Ligand-Modified Boron-Doped Diamond Surface: DFT Insights into the Electronic Properties of Biofunctionalization

 , ,
, , 
Abstract
:1. Introduction
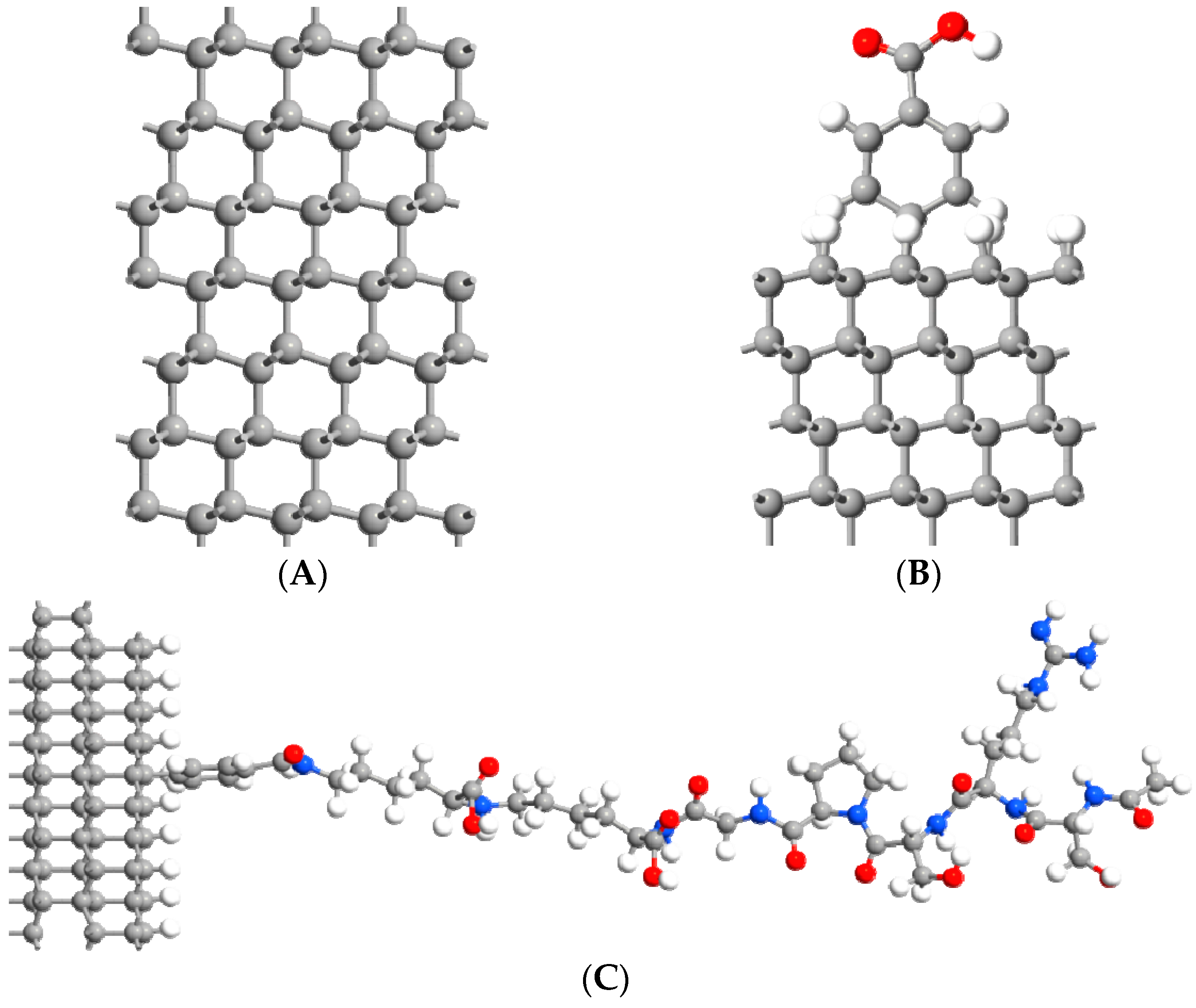
2. Materials and Methods
3. Results and Discussion
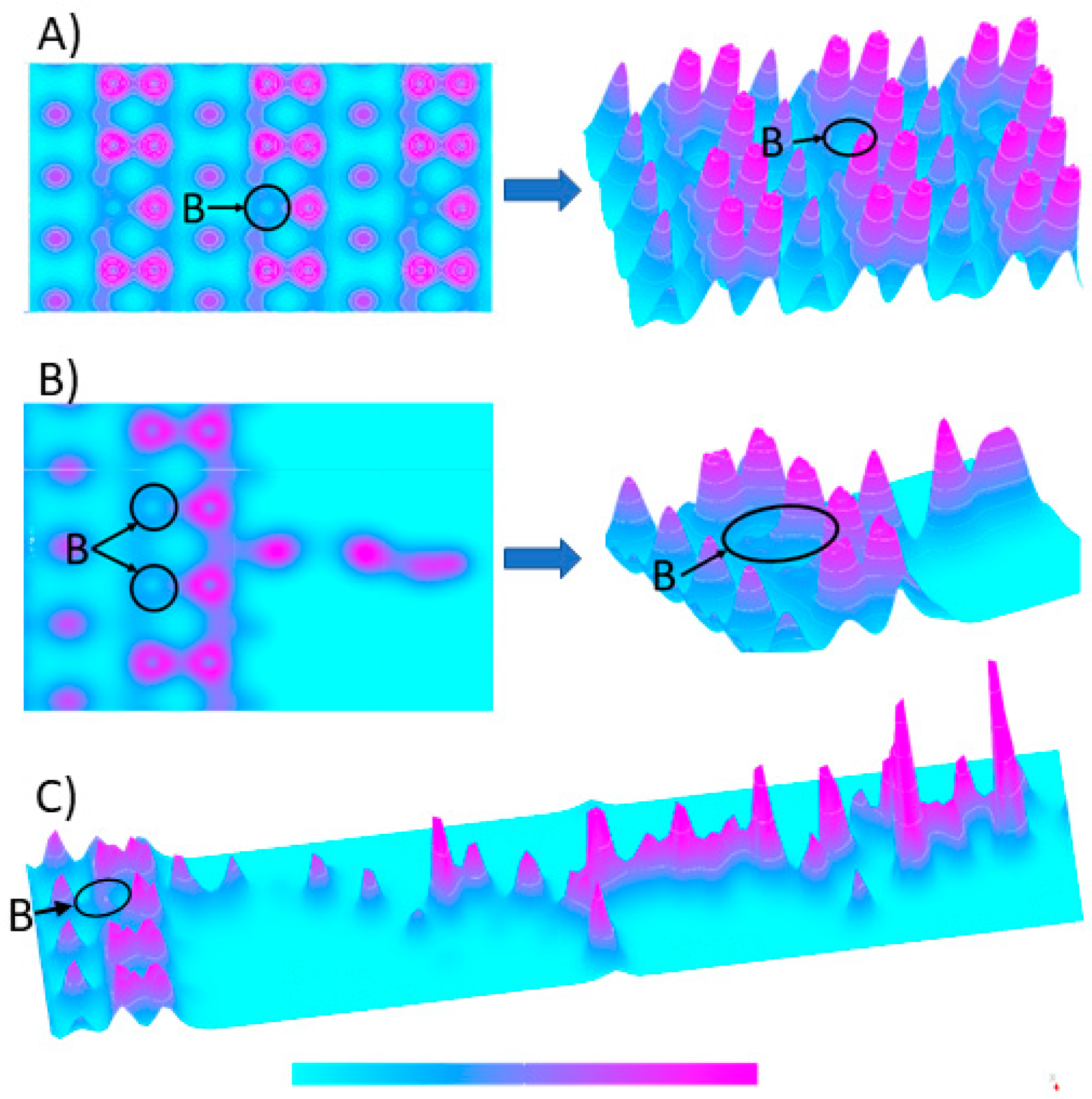
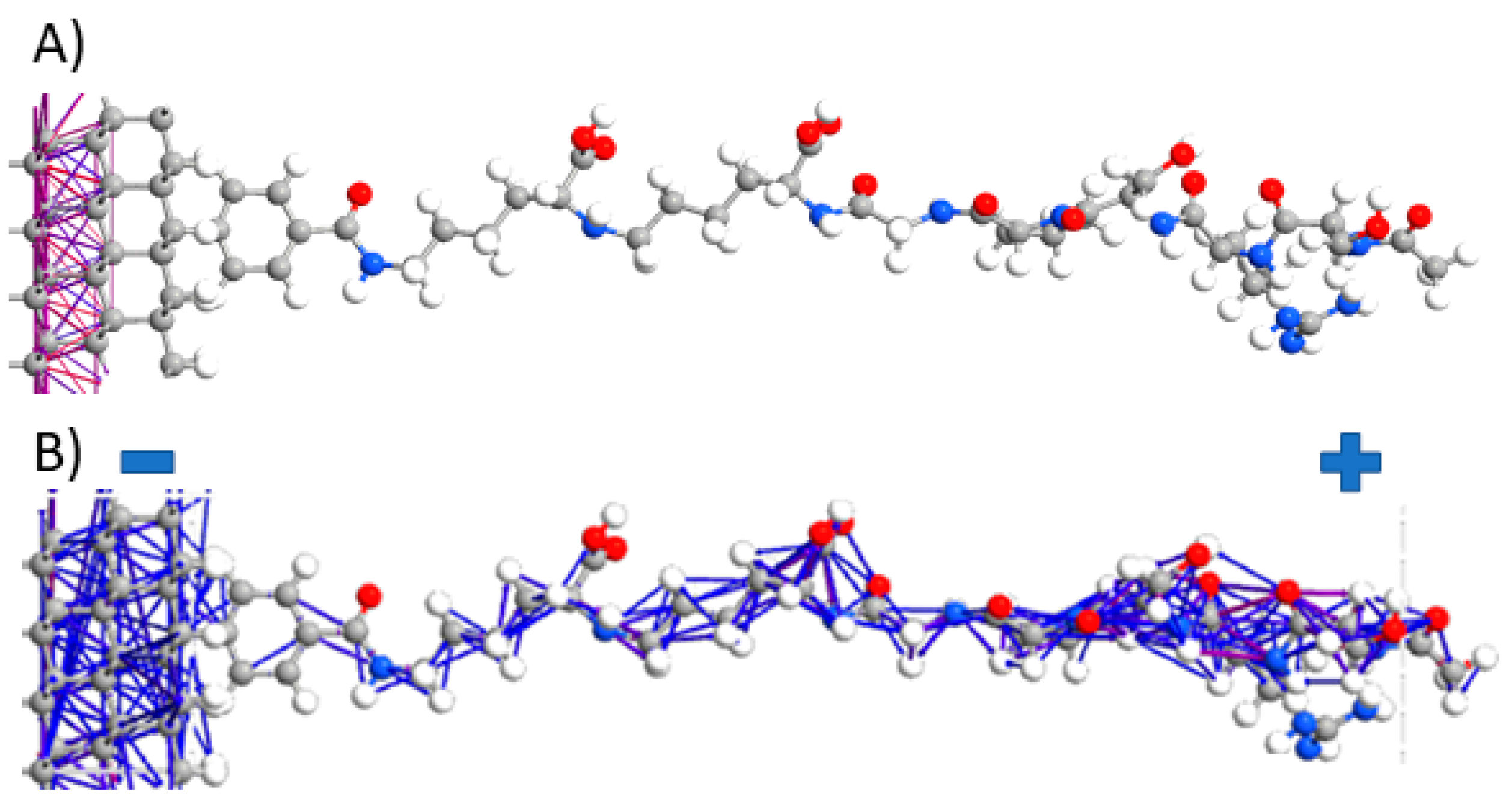
3.1. Electron Density Distribution at the BDDE-Analyte Junction
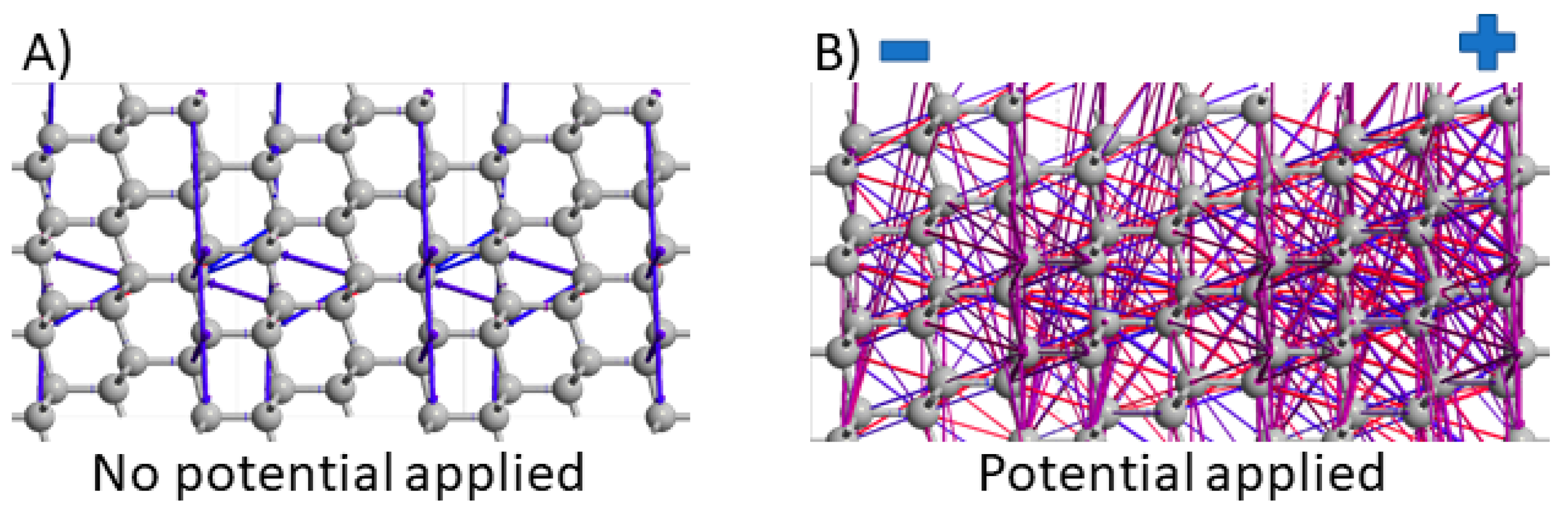
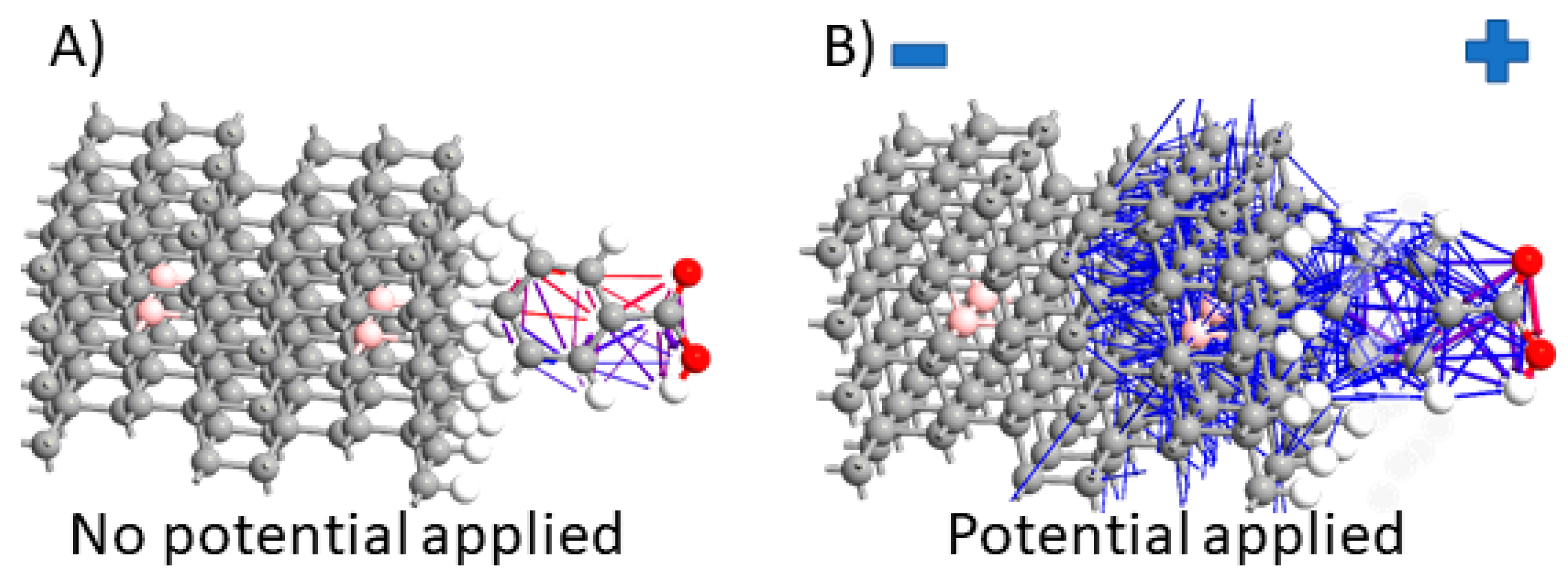
3.2. Electron Pathways Study during Device Operation
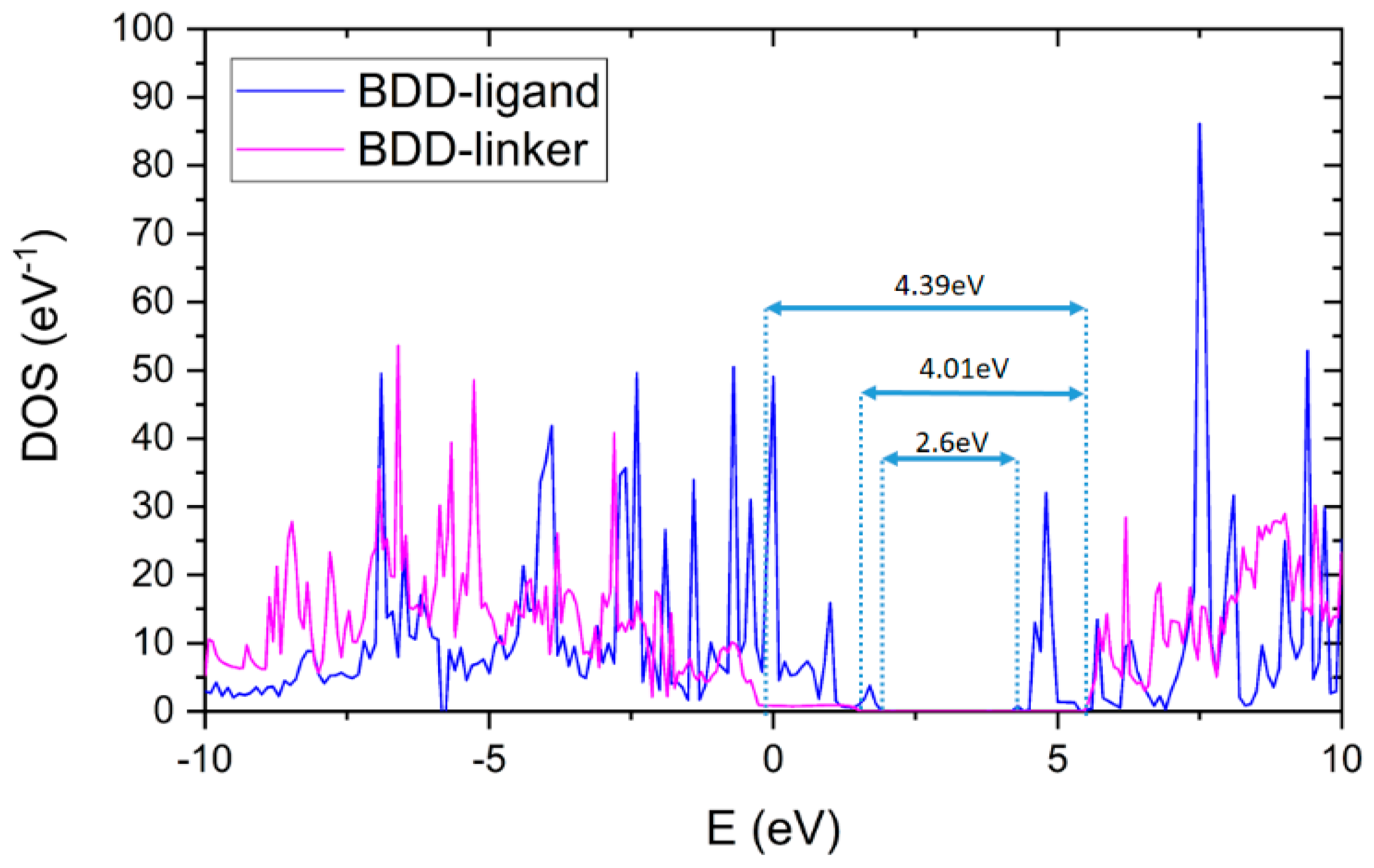
3.3. Device—Density of States
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Guo, Z.; Chen, L.; Zeng, H.; Gomez, J.A.; Plowden, J.; Fujita, T.; Katz, J.M.; Donis, R.O.; Sambhara, S. NS1 Protein of Influenza A Virus Inhibits the Function of Intracytoplasmic Pathogen Sensor, RIG-I. Am. J. Respir. Cell Mol. Biol. 2007, 36, 263–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartree, D.R. The Wave Mechanics of an Atom with a Non-Coulomb Central Field. Part II. Some Results and Discussion. Math. Proc. Camb. Philos. Soc. 1928, 24, 111–132. [Google Scholar] [CrossRef]
- Liu, X.-Y.; Andersson, D.A.; Uberuaga, B.P. First-principles DFT modeling of nuclear fuel materials. J. Mater. Sci. 2012, 47, 7367–7384. [Google Scholar] [CrossRef]
- Tian; Liu DFT Description on Electronic Structure and Optical Absorption Properties of Anionic S-Doped Anatase TiO2. J. Phys. Chem. B 2006, 110, 17866–17871. [CrossRef] [PubMed]
- Gurkan, Y.Y.; Kasapbasi, E.; Cinar, Z. Enhanced solar photocatalytic activity of TiO2 by selenium(IV) ion-doping: Characterization and DFT modeling of the surface. Chem. Eng. J. 2013, 214, 34–44. [Google Scholar] [CrossRef]
- Maron, L.; Eisenstein, O. Do f Electrons Play a Role in the Lanthanide−Ligand Bonds? A DFT Study of Ln(NR2)3; R = H, SiH3. J. Phys. Chem. A 2000, 104, 7140–7143. [Google Scholar] [CrossRef]
- Maron, L.; Eisenstein, O. DFT modeling of ligands in lanthanide chemistry: Is Ln[N(SiH3)2]3 a model for Ln[N(SiMe3)2]3? New J. Chem. 2001, 25, 255–258. [Google Scholar] [CrossRef]
- Petit, L.; Daul, C.; Adamo, C.; Maldivi, P. DFT modeling of the relative affinity of nitrogen ligands for trivalent f elements: An energetic point of view. New J. Chem. 2007, 31, 1738–1745. [Google Scholar] [CrossRef]
- Bogdanowicz, R. Characterization of Optical and Electrical Properties of Transparent Conductive Boron-Doped Diamond thin Films Grown on Fused Silica. Metrol. Meas. Syst. 2014, 21, 685–698. [Google Scholar] [CrossRef] [Green Version]
- Sobaszek, M.; Skowroński, Ł.; Bogdanowicz, R.; Siuzdak, K.; Cirocka, A.; Zięba, P.; Gnyba, M.; Naparty, M.; Gołuński, Ł.; Płotka, P. Optical and electrical properties of ultrathin transparent nanocrystalline boron-doped diamond electrodes. Opt. Mater. 2015, 42, 24–34. [Google Scholar] [CrossRef]
- Ficek, M.; Sobaszek, M.; Gnyba, M.; Ryl, J.; Gołuński, Ł.; Smietana, M.; Jasiński, J.; Caban, P.; Bogdanowicz, R. Optical and electrical properties of boron doped diamond thin conductive films deposited on fused silica glass substrates. Appl. Surf. Sci. 2016, 387, 846–856. [Google Scholar] [CrossRef]
- Tang, L.; Tsai, C.; Gerberich, W.W.; Kruckeberg, L.; Kania, D.R. Biocompatibility of chemical-vapour-deposited diamond. Biomaterials 1995, 16, 483–488. [Google Scholar] [CrossRef]
- Falina, S.; Kawai, S.; Oi, N.; Yamano, H.; Kageura, T.; Suaebah, E.; Inaba, M.; Shintani, Y.; Syamsul, M.; Kawarada, H. Role of Carboxyl and Amine Termination on a Boron-Doped Diamond Solution Gate Field Effect Transistor (SGFET) for pH Sensing. Sensors 2018, 18, 2178. [Google Scholar] [CrossRef] [PubMed]
- Karaya, R.; Baba, I.; Mori, Y.; Matsumoto, T.; Nakajima, T.; Tokuda, N.; Kawae, T. B-doped diamond field-effect transistor with ferroelectric vinylidene fluoride–trifluoroethylene gate insulator. Jpn. J. Appl. Phys. 2017, 56, 10PF06. [Google Scholar] [CrossRef]
- Zkria, A.; Gima, H.; Yoshitake, T. Application of nitrogen-doped ultrananocrystalline diamond/hydrogenated amorphous carbon composite films for ultraviolet detection. Appl. Phys. A 2017, 123, 167. [Google Scholar] [CrossRef]
- Abubakr, E.; Zkria, A.; Katamune, Y.; Ohmagari, S.; Imokawa, K.; Ikenoue, H.; Yoshitake, T. Formation of low resistivity layers on singlecrystalline diamond by excimer laser irradiation. Diam. Relat. Mater. 2019, 95, 166–173. [Google Scholar] [CrossRef]
- Fujimori, N.; Nakahata, H.; Imai, T. Properties of Boron-Doped Epitaxial Diamond Films. Jpn. J. Appl. Phys. 1990, 29, 824. [Google Scholar] [CrossRef]
- Carlisle, J.A. Precious biosensors. Nat. Mater. 2004, 3, 668. [Google Scholar] [CrossRef] [PubMed]
- Härtl, A.; Schmich, E.; Garrido, J.A.; Hernando, J.; Catharino, S.C.R.; Walter, S.; Feulner, P.; Kromka, A.; Steinmüller, D.; Stutzmann, M. Protein-modified nanocrystalline diamond thin films for biosensor applications. Nat. Mater. 2004, 3, 736. [Google Scholar] [CrossRef] [PubMed]
- Boukhvalov, D.W. DFT modeling of the covalent functionalization of graphene: From ideal to realistic models. RSC Adv. 2013, 3, 7150–7159. [Google Scholar] [CrossRef]
- Jaramillo-Botero, A.; Marmolejo-Tejada, J.M. All-Armchair Graphene Nanoribbon Field-Effect Uridine Diphosphate Glucose Sensor: First-Principles In-Silico Design and Characterization. IEEE Sens. J. 2019, 19, 3975–3983. [Google Scholar] [CrossRef]
- Tian, Y.; Larsson, K. Effect by Diamond Surface Modification on Biomolecular Adhesion. Mater. Basel Switz. 2019, 12. [Google Scholar]
- Dec, B.; Bogdanowicz, R. DFT studies of refractive index of boron-doped diamond. Photonics Lett. Pol. 2018, 10, 39–41. [Google Scholar] [CrossRef]
- Sobaszek, M.; Siuzdak, K.; Skowroński, L.; Bogdanowicz, R.; Pluciński, J. Optically transparent boron-doped nanocrystalline diamond films for spectroelectrochemical measurements on different substrates. IOP Conf. Ser. Mater. Sci. Eng. 2016, 104, 012024. [Google Scholar] [CrossRef] [Green Version]
- Dec, B.; Bogdanowicz, R.; Pyrchla, K. Ab-initio study of electrical and optical properties of allylamine. Photonics Lett. Pol. 2018, 10, 94–96. [Google Scholar] [CrossRef]
- Kohn, W.; Becke, A.D.; Parr, R.G. Density Functional Theory of Electronic Structure. J. Phys. Chem. 1996, 100, 12974–12980. [Google Scholar] [CrossRef] [Green Version]
- Smidstrup, S.; Stradi, D.; Wellendorff, J.; Khomyakov, P.A.; Vej-Hansen, U.G.; Lee, M.E.; Ghosh, T.; Jónsson, E.; Jónsson, H.; Stokbro, K. First-principles Green's-function method for surface calculations: A pseudopotential localized basis set approach. Phys. Rev. B 2017, 96, 195309. [Google Scholar] [CrossRef] [Green Version]
- Griebel, M.; Hamaekers, J. Molecular dynamics simulations of the elastic moduli of polymer–carbon nanotube composites. Comput. Methods Appl. Mech. Eng. 2004, 193, 1773–1788. [Google Scholar] [CrossRef]
- Griebel, M.; Knapek, S.; Zumbusch, G. Numerical Simulation in Molecular Dynamics: Numerics, Algorithms, Parallelization, Applications; Springer Science & Business Media: Heidelberg, Germany, 2007; ISBN 978-3-540-68095-6. [Google Scholar]
- Ekimov, E.A.; Sidorov, V.A.; Bauer, E.D.; Mel’nik, N.N.; Curro, N.J.; Thompson, J.D.; Stishov, S.M. Superconductivity in diamond. Nature 2004, 428, 542–545. [Google Scholar] [CrossRef] [Green Version]
- Zhao, S.; Larsson, K. Theoretical Study of the Energetic Stability and Geometry of Terminated and B-Doped Diamond (111) Surfaces. J. Phys. Chem. C 2014, 118, 1944–1957. [Google Scholar] [CrossRef]
- Schlipf, M.; Gygi, F. Optimization algorithm for the generation of ONCV pseudopotentials. Comput. Phys. Commun. 2015, 196, 36–44. [Google Scholar] [CrossRef] [Green Version]
- Chelikowsky, J.R.; Louie, S.G. First-principles linear combination of atomic orbitals method for the cohesive and structural properties of solids: Application to diamond. Phys. Rev. B 1984, 29, 3470–3481. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Fuchs, M.; Scheffler, M. Ab initio pseudopotentials for electronic structure calculations of poly-atomic systems using density-functional theory. Comput. Phys. Commun. 1999, 119, 67–98. [Google Scholar] [CrossRef] [Green Version]
- Liu, D.C.; Nocedal, J. On the limited memory BFGS method for large scale optimization. Math. Program. 1989, 45, 503–528. [Google Scholar] [CrossRef] [Green Version]
- Turner, S.; Lu, Y.-G.; Janssens, S.D.; Da Pieve, F.; Lamoen, D.; Verbeeck, J.; Haenen, K.; Wagner, P.; Van Tendeloo, G. Local boron environment in B-doped nanocrystalline diamond films. Nanoscale 2012, 4, 5960. [Google Scholar] [CrossRef] [PubMed]
- Brunetta, C.D.; Karuppannan, B.; Rosmus, K.A.; Aitken, J.A. The crystal and electronic band structure of the diamond-like semiconductor Ag2ZnSiS4. J. Alloys Compd. 2012, 516, 65–72. [Google Scholar] [CrossRef]
- Lejaeghere, K.; Bihlmayer, G.; Björkman, T.; Blaha, P.; Blügel, S.; Blum, V.; Caliste, D.; Castelli, I.E.; Clark, S.J.; Corso, A.D.; et al. Reproducibility in density functional theory calculations of solids. Science 2016, 351, aad3000. [Google Scholar] [CrossRef]
- Comparing Solid State DFT Codes, Basis Sets and Potentials. Available online: https://molmod.ugent.be/deltacodesdft (accessed on 25 July 2019).
- Lejaeghere, K.; Speybroeck, V.; Oost, G.; Cottenier, S. Error Estimates for Solid-State Density-Functional Theory Predictions: An Overview by Means of the Ground-State Elemental Crystals. Crit. Rev. Solid State Mater. Sci. 2013, 39, 1–24. [Google Scholar] [Green Version]
- Bogdanowicz, R.; Fabiańska, A.; Golunski, L.; Sobaszek, M.; Gnyba, M.; Ryl, J.; Darowicki, K.; Ossowski, T.; Janssens, S.D.; Haenen, K.; et al. Influence of the boron doping level on the electrochemical oxidation of the azo dyes at Si/BDD thin film electrodes. Diam. Relat. Mater. 2013, 39, 82–88. [Google Scholar] [CrossRef]
- Cui, J.B.; Ristein, J.; Ley, L. Electron Affinity of the Bare and Hydrogen Covered Single Crystal Diamond (111) Surface. Phys. Rev. Lett. 1998, 81, 429–432. [Google Scholar] [CrossRef]
- Dai, Y.; Huang, B.; Dai, D. The role of dangling-bond, hydrogen and adsorbate in diamond surface conduction. Diam. Relat. Mater. 2003, 12, 15–19. [Google Scholar] [CrossRef]
- Chua, L.-L.; Zaumseil, J.; Chang, J.-F.; Ou, E.C.-W.; Ho, P.K.-H.; Sirringhaus, H.; Friend, R.H. General observation of n-type field-effect behaviour in organic semiconductors. Nature 2005, 434, 194–199. [Google Scholar] [CrossRef] [PubMed]
- Cornil, J.; Beljonne, D.; Calbert, J.-P.; Brédas, J.-L. Interchain Interactions in Organic π-Conjugated Materials: Impact on Electronic Structure, Optical Response, and Charge Transport. Adv. Mater. 2001, 13, 1053–1067. [Google Scholar] [CrossRef]
- Coropceanu, V.; Cornil, J.; da Silva Filho, D.A.; Olivier, Y.; Silbey, R.; Brédas, J.-L. Charge Transport in Organic Semiconductors. Chem. Rev. 2007, 107, 926–952. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Larsson, K. First Principle Study of the Attachment of Graphene onto Different Terminated Diamond (111) Surfaces. Available online: https://www.hindawi.com/journals/acmp/2019/9098256/ (accessed on 28 August 2019).
- Ashcheulov, P.; Šebera, J.; Kovalenko, A.; Petrák, V.; Fendrych, F.; Nesládek, M.; Taylor, A.; Vlčková Živcová, Z.; Frank, O.; Kavan, L.; et al. Conductivity of boron-doped polycrystalline diamond films: Influence of specific boron defects. Eur. Phys. J. B 2013, 86, 443. [Google Scholar] [CrossRef]
- Strange, M.; Thygesen, K.S. Towards quantitative accuracy in first-principles transport calculations: The GW method applied to alkane/gold junctions. Beilstein J. Nanotechnol. 2011, 2, 746–754. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Liu, J.; Tang, T. The Influence of B, N and Si Doping on the CH3 Adsorption on the Diamond Surface Based on DFT Calculations. Crystals 2019, 9, 427. [Google Scholar] [CrossRef]
- Watanabe, T.; Teraji, T.; Ito, T.; Kamakura, Y.; Taniguchi, K. Monte Carlo simulations of electron transport properties of diamond in high electric fields using full band structure. J. Appl. Phys. 2004, 95, 4866–4874. [Google Scholar] [CrossRef]
- Kamakura, Y.; Fujita, R.; Konaga, K.; Ueoka, Y.; Mori, N.; Kotani, T. Full band Monte Carlo simulation of impact ionization in wide bandgap semiconductors based on ab initio calculation. In Proceedings of the 2016 International Conference on Simulation of Semiconductor Processes and Devices (SISPAD), Nuremberg, Germany, 6–8 September 2016; pp. 47–51. [Google Scholar]
- Barnard, A.S.; Russo, S.P.; Snook, I.K. Ab initio modelling of band states in doped diamond. Philos. Mag. 2003, 83, 1163–1174. [Google Scholar] [CrossRef]
- Collins, A.T.; Lightowlers, E.C. Photothermal Ionization and Photon-Induced Tunneling in the Acceptor Photoconductivity Spectrum of Semiconducting Diamond. Phys. Rev. 1968, 171, 843–855. [Google Scholar] [CrossRef]
- Rivero, P.; Shelton, W.; Meunier, V. Surface properties of hydrogenated diamond in the presence of adsorbates: A hybrid functional DFT study. Carbon 2016, 110, 469–479. [Google Scholar] [CrossRef] [Green Version]
- Liberman, D.A. Slater transition-state band-structure calculations. Phys. Rev. B 2000, 62, 6851–6853. [Google Scholar] [CrossRef]
- Ullah, M.; Ahmed, E.; Hussain, F.; Rana, A.M.; Raza, R.; Ullah, H. Electronic structure calculations of oxygen-doped diamond using DFT technique. Microelectron. Eng. 2015, 146, 26–31. [Google Scholar] [CrossRef]
- Tran, F.; Blaha, P. Accurate Band Gaps of Semiconductors and Insulators with a Semilocal Exchange-Correlation Potential. Phys. Rev. Lett. 2009, 102, 226401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heyd, J.; Scuseria, G.E.; Ernzerhof, M. Hybrid functionals based on a screened Coulomb potential. J. Chem. Phys. 2003, 118, 8207–8215. [Google Scholar] [CrossRef] [Green Version]
- Xiao, H.; Tahir-Kheli, J.; Goddard, W.A. Accurate Band Gaps for Semiconductors from Density Functional Theory. J. Phys. Chem. Lett. 2011, 2, 212–217. [Google Scholar] [CrossRef] [Green Version]
- Heyd, J.; Scuseria, G.E. Efficient hybrid density functional calculations in solids: Assessment of the Heyd–Scuseria–Ernzerhof screened Coulomb hybrid functional. J. Chem. Phys. 2004, 121, 1187–1192. [Google Scholar] [CrossRef]
- O’Donnell, K.M.; Martin, T.L.; Allan, N.L. Light Metals on Oxygen-Terminated Diamond (100): Structure and Electronic Properties. Chem. Mater. 2015, 27, 1306–1315. [Google Scholar] [CrossRef] [Green Version]
- Dean, P.J.; Lightowlers, E.C.; Wight, D.R. Intrinsic and Extrinsic Recombination Radiation from Natural and Synthetic Aluminum-Doped Diamond. Phys. Rev. 1965, 140, A352–A368. [Google Scholar] [CrossRef]
- Ristein, J. Electronic properties of diamond surfaces — blessing or curse for devices? Diam. Relat. Mater. 2000, 9, 1129–1137. [Google Scholar] [CrossRef]
- Gairola, S.C. Electron and Phonon in Neutron Scattering of Semiconductor Crystals. Int. J. Theor. Phys. 2013, 12, 4256–4264. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Atom Type | SG15 (Medium) [meV] | SG15 (High) [meV] | FHI (DPZ) [meV] |
|---|---|---|---|
| Boron | 1.65 | 1.88 | 3.67 |
| Carbon | 0.66 | 2.80 | 5.89 |
| Hydrogen | 0.96 | 0.19 | 0.54 |
| Nitrogen | 6.57 | 1.04 | 2.45 |
| Oxygen | 9.37 | 1.68 | 24.28 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dec, B.; Sobaszek, M.; Jaramillo-Botero, A.; Goddard, W.A., III; Bogdanowicz, R. Ligand-Modified Boron-Doped Diamond Surface: DFT Insights into the Electronic Properties of Biofunctionalization. Materials 2019, 12, 2910. https://doi.org/10.3390/ma12182910
Dec B, Sobaszek M, Jaramillo-Botero A, Goddard WA III, Bogdanowicz R. Ligand-Modified Boron-Doped Diamond Surface: DFT Insights into the Electronic Properties of Biofunctionalization. Materials. 2019; 12(18):2910. https://doi.org/10.3390/ma12182910
Chicago/Turabian StyleDec, Bartłomiej, Michał Sobaszek, Andrés Jaramillo-Botero, William Andrew Goddard, III, and Robert Bogdanowicz. 2019. "Ligand-Modified Boron-Doped Diamond Surface: DFT Insights into the Electronic Properties of Biofunctionalization" Materials 12, no. 18: 2910. https://doi.org/10.3390/ma12182910
APA StyleDec, B., Sobaszek, M., Jaramillo-Botero, A., Goddard, W. A., III, & Bogdanowicz, R. (2019). Ligand-Modified Boron-Doped Diamond Surface: DFT Insights into the Electronic Properties of Biofunctionalization. Materials, 12(18), 2910. https://doi.org/10.3390/ma12182910