First-Principles Investigation of Adsorption of Ag on Defected and Ce-doped Graphene

Abstract
:1. Introduction
2. Theoretical Method
3. Results and Discussion
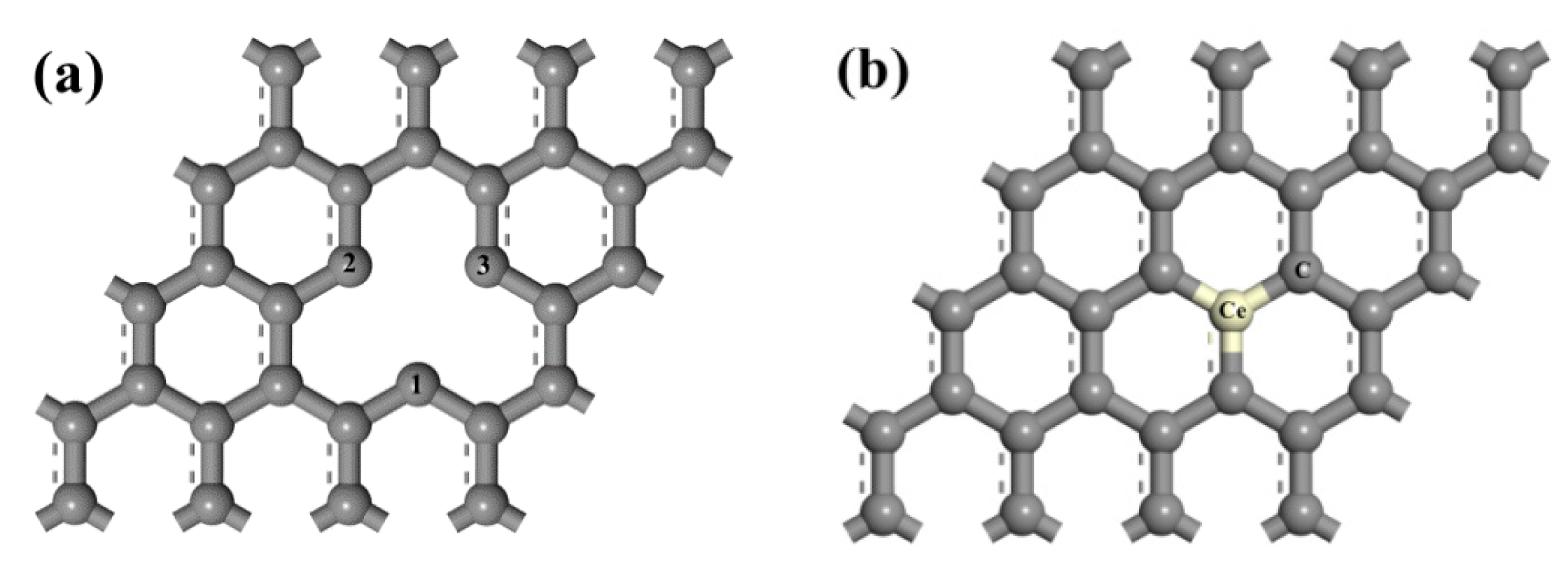
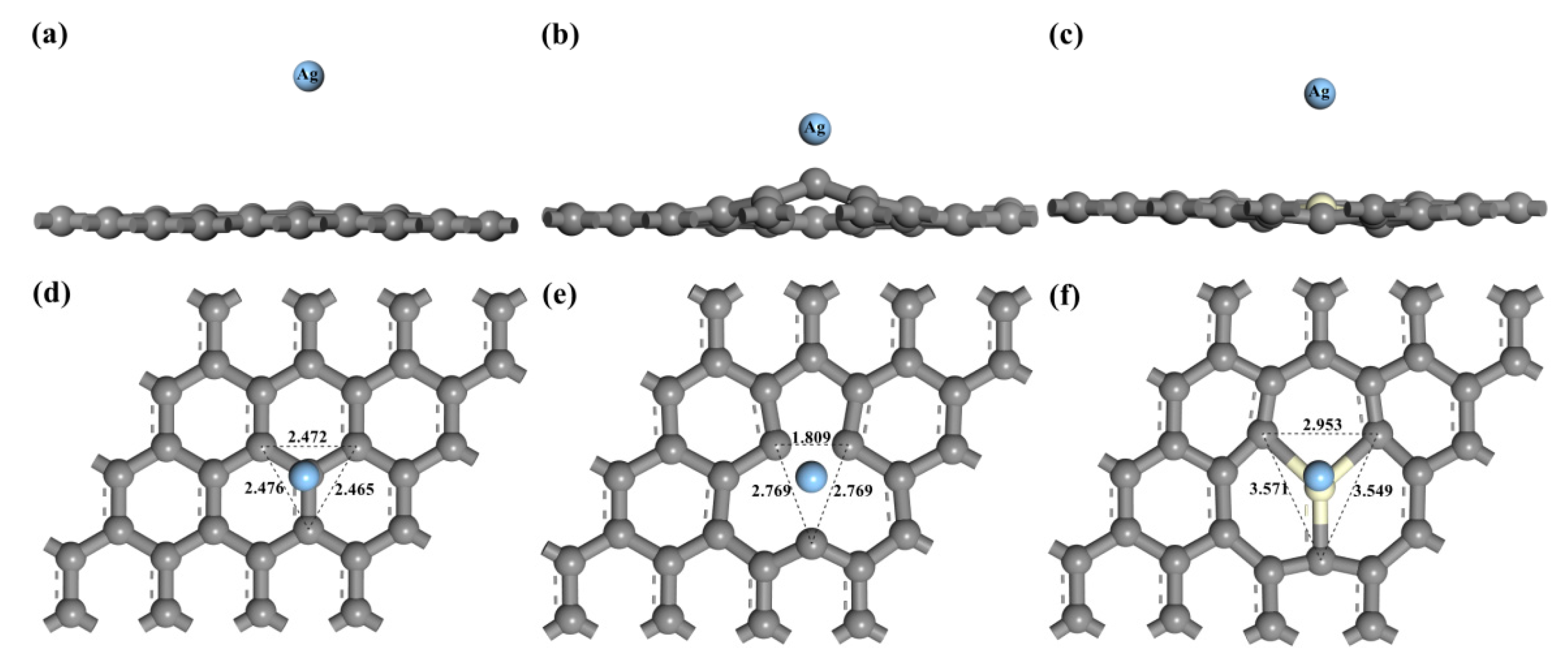
3.1. Adsorption Energy and Charge Transfer
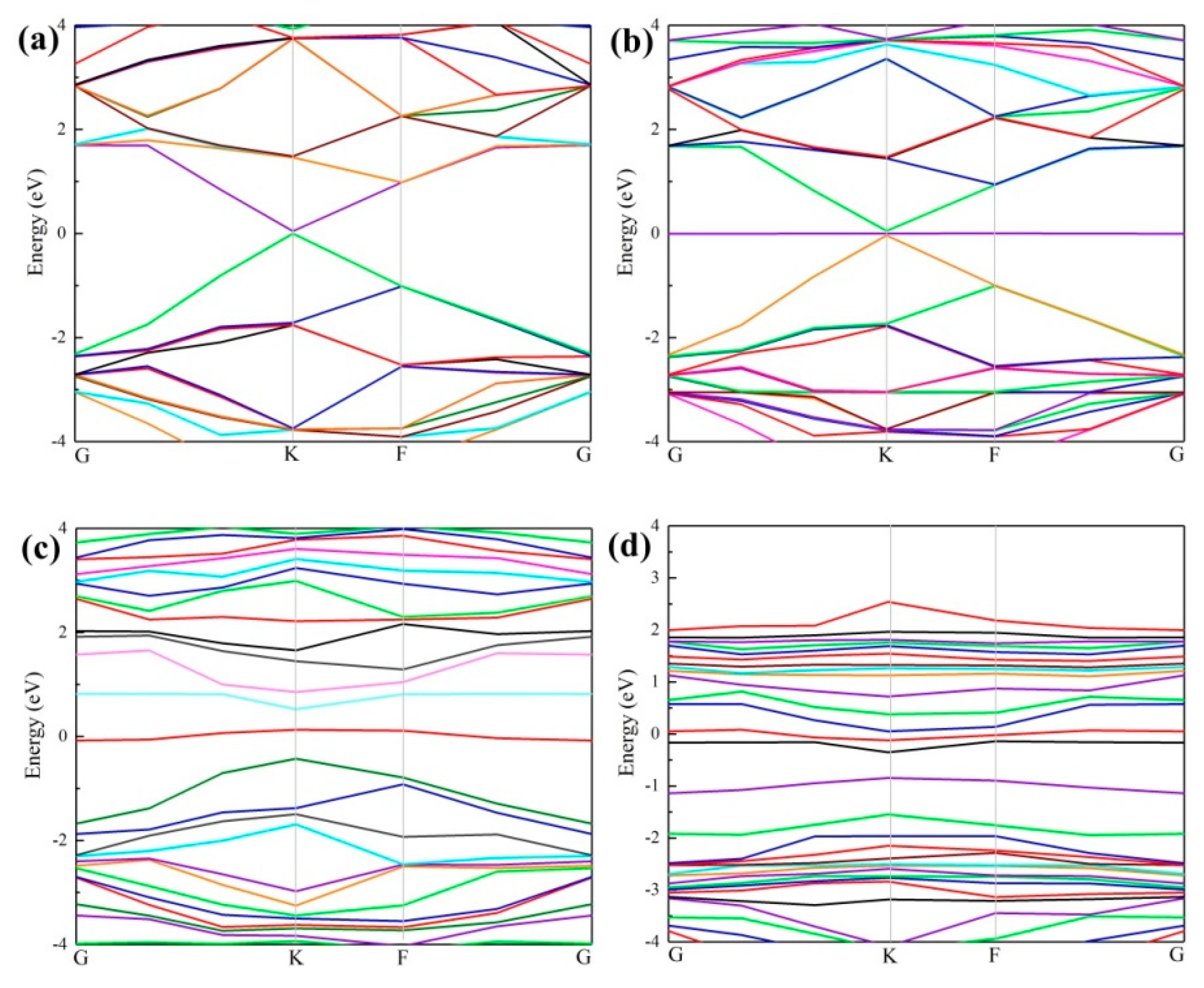
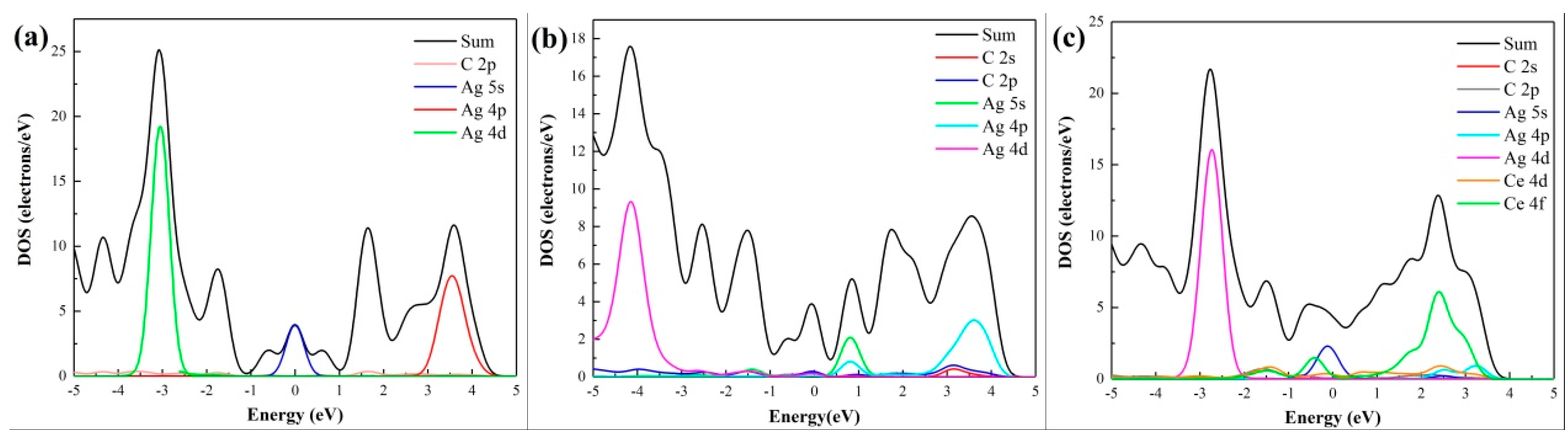
3.2. Density of States
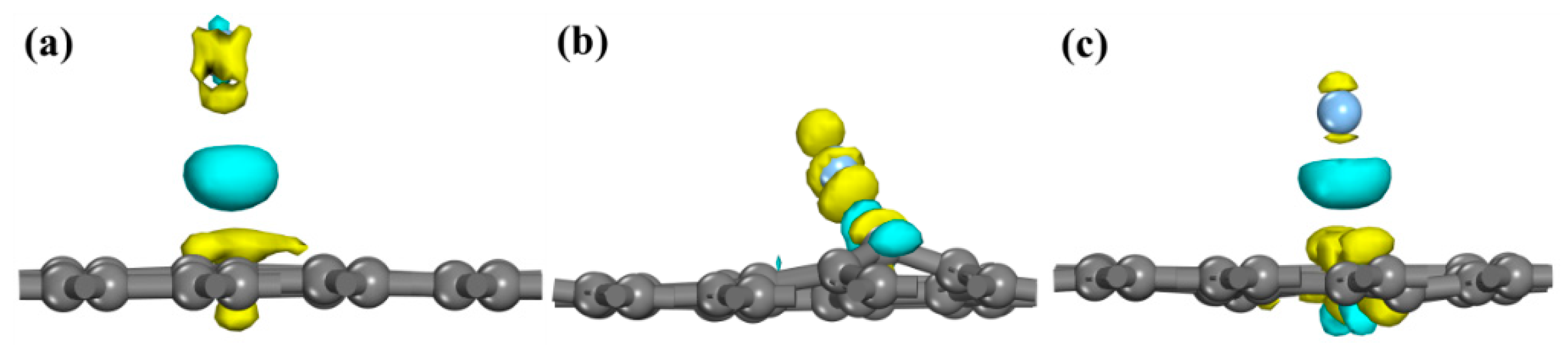
3.3. Population Analysis
4. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Lai, Z.; Xue, S.; Han, X.; Gu, L.; Gu, W. Study on Microstructure and Property of Brazed Joint of AgCuZn-X(Ga, Sn, In, Ni) Brazing Alloy. Rare Metal Mater. Eng. 2010, 39, 397–400. [Google Scholar]
- Zhang, L.; Feng, J.; Zhang, B.; Jing, X. Ag–Cu–Zn alloy for brazing TiC cermet/steel. Mater. Lett. 2005, 59, 110–113. [Google Scholar] [CrossRef]
- Wang, H.; Xue, S.-B. Effect of Ag on the properties of solders and brazing filler metals. J. Mater. Sci. Mater. Electron. 2015, 27, 1–13. [Google Scholar] [CrossRef]
- Khorram, A.; Ghoreishi, M. Comparative study on laser brazing and furnace brazing of Inconel 718 alloys with silver based filler metal. Opt. Laser Technol. 2015, 68, 165–174. [Google Scholar] [CrossRef]
- Chen, Y.; Yun, D.; Sui, F.; Long, W.; Zhang, G.; Liu, S. Influence of sulphur on the microstructure and properties of Ag–Cu–Zn brazing filler metal. Mater. Sci. Technol. 2013, 29, 1267–1271. [Google Scholar] [CrossRef]
- Sui, F.; Long, W.; Liu, S.; Zhang, G.; Bao, L.; Li, H.; Chen, Y. Effect of calcium on the microstructure and mechanical properties of brazed joint using Ag–Cu–Zn brazing filler metal. Mater. Des. 2013, 46, 605–608. [Google Scholar] [CrossRef]
- Demianová, K.; Behúlová, M.; Milan, O.; Turňa, M.; Sahul, M. Brazing of Aluminum Tubes Using Induction Heating. Adv. Mater. Res. 2012, 1405–1409. [Google Scholar] [CrossRef]
- Shao, L.; Chen, G.; Ye, H.; Wu, Y.; Qiao, Z.; Zhu, Y.; Niu, H. Sulfur dioxide adsorbed on graphene and heteroatom-doped graphene: A first-principles study. Eur. Phys. J. B 2013, 86, 54. [Google Scholar] [CrossRef]
- He, Y.L.; Liu, D.X.; Qu, Y.; Yao, Z. Adsorption of Hydrogen Molecule on the Intrinsic and Al-Doped Graphene: A First Principle Study. Adv. Mater. Res. 2012, 507, 61–64. [Google Scholar] [CrossRef] [Green Version]
- Sen, D.; Thapa, R.; Chattopadhyay, K. Small Pd cluster adsorbed double vacancy defect graphene sheet for hydrogen storage: A first-principles study. Int. J. Hydrog. Energy 2013, 38, 3041–3049. [Google Scholar] [CrossRef]
- Thirumal, V.; Pandurangan, A.; Jayavel, R.; Ilangovan, R. Synthesis and characterization of boron doped graphene nanosheets for supercapacitor applications. Synth. Metals 2016, 220, 524–532. [Google Scholar] [CrossRef]
- Cai, Z.; Xiong, H.; Zhu, Z.; Huang, H.; Li, L.; Huang, Y.; Yu, X. Electrochemical synthesis of graphene/polypyrrole nanotube composites for multifunctional applications. Synth. Metals 2017, 227, 100–105. [Google Scholar] [CrossRef]
- Song, X.-R.; Li, H.-J.; Zeng, X. Brazing of C/C composites to Ti6Al4V using multiwall carbon nanotubes reinforced TiCuZrNi brazing alloy. J. Alloys Compd. 2016, 664, 175–180. [Google Scholar] [CrossRef]
- Wang, X.; Xing, W.; Zhang, P.; Song, L.; Yang, H.; Hu, Y. Covalent functionalization of graphene with organosilane and its use as a reinforcement in epoxy composites. Compos. Sci. Technol. 2012, 72, 737–743. [Google Scholar] [CrossRef]
- Qi, J.L.; Wang, Z.Y.; Lin, J.H.; Zhang, T.Q.; Zhang, A.T.; Cao, J.; Zhang, L.X.; Feng, J.C. Graphene-enhanced Cu composite interlayer for contact reaction brazing aluminum alloy 6061. Vacuum 2017, 136, 142–145. [Google Scholar] [CrossRef]
- Chang, K.; Chen, W. L-cysteine-assisted synthesis of layered MoS2/graphene composites with excellent electrochemical performances for lithium ion batteries. ACS Nano 2011, 5, 4720–4728. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Guqiao, D. Research progress in graphene reinforced metal matrix composites. Electron. Compon. Mater. 2017, 36, 78. [Google Scholar]
- Ma, D.; Wu, P. Improved microstructure and mechanical properties for Sn58Bi0.7Zn solder joint by addition of graphene nanosheets. J. Alloys Compd. 2016, 671, 127–136. [Google Scholar] [CrossRef]
- Huang, Y.; Xiu, Z.; Wu, G.; Tian, Y.; He, P. Sn–3.0Ag–0.5Cu nanocomposite solders reinforced by graphene nanosheets. J. Mater. Sci. Mater. Electron. 2016, 27, 6809–6815. [Google Scholar] [CrossRef]
- Huang, Y.; Xiu, Z.; Wu, G.; Tian, Y.; He, P.; Gu, X.; Long, W. Improving shear strength of Sn-3.0Ag-0.5Cu/Cu joints and suppressing intermetallic compounds layer growth by adding graphene nanosheets. Mater. Lett. 2016, 169, 262–264. [Google Scholar] [CrossRef]
- Hu, X.; Chan, Y.; Zhang, K.; Yung, W.K.C. Effect of graphene doping on microstructural and mechanical properties of Sn–8Zn–3Bi solder joints together with electromigration analysis. J. Alloys Compd. 2013, 580, 162–171. [Google Scholar] [CrossRef]
- Liu, X.; Han, Y.; Jing, H.; Wei, J.; Xu, L. Effect of graphene nanosheets reinforcement on the performance of Sn-Ag-Cu lead-free solder. Mater. Sci. Eng. A 2013, 562, 25–32. [Google Scholar] [CrossRef]
- Chen, F.; Gupta, N.; Behera, R.K.; Rohatgi, P.K. Graphene-Reinforced Aluminum Matrix Composites: A Review of Synthesis Methods and Properties. JOM 2018, 70, 837–845. [Google Scholar] [CrossRef]
- Wang, S.; Zhang, Y.; Abidi, N.; Cabrales, L. Wettability and Surface Free Energy of Graphene Films. Langmuir 2009, 25, 11078–11081. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Michaelides, A.; Alfè, D.; Schimka, L.; Kresse, G.; Wang, E. Adsorption and diffusion of water on graphene from first principles. Phys. Rev. B 2011, 84, 033402. [Google Scholar] [CrossRef]
- Kysilka, J.; Rubes, M.; Grajciar, L.; Nachtigall, P.; Bludsky, O. Accurate Description of Argon and Water Adsorption on Surfaces of Graphene-Based Carbon Allotropes. J. Phys. Chem. A 2011, 115, 11387–11393. [Google Scholar] [CrossRef] [PubMed]
- Błoński, P.; Otyepka, M. First-principles study of the mechanism of wettability transition of defective graphene. Nanotechnology 2017, 28, 64003. [Google Scholar] [CrossRef]
- Ashraf, A.; Wu, Y.; Wang, M.C.; Yong, K.; Sun, T.; Jing, Y.; Haasch, R.T.; Aluru, N.R.; Nam, S. Doping-Induced Tunable Wettability and Adhesion of Graphene. Nano Lett. 2016, 16, 4708–4712. [Google Scholar] [CrossRef]
- Dazhi, F.; Guili, L.; Shuang, Z. Effects of vacancy and deformation on an Al atom adsorbed on graphene. Chin. J. Phys. 2018, 56, 689–695. [Google Scholar] [CrossRef]
- Liu, Y.; An, L.; Gong, L. First-principles study of Cu adsorption on vacancy-defected/Au-doped graphene. Mod. Phys. Lett. B 2018, 32, 1850139. [Google Scholar] [CrossRef]
- Wu, C.M.L.; Yu, D.Q.; Law, C.M.T.; Wang, L. Properties of lead-free filler alloys with rare earth element additions. Mater. Sci. Eng. R 2004, 44, 1–44. [Google Scholar] [CrossRef]
- Yang, C.; Xu, J.; Ding, W.; Chen, Z.; Fu, Y. Effect of cerium on microstructure, wetting and mechanical properties of Ag-Cu-Ti filler alloy. J. Rare Earths 2009, 27, 1051–1055. [Google Scholar] [CrossRef]
- Clark, S.J.; Segall, M.D.; Pickard, C.J.; Hasnip, P.J.; Payne, M.C. First principles methods using castep. Z. Kristallogr. 2005, 220, 567–570. [Google Scholar] [CrossRef]
- Milman, V.; Refson, K.; Clark, S.; Pickard, C.; Yates, J.; Gao, S.-P.; Hasnip, P.; Probert, M.; Perlov, A.; Segall, M.; et al. Electron and vibrational spectroscopies using DFT, plane waves and pseudopotentials: CASTEP implementation. J. Mol. Struct. THEOCHEM 2010, 954, 22–35. [Google Scholar] [CrossRef]
- Becke, A.D. A new mixing of Hartree–Fock and local density-functional theories. J. Chem. Phys. 1993, 98, 1372–1377. [Google Scholar] [CrossRef]
- Car, R.; Parrinello, M. Unified Approach for Molecular Dynamics and Density-Functional Theory. Phys. Rev. Lett. 1985, 55, 2471–2474. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P.; Chevary, J.A.; Vosko, S.H.; Jackson, K.A.; Pederson, M.R.; Singh, D.J.; Fiolhais, C. Erratum: Atoms, molecules, solids, and surfaces: Applications of the generalized gradient approximation for exchange and correlation. Phys. Rev. B 1993, 48, 4978. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Wang, Y. Generalized gradient approximation for the exchange-correlation hole of a many-electron system. Phys. Rev. B 1996, 54, 16533–16539. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Wan, W.; Wang, H. First-Principles Investigation of Adsorption and Diffusion of Ions on Pristine, Defective and B-doped Graphene. Materials 2015, 8, 6163–6178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Granatier, J.; Lazar, P.; Prucek, R.; Šafářová, K.; Zbořil, R.; Otyepka, M.; Hobza, P. Interaction of Graphene and Arenes with Noble Metals. J. Phys. Chem. C 2012, 116, 14151–14162. [Google Scholar] [CrossRef]
- Del Castillo, R.M.; Sansores, L.E. Study of the electronic structure of Ag, Au, Pt and Pd clusters adsorption on graphene and their effect on conductivity. Eur. Phys. J. B 2015, 88, 248. [Google Scholar] [CrossRef]
- Amft, M.; Lebègue, S.; Eriksson, O.; Skorodumova, N.V. Adsorption of Cu, Ag, and Au atoms on graphene including van der Waals interactions. J. Phys. Condens. Matter 2011, 23, 395001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.; He, C.; Li, T.; Gong, S.; Zhao, L.; Tao, J. First-principles study on the electronic and magnetic properties of armchair graphane/graphene heterostructure nanoribbons. Solid State Commun. 2015, 211, 23–28. [Google Scholar] [CrossRef]
- Jiang, Z.; Zhang, Y.; Störmer, H.L.; Kim, P. Quantum Hall States near the Charge-Neutral Dirac Point in Graphene. Phys. Rev. Lett. 2007, 99, 106802. [Google Scholar] [CrossRef]
- Yu, W.J.; Liao, L.; Chae, S.H.; Lee, Y.H.; Duan, X. Toward Tunable Band Gap and Tunable Dirac Point in Bilayer Graphene with Molecular Doping. Nano Lett. 2011, 11, 4759–4763. [Google Scholar] [CrossRef] [Green Version]
- De Lara-Castells, M.P.; Mitrushchenkov, A.O.; Stoll, H. Combining density functional and incremental post-Hartree-Fock approaches for van der Waals dominated adsorbate-surface interactions: Ag2/graphene. J. Chem. Phys. 2015, 143, 102804. [Google Scholar] [CrossRef]
- Ashrafian, S.; Jahanshahi, M.; Ganji, M.D.; Agheb, R. Greatly enhanced adsorption of platinum on periodic graphene nanobuds: A first-principles study. Appl. Surf. Sci. 2015, 351, 1105–1115. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type of Graphene | D/Å | Ead/eV | Q/e | ∆h/Å | |||
|---|---|---|---|---|---|---|---|
| T | B | H | |||||
| Intrinsic graphene | 1.564 | −0.011 | −0.009 | −0.010 | 0.09 | 0.055 | |
| VG | 1.025 | −2.358 | −2.226 | −2.439 | 0.78 | 0.380 | |
| Ce-doped graphene | 1.530 | TCe: −2.241 | TC: −2.230 | −2.226 | −2.218 | Ce: 1.96 | 0.147 |
| Ag: −0.17 | |||||||
| Adsorption System | Bond | Population | Length/10−1 nm | P/charge·nm−1 |
|---|---|---|---|---|
| VG | Ag–C1 | 0.54 | 2.14657 | 2.516 |
| Ag–C2 | −0.06 | 2.51845 | −0.238 | |
| Ag–C2 | −0.06 | 2.52454 | −0.238 | |
| Ce-G | Ag–Ce | 0.28 | 2.99968 | 0.933 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fan, Z.; Hu, M.; Liu, J.; Luo, X.; Zhang, K.; Tang, Z. First-Principles Investigation of Adsorption of Ag on Defected and Ce-doped Graphene. Materials 2019, 12, 649. https://doi.org/10.3390/ma12040649
Fan Z, Hu M, Liu J, Luo X, Zhang K, Tang Z. First-Principles Investigation of Adsorption of Ag on Defected and Ce-doped Graphene. Materials. 2019; 12(4):649. https://doi.org/10.3390/ma12040649
Chicago/Turabian StyleFan, Zhou, Min Hu, Jianyi Liu, Xia Luo, Kun Zhang, and Zhengchao Tang. 2019. "First-Principles Investigation of Adsorption of Ag on Defected and Ce-doped Graphene" Materials 12, no. 4: 649. https://doi.org/10.3390/ma12040649
APA StyleFan, Z., Hu, M., Liu, J., Luo, X., Zhang, K., & Tang, Z. (2019). First-Principles Investigation of Adsorption of Ag on Defected and Ce-doped Graphene. Materials, 12(4), 649. https://doi.org/10.3390/ma12040649