Monte Carlo Study of Electronic Transport in Monolayer InSe


Abstract
:1. Introduction
2. Structure and Computational Methodology
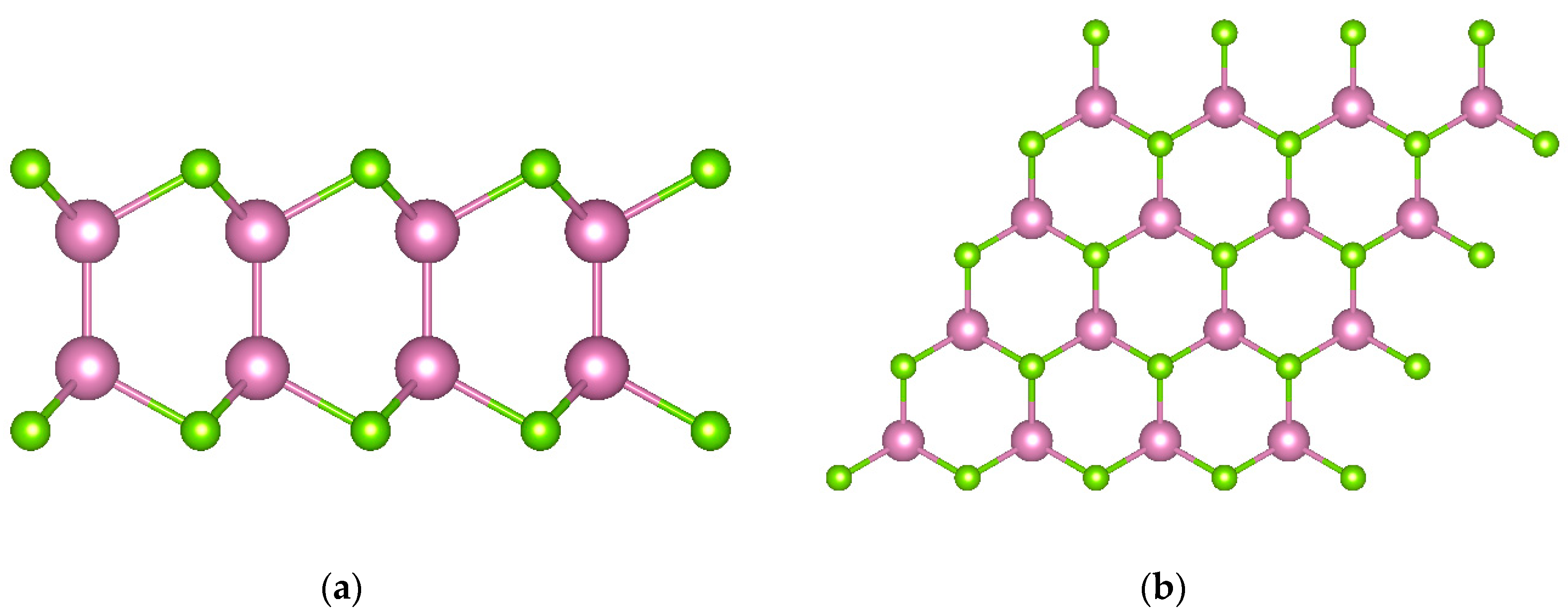
2.1. Structure
2.2. Theoretical Model
2.2.1. Band Structure and Phonon Spectrum
2.2.2. Electron-Phonon Interaction and Scattering Rates
2.2.3. Monte Carlo Simulations
3. Results
3.1. Electronic Band Structure and Phonon Spectrum
3.2. Scattering Rates
3.3. Mobility Calculations
3.4. Velocity-Field Calculations
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bolotin, K.I.; Sikes, K.J.; Jiang, Z.; Klima, M.; Fudenberg, G.; Hone, J.; Kim, P.; Stormer, H.L. Ultrahigh mobility in suspended graphene. Solid State Commun. 2008, 146, 351–355. [Google Scholar] [CrossRef] [Green Version]
- Morozov, S.V.; Novoselov, K.S.; Katsnelson, M.I.; Schedin, F.; Elias, D.C.; Jaszczak, J.A.; Geim, A.K. Giant intrinsic carrier mobilities in graphene and its bilayer. Phys. Rev. Lett. 2008, 100, 016602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaddemane, G.; Vandenberghe, W.G.; Van de Put, M.L.; Chen, S.; Tiwari, S.; Chen, E.; Fischetti, M.V. Theoretical studies of electronic transport in monolayer and bilayer phosphorene: A critical overview. Phys. Rev. B 2008, 98, 115416. [Google Scholar] [CrossRef] [Green Version]
- Castellanos-Gomez, A.; Vicarelli, L.; Prada, E.; Island, J.O.; Narasimha-Acharya, K.L.; Blanter, S.I.; Groenendijk, D.J.; Buscema, M.; Steele, G.A.; Alvarez, J.V.; et al. Isolation and characterization of few-layer black phosphorus. 2D Mater. 2014, 1, 025001. [Google Scholar] [CrossRef]
- Li, L.; Yu, Y.; Ye, G.J.; Ge, Q.; Ou, X.; Wu, H.; Feng, D.; Chen, X.H.; Zhang, Y. Black phosphorus field-effect transistors. Nat. Nanotechnol. 2014, 9, 372–377. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Neal, A.T.; Zhu, Z.; Luo, Z.; Xu, X.; Tomanek, D.; Ye, P.D. Phosphorene: An unexplored 2D semiconductor with high hole mobility. ACS Nano 2014, 8, 4033–4041. [Google Scholar] [CrossRef] [Green Version]
- Qiao, J.; Kong, X.; Hu, Z.-H.; Yang, F.; Ji, W. High-mobility transport anisotropy and linear dichrosim in few-layer black phosphorus. Nat. Commun. 2014, 5, 4475. [Google Scholar] [CrossRef] [Green Version]
- Gaddemane, G.; Vandenberghe, W.G.; Van De Put, M.L.; Chen, E.; Fischetti, M.V. Monte-Carlo study of electronic transport in non-σh-symmetric two-dimensional materials: Silicene and germanene. J. Appl. Phys. 2018, 124, 044306. [Google Scholar] [CrossRef]
- Houssa, M.; Scalise, E.; Sankaran, K.; Pourtois, G.; Afanas’ev, V.V.; Stesmans, A. Electronic properties of hydrogenated silicene and germanener. Appl. Phys. Lett. 2011, 99, 223107. [Google Scholar] [CrossRef] [Green Version]
- Roome, N.J.; Carey, J.D. Beyond graphene: Stable elemental monolayers of silicene and germanene. ACS Appl. Mater. Interfaces 2014, 6, 7743–7750. [Google Scholar] [CrossRef] [Green Version]
- Khatami, M.M.; Gaddemane, G.; Van de Put, M.L.; Fischetti, M.V.; Moravvej-Farshi, M.K.; Pourfath, M.; Vandenberghe, W.G. Electronic transport properties of silicane determined from first principles. Materials 2019, 12, 2935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Restrepo, O.D.; Mishra, R.; Goldberger, J.E.; Windl, W. Tunable gaps and enhanced mobilities in strain-engineered silicane. J. Appl. Phys. 2014, 115, 033711. [Google Scholar] [CrossRef]
- Low, K.L.; Huang, W.; Yeo, Y.-C.; Liang, G. Ballistic transport performance of silicane and germanene transistors. IEEE Trans. Electron Devices 2014, 61, 1590–1598. [Google Scholar] [CrossRef]
- Davila, M.E.; Xian, L.; Cahangirov, S.; Rubio, A.; Lay, G.L. Germanene: A novel two-dimensional germanium allotrope akin to graphene and silicene. New J. Phys. 2014, 16, 095002. [Google Scholar] [CrossRef]
- Rawat, A.; Jena, N.; Dimple; De Sarkar, A. A comprehensive study on carrier mobility and artificial photosynthetic properties in group VI B transistion metal dichalcogenide monolayers. J. Mater. Chem. A 2018, 6, 8693–8704. [Google Scholar] [CrossRef]
- Sohier, T.; Campi, D.; Marzari, N.; Gibertini, M. Mobility of two-dimensional materials from first principles in an accurate and automated framework. Phy. Rev. Mater. 2018, 2, 114010. [Google Scholar] [CrossRef] [Green Version]
- Kaasbjerg, K.; Thygesen, K.S.; Jacobsen, K.W. Phonon-limited mobility in n-type single-layer MoS2 from first principles. Phy. Rev. B 2012, 85, 115317. [Google Scholar] [CrossRef] [Green Version]
- Jin, Z.; Li, X.; Mullen, J.T.; Kim, K.W. Intrinsic transport properties of electrons and holes in monolayer transistion-metal dichalcogenides. Phy. Rev. B 2014, 90, 045422. [Google Scholar] [CrossRef] [Green Version]
- Li, W. Electrical transport limited by electron-phonon coupling from Boltzmann transport equation: An ab initio study of Si, Al, and MoS2. Phy. Rev. B 2015, 92, 075405. [Google Scholar] [CrossRef]
- Gunst, T.; Markussen, T.; Stokbro, K.; Brandbyge, M. First-principles method for electron-phonon coupling and electron mobility: Applications to two-dimensional materials. Phy. Rev. B 2016, 91, 035414. [Google Scholar] [CrossRef] [Green Version]
- Pandey, T.; Parker, D.S.; Lindsey, L. Ab initio phonon thermal transport in monolayer InSe, GaSe, GaS and alloys. Nanotechnology 2017, 28, 455706. [Google Scholar] [CrossRef] [PubMed]
- Mermin, N.D.; Wagner, H. Absence of ferromagnetism or antiferromagnetism in one-or two-dimensional isotropic heisenberg models. Phys. Rev. Lett. 1966, 17, 1133. [Google Scholar] [CrossRef]
- Hohenberg, P.C. Existence of long-range order in one and two dimensions. Phys. Rev. 1967, 158, 383. [Google Scholar] [CrossRef]
- Fischetti, M.V.; Vandenberghe, W.G. Mermin-Wagner theorem, flexural modes, and degraded carrier mobility in two-dimensional crystals with broken horizontal mirror symmetry. Phys. Rev. B 2016, 93, 155413. [Google Scholar] [CrossRef] [Green Version]
- Sukrit, S.; Goble, N.J.; Rajesh Kumar, U.; Sankar, R.; Bogorad, Z.A.; Chou, F.C.; Chen, Y.T.; Gao, Z.P.A. Intrinsic electron mobility exceeding 103 cm2/(Vs) in multilayer InSe FETs. Nano Lett. 2015, 15, 3815–3819. [Google Scholar]
- Feng, W.; Zheng, W.; Cao, W.; Hu, P. Back gater multilayer InSe transistors with enhanced carrier mobilities via the supresssion of carrier scattering from a dielectric interface. Adv. Mater. 2014, 26, 6587–6593. [Google Scholar] [CrossRef]
- Debbichi, L.; Eriksson, O.; Lebégue, S. Two-dimensional Indium Selenides compounds: An ab initio study. J. Phy. Chem. Lett. 2015, 6, 3098–3103. [Google Scholar] [CrossRef]
- Sang, D.K.; Wang, H.; Qiu, M.; Cao, R.; Guo, Z.; Zhao, J.; Li, Y.; Xiao, Q.; Fan, D.; Zhang, H. Two dimensional β-InSe with layer-dependent properties: Band alignment, work function and optical properties. Nanomaterials 2019, 9, 82. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Han, L.; Wu, L.; Zhang, T.; Li, S.; Lu, P. Strain effect on thermoelectric performance of InSe monolayer. Nanoscale Res. Lett. 2019, 14, 287. [Google Scholar] [CrossRef]
- Kang, J.; Wells, S.A.; Sangwan, V.K.; Lam, D.; Liu, X.; Luxa, J.; Sofer, Z.; Hersam, M.C. Solution-based processing of optically active Indium Selenide. Adv. Mater. 2018, 30, 1802990. [Google Scholar] [CrossRef]
- Boukhvalov, D.W.; Gürbulak, B.; Duman, S.; Wang, L.; Politano, A.; Caputi, L.S.; Chiarello, G.; Cupolillo, A. The advent of Indium Selenide: Synthesis, electronic properties, ambient stability and applications. Nanomaterials 2017, 7, 372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Poncé, S.; Giustino, F. Dimensional crossover in the carrier mobility of Two-Dimensional semiconductors: The case of InSe. Nano Lett. 2019, 19, 1774–1781. [Google Scholar] [CrossRef] [PubMed]
- Hu, T.; Zhou, J.; Dong, J. Strain induced new phase and indirect–direct band gap transition of monolayer InSe. Phys. Chem. Chem. Phys. 2017, 19, 21722. [Google Scholar] [CrossRef] [PubMed]
- Peng, Q.; Xiong, R.; Sa, B.; Zhou, J.; Wen, C.; Wu, B.; Anpo, M.; Sun, Z. Computational mining of photocatalysts for water splitting hydrogen production: Two-dimensional InSe-family monolayers. Catal. Sci. Technol. 2017, 7, 2744. [Google Scholar] [CrossRef]
- Sun, C.; Xiang, H.; Xu, B.; Xia, Y.; Yin, J.; Liu, Z. Ab initio study of carrier mobility of few-layer InSe. Appl. Phys. Express 2016, 9, 035203. [Google Scholar] [CrossRef]
- Mounet, N.; Gibertini, M.; Schwaller, P.; Campi, D.; Merkys, A.; Marrazzo, A.; Sohier, T.; Castelli, I.E.; Cepellotti, A.; Pizzi, G.; et al. Two-dimensional materials from high-throughput computational exfoliation of experimentally known compounds. Nat. Nanotechnol. 2018, 13, 246–252. [Google Scholar] [CrossRef] [Green Version]
- Giannozzi, P.; Baroni, S.; Bonini, N.; Calandra, M.; Car, R.; Cavazzoni, C.; Ceresoli, D.; Chiarotti, G.L.; Cococcioni, M.; Dabo, I.; et al. Quantum Espresso: A modular and open-source software project for quantum simulations of materials. J. Phys. Condens. Matter 2009, 21, 395502. [Google Scholar] [CrossRef]
- Giannozzi, P.; Andreussi, O.; Brumme, T.; Bunau, O.; Nardelli, M.B.; Calandra, M.; Car, R.; Cavazzoni, C.; Ceresoli, D.; Cococcioni, M.; et al. Advanced capabilities for materials modelling with QUANTUM ESPRESSO. J. Phys. Condens. Matter 2017, 29, 465901. [Google Scholar] [CrossRef] [Green Version]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation made simple. Phys. Rev. Lett. 2019, 77, 3865. [Google Scholar] [CrossRef] [Green Version]
- Perdew, J.P.; Wang, Y. Accurate and simple analytic representation of the electron gas correlation energy. Phys. Rev. B 1992, 45, 13244. [Google Scholar] [CrossRef]
- Hamann, D.R. Optimized norm-conserving Vanderbilt pseudopotentials. Phy. Rev. B 2013, 88, 085117. [Google Scholar] [CrossRef] [Green Version]
- Baroni, S.; De Gironcoli, S.; Corso, A.D.; Giannozzi, P. Phonons and related crystal properties from density-functional perturbation theory. Rev. Mod. Phys. 2001, 73, 515. [Google Scholar] [CrossRef] [Green Version]
- Poncé, S.; Margine, E.R.; Verdi, C.; Guistino, F. EPW: Electron-phonon coupling, transport and superconducting properties using maximally localized Wannier functions. Comput. Phys. Commun. 2016, 209, 116–133. [Google Scholar] [CrossRef] [Green Version]
- Poncé, S.; Li, W.; Reichardt, S.; Giustino, F. First-principles calculations of charge carrier mobility and conductivity in bulk semiconductors and two-dimensional materials. arXiv 2019, arXiv:1908.01733. [Google Scholar]
- Verdi, C.; Giustino, F. Fröhlich electron-phonon vertex from first principles. Phys. Rev. Lett. 2015, 115, 176401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fröhlich, H. Electrons in lattice fields. Adv. Phys. 1954, 3, 325–361. [Google Scholar] [CrossRef]
- Gilat, G.; Raubenheimer, L.J. Accurate numerical method for calculating frequency-distribution functions in solids. Phys. Rev. 1966, 144, 390. [Google Scholar] [CrossRef]
- Jacoboni, C.; Reggiani, L. The Monte Carlo method for the solution of charge transport in semiconductors with applications to covalent materials. Rev. Mod. Phys. 1983, 55, 645. [Google Scholar] [CrossRef]
- Michel, K.H.; Verberck, B. Theory of elastic and piezoelectric effects in two-dimensional hexagonal boron nitride. Phy. Rev. B 2009, 80, 224301. [Google Scholar] [CrossRef]
- Kohn, W. Image of the fermi surface in the vibration spectrum of a metal. Phy. Rev. Lett. 1959, 2, 393. [Google Scholar] [CrossRef]
- Li, W.; Li, J. Piezoelectricity in two-dimensional group III monochalcogenides. Nano Res. 2015, 8, 3796–3802. [Google Scholar] [CrossRef] [Green Version]
- Li, S.S. Semiconductor Physical Electronics; Springer: New York, NY, USA, 2007. [Google Scholar]
- Gaddemane, G.; Gopalan, S.; Van de Put, M.; Fischetti, M.V. Limitations of ab initio methods to predict the electronic-transport properties of two-dimensional materials: The computational example of 2H-phase transistion metal dichalcogenides. arXiv 2019, arXiv:1912.03795. [Google Scholar]
- Jacoboni, C.; Canali, C.; Ottavani, G.; Quaranta, A.A. A review of some charge transport properties of silicon. Solid State Electron. 1977, 20, 77–89. [Google Scholar] [CrossRef]
- Zhou, H.; Cai, Y.; Zhang, G.; Zhang, Y. Unusual phonon behaviour and ultra-low thermal conductance of monolayer InSe. Nanoscale 2018, 10, 480–487. [Google Scholar] [CrossRef] [PubMed]
- Gamiz, F.; Fischetti, M.V. Monte Carlo simulation of double-gate silicon-on-insulator inversion layers: The role of volume inversion. J. Appl. Phys. 2001, 89, 5478–5487. [Google Scholar] [CrossRef]
- Tamalampudi, S.R.; Lu, Y.; Rajesh Kumar, U.; Sankar, R.; Liao, C.D.; Karukanara Moorthy, B.; Cheng, C.H.; Chou, F.C.; Chen, Y.T. High Performance and bendable few-layered InSe photodetectors with broad spectral response. Nano Lett. 2016, 14, 1263–1272. [Google Scholar]
- Zhang, Y.; Fischetti, M.V.; Sorée, B.; O’Regan, T. Theory of the hole mobility in strained Ge and III-V p-channel inversion layers with high-κ insulators. J. Appl. Phys. 2010, 108, 123713. [Google Scholar] [CrossRef] [Green Version]
- Wickramaratne, D.; Zahid, F.; Lake, R.K. Electronic and thermoelectric properties of van der Waals materials with ring-shapped valence bands. J. Appl. Phys. 2015, 118, 075101. [Google Scholar] [CrossRef] [Green Version]
- Island, J.O.; Steele, G.A.; van der Zant, H.S.J.; Castellanos-Gomez, A. Environmental instability of few-layer black phosphorus. 2D Mater. 2015, 2, 011002. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | This Work | Previous First-Principles Results |
|---|---|---|
| Lattice Constant (Å) | 4.05 | 4.09 [33], 3.95 [29], 4.04 [34], 4.09 [35] |
| In-In bond length (Å) | 2.82 | 2.82 [33], 2.81 [34], 2.83 [35] |
| In-Se bond length (Å) | 2.67 | 2.69 [33], 2.62 [29], 2.69 [34] |
| Band gap (eV) | 1.45 | 1.41 [33], 1.67 [29], 2.18 [34], 1.44 [35] |
| Parameters | QE |
|---|---|
| Kinetic energy (Ek) cutoff | 60 Ry |
| Charge density cutoff | 240 Ry |
| Ionic minimization threshold | 10−6 Ry |
| Self-consistent field threshold | 10−12 Ry |
| k-point mesh | 12 × 12 × 1 |
| Type of Scattering Processes | µe (cm2V−1s−1) | |
|---|---|---|
| GGA-PBE | LDA | |
| Total | 110 | 154 |
| Long-range interaction only | 188 | 225 |
| Short-range interaction only | 365 | 465 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gopalan, S.; Gaddemane, G.; Van de Put, M.L.; Fischetti, M.V. Monte Carlo Study of Electronic Transport in Monolayer InSe. Materials 2019, 12, 4210. https://doi.org/10.3390/ma12244210
Gopalan S, Gaddemane G, Van de Put ML, Fischetti MV. Monte Carlo Study of Electronic Transport in Monolayer InSe. Materials. 2019; 12(24):4210. https://doi.org/10.3390/ma12244210
Chicago/Turabian StyleGopalan, Sanjay, Gautam Gaddemane, Maarten L. Van de Put, and Massimo V. Fischetti. 2019. "Monte Carlo Study of Electronic Transport in Monolayer InSe" Materials 12, no. 24: 4210. https://doi.org/10.3390/ma12244210
APA StyleGopalan, S., Gaddemane, G., Van de Put, M. L., & Fischetti, M. V. (2019). Monte Carlo Study of Electronic Transport in Monolayer InSe. Materials, 12(24), 4210. https://doi.org/10.3390/ma12244210