino, M.; Pardo, A. Role of PEO coatings in long-term biodegradation of a Mg alloy. Appl. Surf. Sci. 2016, 389, 810–823. [Google Scholar] [CrossRef]
- Lu, J.; He, X.; Li, H.; Song, R. Microstructure and Corrosion Resistance of PEO Coatings Formed on KBM10 Mg Alloy Pretreated with Nd(NO3)3. Materials 2018, 11, 1062. [Google Scholar] [CrossRef]
- Li, L.; Gao, J.; Wang, Y.; Srinivasan, P.B.; Liang, J.; Blawert, C.; Störmer, M.; Dietzel, W.; Asoh, H.; Ono, S.; et al. Effect of current density on the microstructure and corrosion behaviour of plasma electrolytic oxidation treated AM50 magnesium alloy. Surf. Coat Technol. 2015, 272, 182–189. [Google Scholar] [CrossRef]
- Hussein, R.; Northwood, D.; Nie, X. The effect of processing parameters and substrate composition on the corrosion resistance of plasma electrolytic oxidation (PEO) coated magnesium alloys. Surf. Coat. Technol. 2013, 237, 357–368. [Google Scholar] [CrossRef]
- Ono, S.; Moronuki, S.; Mori, Y.; Koshi, A.; Liao, J.; Asoh, H. Effect of Electrolyte Concentration on the Structure and Corrosion Resistance of Anodic Films Formed on Magnesium through Plasma Electrolytic Oxidation. Electrochim. Acta 2017, 240, 415–423. [Google Scholar] [CrossRef]
- Asoh, H.; Matsuoka, S.; Sayama, H.; Ono, S. Anodizing under sparking of AZ31B magnesium alloy in Na3PO4 solution. J. Jpn. Inst. Light Met. 2010, 60, 608–614. [Google Scholar] [CrossRef]
- Kanter, B.; Vikman, A.; Brückner, T.; Schamel, M.; Gbureck, U.; Ignatius, A. Bone regeneration capacity of magnesium phosphate cements in a large animal model. Acta Biomater. 2018, 69, 352–361. [Google Scholar] [CrossRef] [PubMed]
- Anawati; Asoh, H.; Ono, S. Enhanced uniformity of apatite coating on a PEO film formed on AZ31 Mg alloy by an alkali pretreatment. Surf. Coatings Technol. 2015, 272, 182–189. [Google Scholar] [CrossRef]
- Han, Y.; Hong, S.H.; Xu, K. Structure and in vitro bioactivity of titania-based films by micro-arc oxidation. Surf. Coatings Technol. 2003, 168, 249–258. [Google Scholar] [CrossRef]
- Gu, X.; Li, N.; Zhou, W.; Zheng, Y.; Zhao, X.; Cai, Q.; Ruan, L. Corrosion resistance and surface biocompatibility of a microarc oxidation coating on a Mg–Ca alloy. Acta Biomater. 2011, 7, 1880–1889. [Google Scholar] [CrossRef] [PubMed]
- Kannan, M.B.; Mathan, B.K. Electrochemical deposition of calcium phosphates on magnesium and its alloys for improved biodegradation performance: A review. Surf. Coat. Technol. 2016, 301, 36–41. [Google Scholar] [CrossRef]
- Gao, Y.; Yerokhin, A.; Matthews, A. Deposition and evaluation of duplex hydroxyapatite and plasma electrolytic oxidation coatings on magnesium. Surf. Coat. Technol. 2015, 269, 170–182. [Google Scholar] [CrossRef]
- Srinivasan, P.B.; Liang, J.; Blawert, C.; Stormer, M.; Dietzel, W. Characterization of calcium containing plasma electrolytic oxidation coatings on AM50 magnesium alloy. Appl. Surf. Sci. 2010, 256, 4017–4022. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Lu, X.; Blawert, C.; Di, S.; Zheludkevich, M.L. Microstructure and corrosion behavior of Ca/P coatings prepared on magnesium by plasma electrolytic oxidation. Surf. Coat. Technol. 2017, 319, 359–369. [Google Scholar] [CrossRef]
- Lederer, S.; Sankaran, S.; Smith, T.; Fürbeth, W. Formation of bioactive hydroxyapatite-containing titania coatings on CP-Ti 4+ alloy generated by plasma electrolytic oxidation. Surf. Coat. Technol. 2019, 363, 66–74. [Google Scholar] [CrossRef]
- Adeleke, S.; Ramesh, S.; Bushroa, A.; Ching, Y.; Sopyan, I.; Maleque, M.; Krishnasamy, S.; Chandran, H.; Misran, H.; Sutharsini, U. The properties of hydroxyapatite ceramic coatings produced by plasma electrolytic oxidation. Ceram. Int. 2018, 44, 1802–1811. [Google Scholar] [CrossRef]
- Anawati, A.; Asoh, H.; Ono, S. Effects of alloying element ca on the corrosion behavior and bioactivity of anodic films formed on AM60 mg alloys. Materials 2017, 10, 11. [Google Scholar] [CrossRef] [PubMed]
- Anawati, A.; Asoh, H.; Ono, S. Degradation Behavior of Coatings Formed by the Plasma Electrolytic Oxidation Technique on AZ61 Magnesium Alloys Containing 0, 1 and 2 wt.% Ca. Int. J. Technol. 2018, 9, 622. [Google Scholar] [CrossRef]
- Jun, J.H.; Park, B.K.; Kim, J.M.; Kim, K.T.; Jung, W.J. Effects of Ca Addition on Microstructure and Mechanical Properties of Mg-RE-Zn Casting Alloy. Mater. Sci. Forum 2009, 488, 107–110. [Google Scholar] [CrossRef]
- Jang, Y.; Tan, Z.; Jurey, C.; Xu, Z.; Dong, Z.; Collins, B.; Yun, Y.; Sankar, J. Understanding corrosion behavior of Mg–Zn–Ca alloys from subcutaneous mouse model: Effect of Zn element concentration and plasma electrolytic oxidation. Mater. Sci. Eng. C 2015, 48, 28–40. [Google Scholar] [CrossRef] [PubMed]
- Brady, M.P.; Fayek, M.; Leonard, D.N.; Meyer, H.M.; Thomson, J.K.; Anovitz, L.M.; Rother, G.; Song, G.L.; Davis, B. Tracer Film Growth Study of the Corrosion of Magnesium Alloys AZ31B and ZE10A in 0.01% NaCl Solution. J. Electrochem. Soc. 2017, 164, C367–C375. [Google Scholar] [CrossRef]
- Channing, S. Annual Book of ASTM Standards; ASTM International: Conshohocken, PA, USA, 1991. [Google Scholar]
- Müller, L.; Müller, F.A. Preparation of SBF with different HCO3-content and its influence on the composition of biomimetic apatites. Acta Biomater. 2006, 2, 181–189. [Google Scholar] [CrossRef]
- Lu, X.; Leng, Y. Theoretical analysis of calcium phosphate precipitation in simulated body fluid. Biomaterials 2005, 26, 1097–1108. [Google Scholar] [CrossRef]
- Pekguleryuz, M.; Kainer, K.; Kaya, A. Fundamentals of Magnesium Alloy; Woodhead Publishing Ltd.: Cambridge, UK, 2013. [Google Scholar]
- Massalski, T.; Okamoto, H.; Subramanian, P.; Kacprzak, L. Binary Alloy Phase Diagrams, 2nd ed.; ASM: Materials Park, OH, USA, 1990. [Google Scholar]
- Hosford, W.F. Physical Metallurgy, 2nd ed.; Taylor and Francis Group: Boca Raton, FL, UAS, 2010. [Google Scholar]
- Lunder, O.; Nordien, J.; Nisancioglu, K. Corrosion Resistance of Cast Mg-Al Alloys. Corros. Rev. 1997, 15, 439–470. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Element | AZ61 | AZX611 | AZX612 |
|---|---|---|---|
| Mg | Bal. | Bal. | Bal. |
| Al | 6.50 | 5.8–7.2 | 5.8–7.2 |
| Zn | 0.92 | 0.4–1.5 | 0.4–1.5 |
| Mn | 0.32 | 0.15–0.5 | 0.15–0.5 |
| Cu | ≤0.05 | ≤0.05 | ≤0.05 |
| Ni | ≤0.005 | ≤0.005 | ≤0.005 |
| Fe | ≤0.005 | ≤0.005 | ≤0.005 |
| Si | ≤0.1 | ≤0.1 | ≤0.1 |
| Ca | - | 1.0 | 2.0 |
| Ion | Na+ | K+ | Mg+ | Ca+ | Cl− | HCO3− | HPO42− | SO42− |
|---|---|---|---|---|---|---|---|---|
| Concentration (mM) | 142 | 5 | 1 | 2.5 | 126 | 10 | 1 | 1 |
| Specimen | Rs (Ωcm2) | CPE1 (Ω−1sncm−2) | n1 | R1 (Ωcm2) | CPE2 (Ω−1sncm−2) | n2 | R2 (Ωcm2) | L (H) | RL (Ωcm2) |
|---|---|---|---|---|---|---|---|---|---|
| AZ61 | 28.25 | 9.78 × 10−6 | 0.88 | 1.32 × 103 | −1.00 × 10−3 | 0.81 | −200 | - | - |
| AZX611 | 9.40 | 7.68 × 10−6 | 0.94 | 1.10 × 103 | −1.00 × 10−3 | 0.75 | −1000 | 15 | 700 |
| AZX612 | 25.0 | 8.00 × 10−6 | 0.88 | 1.00 × 103 | −6.00 × 10−4 | 0.75 | −725 | 20 | 950 |
| AZ61 (Coated) | 40.5 | 1.09 × 10−9 | 0.87 | 1.04 × 104 | 3.67 × 10−7 | 0.91 | 3.61 × 103 | - | - |
| AZX611 (Coated) | 61.9 | 1.64 × 10−9 | 0.87 | 8.4 × 103 | 2.48 × 10−7 | 0.96 | 2.61 × 103 | - | - |
| AZX612 (Coated) | 163.8 | 1.97 × 10−9 | 0.82 | 9.45 × 103 | 3.67 × 10−7 | 0.99 | 1.41 × 103 | - | - |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Anawati, A.; Asoh, H.; Ono, S. Corrosion Resistance and Apatite-Forming Ability of Composite Coatings formed on Mg–Al–Zn–Ca Alloys. Materials 2019, 12, 2262. https://doi.org/10.3390/ma12142262
Anawati A, Asoh H, Ono S. Corrosion Resistance and Apatite-Forming Ability of Composite Coatings formed on Mg–Al–Zn–Ca Alloys. Materials. 2019; 12(14):2262. https://doi.org/10.3390/ma12142262
Chicago/Turabian StyleAnawati, Anawati, Hidetaka Asoh, and Sachiko Ono. 2019. "Corrosion Resistance and Apatite-Forming Ability of Composite Coatings formed on Mg–Al–Zn–Ca Alloys" Materials 12, no. 14: 2262. https://doi.org/10.3390/ma12142262
APA StyleAnawati, A., Asoh, H., & Ono, S. (2019). Corrosion Resistance and Apatite-Forming Ability of Composite Coatings formed on Mg–Al–Zn–Ca Alloys. Materials, 12(14), 2262. https://doi.org/10.3390/ma12142262