Synthesis and Luminescence of Optical Memory Active Tetramethylammonium Cyanocuprate(I) 3D Networks

 , and
, and
Abstract
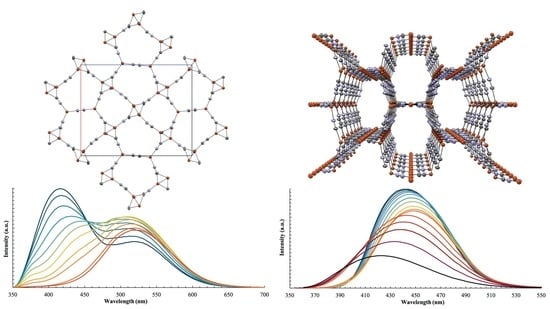
:
1. Introduction
2. Materials and Methods
2.1. General
2.2. Synthesis
2.3. X-Ray Analysis
2.4. Photophysical Studies
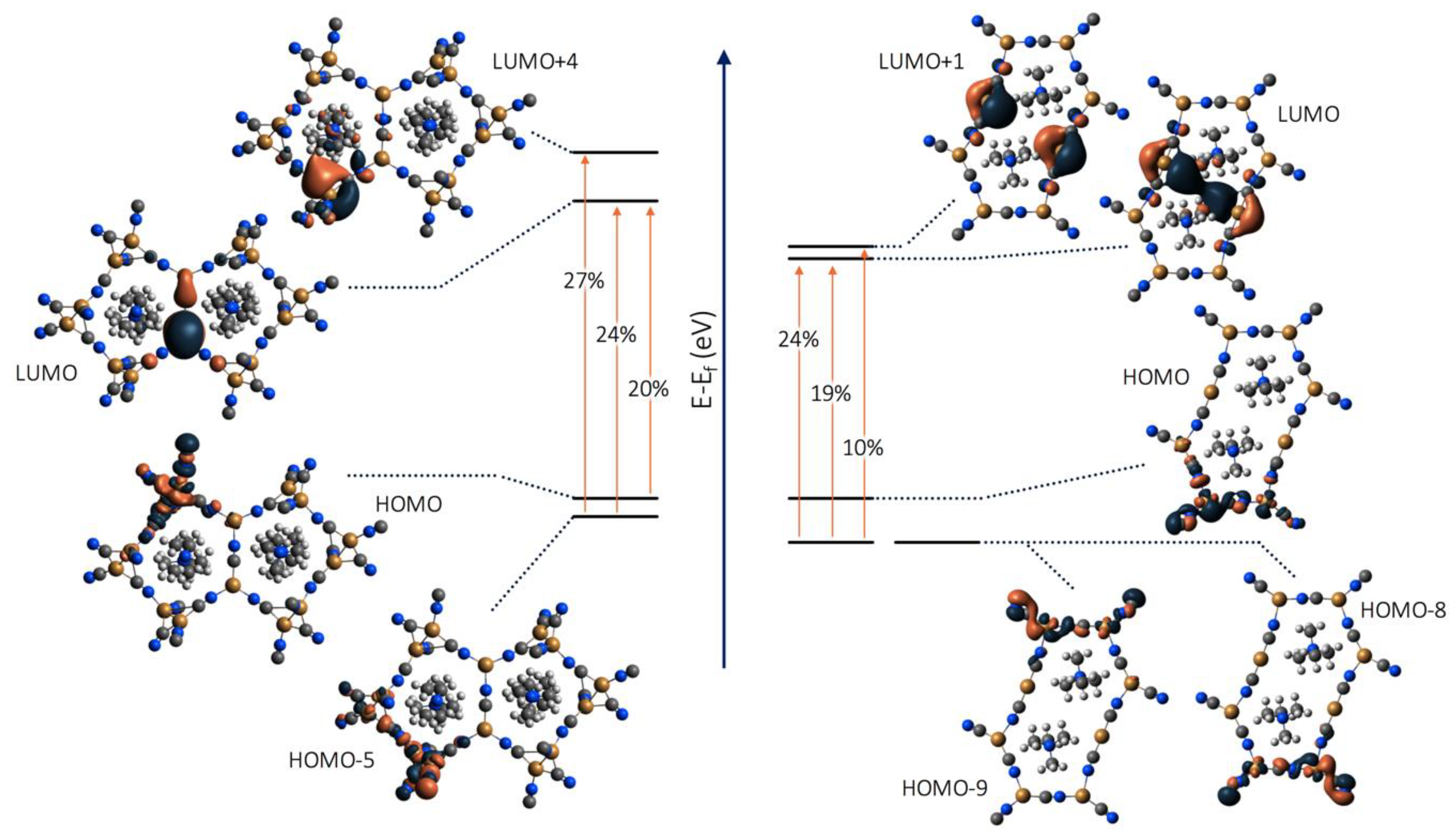
2.5. Molecular Modelling
3. Results
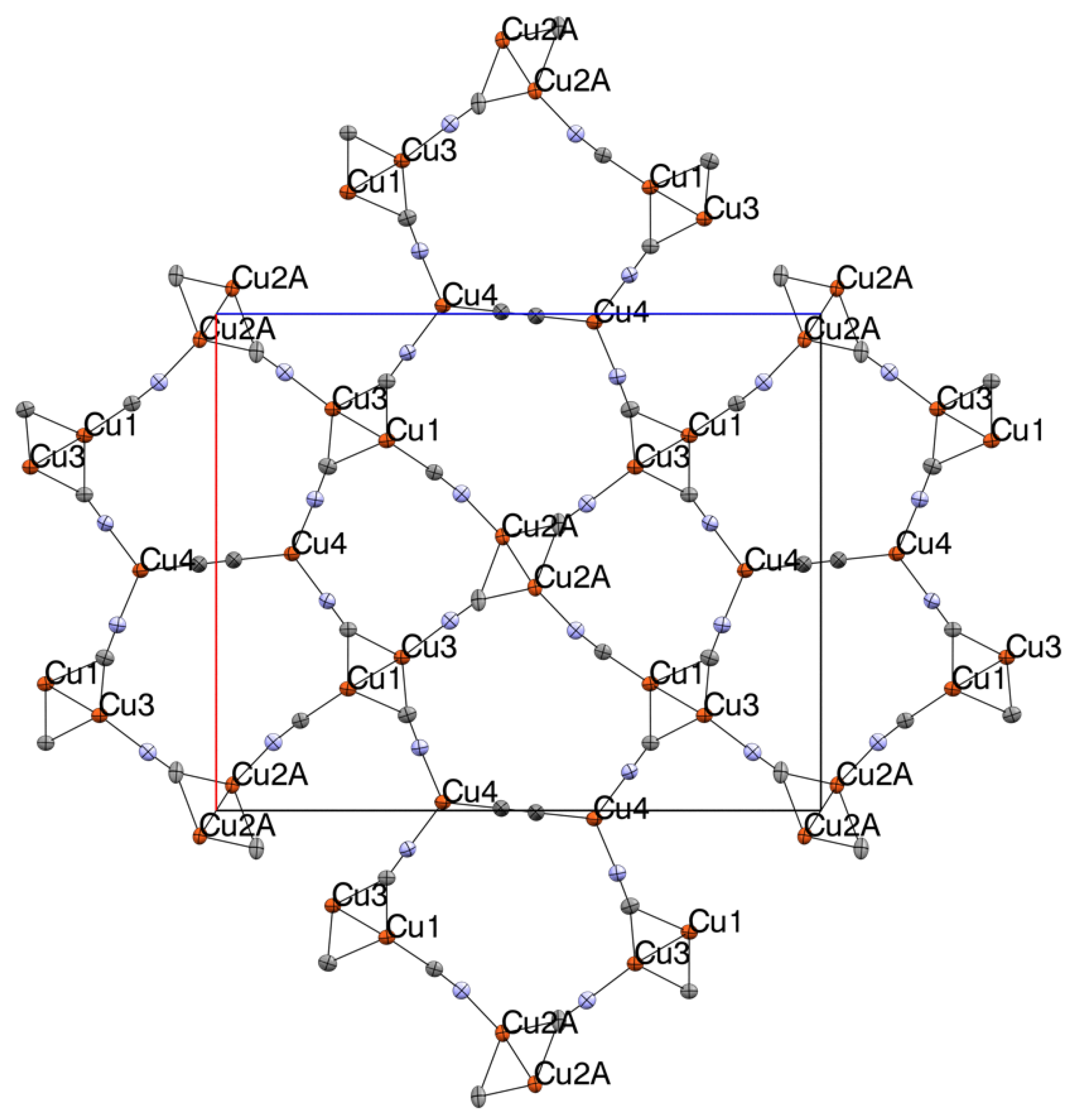
3.1. Structural Studies
3.2. Infrared Characterization
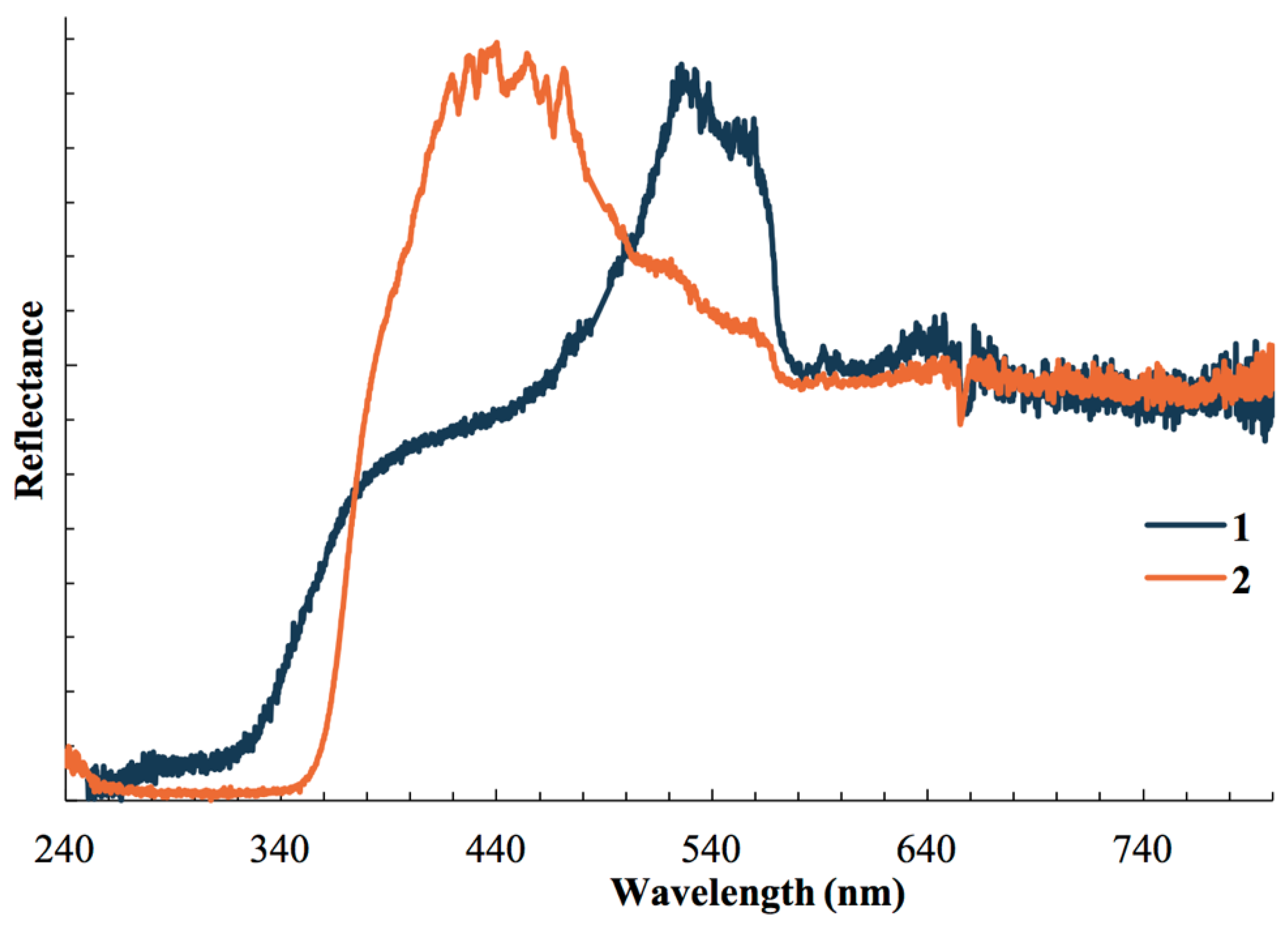
3.3. Photophysical Studies
3.4. Optical Memory Behavior
3.5. Optical Memory Mechanism
| (1) Cu(CN)2− + Cu(CN)2− + hⱱ ➝ 2Cu2+ + 4CN− + 2e− | Eo = −1.103 V |
| (2) Cu2+ + 2e− ➝ Cu | Eo = 0.339 V |
| (3) Cu2+ + Cu ➝ 2Cu+ | Eo = −0.357 V |
| (4) Cu + 2CN− ➝ Cu(CN)2− + e− | Eo = 0.429 V |
| (5) Cu2+ + e− ➝ Cu+ | Eo = 0.161 V |
| (6) Cu+ + 2CN− ➝ Cu(CN)2− | Eo = 0 V |
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Grifasi, F.; Chierotti, M.R.; Garino, C.; Gobetto, R.; Priola, E.; Diana, E.; Turci, F. Solvent-Free Synthesis of Luminescent Copper(I) Coordination Polymers with Thiourea Derivatives. Cryst. Growth Des. 2015, 15, 2929–2939. [Google Scholar] [CrossRef]
- Safko, J.P.; Kuperstock, J.E.; McCullough, S.M.; Noviello, A.M.; Li, X.; Killarney, J.P.; Murphy, C.; Patterson, H.H.; Bayse, C.A.; Pike, R.D. Network Formation and Photoluminescence in Copper(I) Halide Complexes with Substituted Piperazine Ligands. Dalton Trans. 2012, 41, 11663–11674. [Google Scholar] [CrossRef]
- Mi, J.L.; Murray, C.A.; Tronic, T.A.; Dekrafft, K.E.; Ley, A.N.; DeButts, J.C.; Pike, R.D.; Lu, H.; Patterson, H.H. Copper(I) Cyanide Networks: Synthesis, Structure, and Luminescence Behavior. Part 2. Piperazine Ligands and Hexamethylenetetramine. Inorg. Chem. 2008, 47, 6931–6947. [Google Scholar]
- Miller, K.M.; McCullough, S.M.; Lepekhina, E.A.; Thibau, I.J.; Pike, R.D.; Li, X.; Killarney, J.P.; Patterson, H.H. Copper(I) Thiocyanate-Amine Networks: Synthesis, Structure, and Luminescence Behavior. Inorg. Chem. 2011, 50, 7239–7249. [Google Scholar] [CrossRef] [PubMed]
- Tronic, T.A.; DeKrafft, K.E.; Lim, M.J.; Ley, A.N.; Pike, R.D. Copper(I) Cyanide Networks: Synthesis, Luminescence Behavior and Thermal Analysis. Part 1. Diimine Ligands. Inorg. Chem. 2007, 46, 8897–8912. [Google Scholar] [CrossRef] [PubMed]
- Deaton, J.C.; Switalski, S.C.; Kondakov, D.Y.; Young, R.H.; Pawlik, T.D.; Giesen, D.J.; Harkins, S.B.; Miller, A.J.M.; Mickenberg, S.F.; Peters, J.C. E-Type Delayed Fluorescence of a Phosphine-Supported Cu2(μ-NAr2)2 Diamond Core: Harvesting Singlet and Triplet Excitons in OLEDs. J. Am. Chem. Soc. 2010, 132, 9499–9508. [Google Scholar] [CrossRef]
- Czerwieniec, R.; Yersin, H. Diversity of Copper(I) Complexes Showing Thermally Activated Delayed Fluorescence: Basic Photophysical Analysis. Inorg. Chem. 2015, 54, 4322–4327. [Google Scholar] [CrossRef]
- Hofbeck, T.; Monkowius, U.; Yersin, H. Highly Efficient Luminescence of Cu(I) Compounds: Thermally Activated Delayed Fluorescence Combined with Short-Lived Phosphorescence. J. Am. Chem. Soc. 2015, 137, 399–404. [Google Scholar] [CrossRef]
- Li, S.-L.; Wang, J.; Zhang, F.-Q.; Zhang, X.-M. Light and Heat Dually Responsive Luminescence in Organic Templated CdSO4-Type Halogeno(cyano)cuprates with Disorder of Halogenide/Cyanide. Cryst. Growth Des. 2017, 17, 746–752. [Google Scholar] [CrossRef]
- Vitale, M.; Ryu, C.K.; Palke, W.E.; Ford, P.C. Ab Initio Studies of the Copper(I) Tetramers Cu4X4L4 (X = I, Br, Cl). Effects of Cluster Structure and of Halide on Photophysical Properties. Inorg. Chem. 1994, 33, 561–566. [Google Scholar] [CrossRef]
- Kyle, K.R.; Palke, W.E.; Ford, P.C. The Photoluminescence Properties of the Copper(I) Clusters Cu4I4A4 (A = Aromatic Amine) in Solution. Coord. Chem. Rev. 1990, 97, 35–46. [Google Scholar] [CrossRef]
- De Angelis, F.; Fantacci, S.; Sgamellotti, A.; Cariati, E.; Ugo, R.; Ford, P.C. Electronic Transitions Involved in the Absorption Spectrum and Dual Luminescence of Tetranuclear Cubane [Cu4I4(pyridine)4] Cluster: A Density Functional Theory/Time-Dependent Density Functional Theory Investigation. Inorg. Chem. 2006, 45, 10576–10584. [Google Scholar] [CrossRef]
- Royzman, D.E.; Noviello, A.M.; Henline, K.M.; Pike, R.D.; Killarney, J.P.; Patterson, H.H.; Crawford, C.; Assefa, Z. Structure, Luminescence, and Vapochromism of Bridged Cationic Copper(I) Dimers and Polymers. J. Inorg. Organomet. Polym. Mater. 2014, 24, 66–77. [Google Scholar] [CrossRef]
- Ley, A.N.; Dunaway, L.E.; Brewster, T.P.; Dembo, M.D.; Harris, T.D.; Baril-Robert, F.; Li, X.; Patterson, H.H.; Pike, R.D. Reversible Luminescent Reaction of Amines with Copper(I) Cyanide. Chem. Commun. 2010, 46, 4565. [Google Scholar] [CrossRef]
- Killarney, J.P.; McKinnon, M.; Murphy, C.; Henline, K.M.; Wang, C.; Pike, R.D.; Patterson, H.H. Amine- and Sulfide-Sensing Copper(I) Iodide Films. Inorg. Chem. Commun. 2014, 40, 18–21. [Google Scholar] [CrossRef]
- Nicholas, A.D.; Otten, B.M.; Ayala, G.; Hutchinson, J.; Wojtas, L.; Omary, M.A.; Pike, R.D.; Patterson, H.H. Light-Induced Photochemical Changes in Copper(I) Thiocyanate Complexes Decorated with Halopyridines: Optical Memory Manifestation. J. Phys. Chem. C 2017, 121, 25430–25439. [Google Scholar] [CrossRef]
- Li, X.; Pan, Z.; Welch, D.A.; Baril-Robert, F.; Pike, R.D.; Patterson, H.H. An Unusual Luminescent Anionic Copper(I) System: Dicyanocuprate(I) Ion in Nano and Bulky States. J. Phys. Chem. C 2012, 116, 26656–26667. [Google Scholar] [CrossRef]
- Nicholas, A.D.; Welch, D.A.; Li, X.; Patterson, H.H. Host lattice Effects on the Design of Different Metallophilic Nanoclusters with Novel Photonic Properties. Inorg. Chim. Acta 2018, 471, 40–49. [Google Scholar] [CrossRef]
- Tyson, D.S.; Bignozzi, C.A.; Castellano, F.N. Metal−Organic Approach to Binary Optical Memory. J. Am. Chem. Soc. 2002, 124, 4562–4563. [Google Scholar] [CrossRef]
- Rath, S.; Heilig, M.; Port, H.; Wrachtrup, J. Periodic Organic Nanodot Patterns for Optical Memory. Nano Lett. 2007, 7, 3845–3848. [Google Scholar] [CrossRef]
- Hollingsworth, G.; Barrie, J.D.; Dunn, B.; Zink, J.I. Optical Memory in Cu+-Doped/Beta-alumina. J. Am. Chem. Soc. 1988, 110, 6569–6570. [Google Scholar] [CrossRef]
- Dvornikov, A.S.; Walker, E.P.; Rentzepis, P.M. Two-Photon Three-Dimensional Optical Storage Memory. J. Phys. Chem. A 2009, 113, 13633–13644. [Google Scholar] [CrossRef]
- Omary, M.A.; Colis, J.C.F.; Larochelle, C.L.; Patterson, H.H. Optical Memory and Multistep Luminescence Thermochromism in Single Crystals of K2Na[Ag(CN)2]3. Inorg. Chem. 2007, 46, 3798–3800. [Google Scholar] [CrossRef]
- Patterson, H.H.; Bourassa, J.; Shankle, G. Light-Induced Electron Transfer in Lead(II)Gold(I) Dicyanide. Inorg. Chim. Acta 1994, 226, 345–348. [Google Scholar] [CrossRef]
- Bandara, H.M.D.; Burdette, S.C. Photoisomerization in Different Classes of Azobenzene. Chem. Soc. Rev. 2012, 41, 1809–1825. [Google Scholar] [CrossRef]
- Crecca, C.R.; Roitberg, A.E. Theoretical Study of the Isomerization Mechanism of Azobenzene and Disubstituted Azobenzene Derivatives. J. Phys. Chem. A 2006, 110, 8188–8203. [Google Scholar] [CrossRef]
- Sato, H.; Sato, F.; Yamagishi, A. Rewritable Optical Memory in Liquid Crystals Containing Photo-Epimerizing Cr(III) Complexes. Chem. Commun. 2013, 49, 4773–4775. [Google Scholar] [CrossRef]
- Pariani, G.; Castagna, R.; Dassa, G.; Hermes, S.; Vailati, C.; Bianco, A.; Bertarelli, C. Diarylethene-Based Photochromic Polyurethanes for Multistate Optical Memories. J. Mater. Chem. 2011, 21, 13223–13231. [Google Scholar] [CrossRef]
- Frolova, L.A.; Rezvanova, A.A.; Lukyanov, B.S.; Sanina, N.A.; Troshin, P.A.; Aldoshin, S.M. Design of Rewritable and Read-Only Non-Volatile Optical Memory Elements using Photochromic Spiropyran-Based Salts as Light-Sensitive Materials. J. Mater. Chem. C 2015, 3, 11675–11680. [Google Scholar] [CrossRef]
- Li, X.; Patterson, H. A Review of Luminescent Anionic Nano System: d10 Metallocyanide Excimers and Exciplexes in Alkali Halide Hosts. Materials (Basel) 2013, 6, 2595–2611. [Google Scholar] [CrossRef]
- Nicholas, A.D.; Bullard, R.M.; Pike, R.D.; Patterson, H.H. Photophysical Investigation of Silver/Gold Dicyanometallates and Tetramethylammonium Networks: An Experimental and Theoretical Investigation. Eur. J. Inorg. Chem. 2019, 2019, 956–962. [Google Scholar] [CrossRef]
- SMART. Apex II Data Collection Software; Bruker: Madison, WI, USA, 2005. [Google Scholar]
- Saint Plus. Saint Plus Data Reduction Software; Bruker: Madison, WI, USA, 2005. [Google Scholar]
- Sheldrick, G.M. SADABS; Bruker: Madison, WI, USA, 2005. [Google Scholar]
- Sheldrick, G.M. A Short History of SHELX. Acta Crystallogr. Sect. A 2008, 64, 112–122. [Google Scholar] [CrossRef]
- Huebschle, C.B.; Sheldrick, G.M.; Dittrich, B. ShelXle: A Qt graphical user interface for SHELXL. J. Appl. Crystallogr. 2011, 44, 1281–1284. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 16, Revision B.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Becke, A.D. Density-Functional Thermochemistry. III. The Role of Exact Exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron Density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Dunning, T.H., Jr.; Hay, P.J. Modern Theoretical Chemistry; Schaefer, H.F., Ed.; Plenum: New York, NY, USA, 1977; Volume 3. [Google Scholar]
- Stoll, H.; Fuentealba, P.; Schwerdtfeger, P.; Flad, J.; Szentpály, L.V.; Preuss, H. Cu and Ag as One-Valence-Electron Atoms: CI Results and Quadrupole Corrections for Cu2, Ag2, CuH, and AgH. J. Chem. Phys. 1984, 81, 2732–2736. [Google Scholar] [CrossRef]
- Veillard, A. (Ed.) Quantum Chemistry: The Challenge of Transition Metals and Coordination Chemistry; Springer: Dordrecht, The Netherlands, 1986; ISBN 978-90-277-2237-9. [Google Scholar]
- Dolg, M.; Wedig, U.; Stoll, H.; Preuss, H. Energy-Adjusted Ab Initio Pseudopotentials for the First Row Transition Elements. J. Chem. Phys. 1987, 86, 866–872. [Google Scholar] [CrossRef]
- Fuentealba, P.; Stoll, H.; von Szentpaly, L.; Schwerdtfeger, P.; Preuss, H. On the Reliability of Semi-Empirical Pseudopotentials: Simulation of Hartree-Fock and Dirac-Fock Results. J. Phys. B At. Mol. Phys. 1983, 16, L323–L328. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. The M06 Suite of Density Functionals for Main Group Thermochemistry, Thermochemical Kinetics, Noncovalent Interactions, Excited States, and Transition Elements: Two New Functionals and Systematic Testing of Four M06-class Functionals and 12 Other Function. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. Density Functionals with Broad Applicability in Chemistry. Acc. Chem. Res. 2008, 41, 157–167. [Google Scholar] [CrossRef]
- Stevens, W.J.; Krauss, M.; Basch, H.; Jasien, P.G. Relativistic Compact Effective Potentials and Efficient, Shared-Exponent Basis Sets for the Third-, Fourth-, and Fifth-Row Atoms. Can. J. Chem. 1992, 70, 612–630. [Google Scholar] [CrossRef]
- Cundari, T.R.; Stevens, W.J. Effective-Core-Potential Methods for the Lanthanides. J. Chem. Phys. 1993, 98, 5555–5565. [Google Scholar] [CrossRef]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An Advanced Semantic Chemical Editor, Visualization, and Analysis Platform. J. Cheminform. 2012, 4, 17. [Google Scholar] [CrossRef]
- Siebel, E.; Schwarz, P.; Fischer, R. The Tetrabutylammonium Ion: A Promising Building Block for the Design of Supramolecular Assemblies? Solid State Ionics 1997, 101–103, 285–292. [Google Scholar] [CrossRef]
- Bowmaker, G.A.; Hartl, H.; Urban, V. Crystal Structures and Vibrational Spectroscopy of [NBu4][Cu(CN)X] (X = Br, I) and [NBu4][Cu3(CN)4]·CH3CN. Inorg. Chem. 2000, 39, 4548–4554. [Google Scholar] [CrossRef]
- Pretsch, T.; Brüdgam, I.; Hartl, H. Synthese und Kristallstruktur von [N(Hex)4]3[Cu2(CN)3]. Z. Anorg. Allg. Chem. 2003, 629, 942–944. [Google Scholar] [CrossRef]
- Pretsch, T.; Hartl, H. Der strukturdirigierende Einfluß vonα,ω-Alkandiammoniumionen auf die Bildung von Cyanocupraten(I). Z. Anorg. Allg. Chem. 2004, 630, 1581–1588. [Google Scholar] [CrossRef]
- Colacio, E.; Kivekäs, R.; Lloret, F.; Sunberg, M.; Suarez-Varela, J.; Bardají, M.; Laguna, A. Architecture Dependence on the Steric Constrains of the Ligand in Cyano-Bridged Copper(I) and Copper(II)−Copper(I) Mixed-Valence Polymer Compounds Containing Diamines: Crystal Structures and Spectroscopic and Magnetic Properties. Inorg. Chem. 2002, 41, 5141–5149. [Google Scholar] [CrossRef]
- Etaiw, S.-E.H.; Abdou, S.N.; Badr El-din, A.S. Structure and Spectral Characterization of Coordination Polymers Constructed by CuCN and Aliphatic Diamines. J. Inorg. Organomet. Polym. Mater. 2015, 25, 1394–1406. [Google Scholar] [CrossRef]
- Ferlay, S.; Dechambenoit, P.; Kyritsakas, N.; Hosseini, M.W. Molecular Tectonics: Tuning the Dimensionality and Topology of Extended Cyanocuprate Networks using a Bisamidinium Cation. Dalton Trans. 2013, 42, 11661–11671. [Google Scholar] [CrossRef]
- Zhao, Y.; Hong, M.; Su, W.; Cao, R.; Zhou, Z.; Chan, A.S.C. Synthesis and Characterization of Two Copper Cyanide Complexes with Hexagonal Cu6 Units. J. Chem. Soc. Dalton Trans. 2000, 0, 1685–1686. [Google Scholar] [CrossRef]
- Pohl, A.H.; Chippindale, A.M.; Hibble, S.J. New Copper (I) Cyanide Networks: Interpenetration, Self-Penetration and Polymorphism. Solid State Sci. 2006, 8, 379–387. [Google Scholar] [CrossRef]
- Chesnut, D.J.; Kusnetzow, A.; Zubieta, J. Solid State Coordination Chemistry of the Copper Cyanide–Organoamine System: Hydrothermal Synthesis and Structural Characterization of [{Cu2(bpy)2(CN)}2Cu5(CN)7]·0.17H2O. J. Chem. Soc. Dalton Trans. 1998, 0, 4081–4084. [Google Scholar] [CrossRef]
- Chesnut, D.J.; Kusnetzow, A.; Birge, R.; Zubieta, J. Ligand Influences on Copper Cyanide Solid-State Architecture: Flattened and Fused “Slinky”, Corrugated Sheet, and Ribbon Motifs in the Copper−Cyanide−Triazolate−Organoamine Family. Inorg. Chem. 1999, 38, 5484–5494. [Google Scholar] [CrossRef]
- Deng, H.; Qiu, Y.; Daiguebonne, C.; Kerbellec, N.; Guillou, O.; Zeller, M.; Batten, S.R. Synthesis of New Copper Cyanide complexes via the Transformation of Organonitrile to Inorganic Cyanide. Inorg. Chem. 2008, 47, 5866–5872. [Google Scholar] [CrossRef]
- Park, K.-M.; Lee, S.; Kang, Y.; Moon, S.-H.; Lee, S.S. An Unusual Anionic Copper(I) Cyanide 3D Framework Encapsulating a Cationic Copper(II) Complex as a Guest. Dalton Trans. 2008, 0, 6521–6523. [Google Scholar] [CrossRef]
- Benmansour, S.; Setifi, F.; Triki, S.; Thétiot, F.; Sala-Pala, J.; Gómez-García, C.J.; Colacio, E. High-Dimensional Mixed-Valence Copper Cyanide Complexes: Syntheses, Structural Characterizations and Magnetism. Polyhedron 2009, 28, 1308–1314. [Google Scholar] [CrossRef]
- Vakulka, A.; Goreshnik, E. Synthesis and Characterization of 2CuCN·DMSO and [CuII(DMSO)6][CuI6(CN)8] 3-D Framework Compounds. Cent. Eur. J. Chem. 2013, 11, 1001–1009. [Google Scholar]
- Grifasi, F.; Priola, E.; Chierotti, M.R.; Diana, E.; Garino, C.; Gobetto, R. Vibrational-Structural Combined Study into Luminescent Mixed Copper(I)/Copper(II) Cyanide Coordination Polymers. Eur. J. Inorg. Chem. 2016, 2016, 2975–2983. [Google Scholar] [CrossRef]
- Zhang, X.-M.; Qing, Y.-L.; Wu, H.-S. Two Cuprous Cyanide Polymorphs: Diamond Net versus 3,4-Connected Net. Inorg. Chem. 2008, 47, 2255–2257. [Google Scholar] [CrossRef]
- Slater, J.C. Atomic Radii in Crystals. J. Chem. Phys. 1964, 41, 3199–3204. [Google Scholar] [CrossRef]
- Minkwitz, R.; Bröchler, R.; Ludwig, R. Tetramethylammonium Difluorobromate(I), (CH3)4N+BrF2−. Inorg. Chem. 1997, 36, 4280–4283. [Google Scholar] [CrossRef]
- Lu, H.; Yson, R.; Li, X.; Larochelle, C.; Patterson, H.H. Luminescent Studies of “Exciplex Tuning” for Nanoclusters of Dicyanocuprate(I) Ions Doped in Potassium Chloride Crystals. J. Phys. Chem. C 2009, 113, 5952–5959. [Google Scholar] [CrossRef]
- Li, X.; Pan, Z.; Baril-Robert, F.; Ahern, J.C.; Patterson, H.H. Novel Luminescent Heterobimetallic Nanoclusters of Copper(I), Silver(I), and Gold(I) Doped in Different Alkali Halide Crystals. J. Phys. Chem. C 2014, 118, 11886–11894. [Google Scholar] [CrossRef]
- Ahern, J.C.; Kanan, S.; Patterson, H.H. Heterogeneous Photocatalysis with Nanoclusters of d10 Metal Ions Doped in Zeolites. GCIC 2015, 35, 59–81. [Google Scholar] [CrossRef]
- Evariste, S.; Khalil, A.M.; Moussa, M.E.; Chan, A.K.-W.; Hong, E.Y.-H.; Wong, H.-L.; Le Guennic, B.; Calvez, G.; Costuas, K.; Yam, V.W.-W.; et al. Adaptive Coordination-Driven Supramolecular Syntheses toward New Polymetallic Cu(I) Luminescent Assemblies. J. Am. Chem. Soc. 2018, 140, 12521–12526. [Google Scholar] [CrossRef]
- Chakkaradhari, G.; Chen, Y.-T.; Karttunen, A.J.; Dau, M.T.; Jänis, J.; Tunik, S.P.; Chou, P.-T.; Ho, M.-L.; Koshevoy, I.O. Luminescent Triphosphine Cyanide d10 Metal Complexes. Inorg. Chem. 2016, 55, 2174–2184. [Google Scholar] [CrossRef]
- Bayse, C.A.; Brewster, T.P.; Pike, R.D. Photoluminescence of 1-D Copper(I) Cyanide Chains: A Theoretical Description. Inorg. Chem. 2009, 48, 174–182. [Google Scholar] [CrossRef]
- Baril-Robert, F.; Guo, Z.; Patterson, H.H. Study of the Energy Transfer Process in the Highly Luminescent Heterometallic Dimers of Ce3+ and d10 [Ag(CN)2]− or d8 [Pt(CN)4]2− Ions. Chem. Phys. Lett. 2009, 471, 258–263. [Google Scholar] [CrossRef]
- Baril-Robert, F.; Palla, V.; Li, X.; Yson, R.; Patterson, H.H. Observation of a Mixed-Metal Transition in a d8–d10 Heterobimetallic Pt–Ag Cyanide System: Experimental and Theoretical Study. Inorg. Chim. Acta 2010, 363, 2637–2642. [Google Scholar] [CrossRef]
- Iwamura, M.; Nozaki, K.; Takeuchi, S.; Tahara, T. Real-Time Observation of Tight Au–Au Bond Formation and Relevant Coherent Motion upon Photoexcitation of [Au(CN)2−] Oligomers. J. Am. Chem. Soc. 2013, 135, 538–541. [Google Scholar] [CrossRef]
- Iwamura, M.; Wakabayashi, R.; Maeba, J.; Nozaki, K.; Takeuchi, S.; Tahara, T. Coherent Vibration and Ultrafast Dynamics upon Bond Formation in Excited Dimers of an Au(I) Complex. Phys. Chem. Chem. Phys. 2016, 18, 5103–5107. [Google Scholar] [CrossRef]
- Rawashdeh-Omary, M.A.; Omary, M.A.; Patterson, H.H.; Fackler, J.P. Excited-State Interactions for [Au(CN)2−]n and [Ag(CN)2−]n Oligomers in Solution. Formation of Luminescent Gold−Gold Bonded Excimers and Exciplexes. J. Am. Chem. Soc. 2001, 123, 11237–11247. [Google Scholar] [CrossRef]
- Rawashdeh-omary, M.A.; Omary, M.A.; Patterson, H.H.; May, R. V Oligomerization of Au(CN)2− and Ag(CN)2− Ions in Solution via Ground-State Aurophilic and Argentophilic Bonding. J. Am. Chem. Soc. 2000, 122, 10371–10380. [Google Scholar] [CrossRef]
- Espenson, J.H. Chemical Kinetics and Reaction Mechanisms, 2nd ed.; McGraw-Hill: New York, NY, USA, 2002; ISBN 978-0072883626. [Google Scholar]
- Srisook, N.; Rizzolo, J.; Shankle, G.E.; Patterson, H.H. Light-Induced Electron Transfer in Tl[Ag(CN)2]: Photochemical Reaction of Luminescent Metal-Metal Exciplexes in the Solid State. Inorg. Chim. Acta 2000, 300–302, 314–318. [Google Scholar] [CrossRef]
- Vanysek, P. Electrochemical Series. In Handbook of Chemistry and Physics; Lide, D.R., Ed.; CRC Press: Boca Raton, FL, USA, 2008; pp. 20–29. ISBN 978-1420066791. [Google Scholar]
- Harris, D.C. Quantitative Chemical Analysis, 7th ed.; W. H. Freeman and Company: New York, NY, USA, 2007. [Google Scholar]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Complex | 1 | 2 | 3 | |
|---|---|---|---|---|
| CCDC deposit number | 1903299 | 1903300 | 1903302 | 1903301 |
| color and habit | Colorless needle | Colorless needle | Colorless needle | Colorless block |
| Size, mm | 0.130 × 0.040 × 0.020 | 0.270 × 0.020 × 0.020 | 0.310 × 0.020 × 0.020 | 0.290 × 0.150 × 0.110 |
| formula | C14H24Cu4N8O0.25 | C8H12Cu3N5 | C16H24Cu6N10 | C7H12Cu2N4 |
| formula weight | 563.07 | 368.85 | 737.69 | 279.29 |
| space group | Pnma | C2/c | P-1 | P-1 |
| a, Å | 16.1310(4) | 15.1615(2) | 8.7275(2) | 8.501(3) |
| b, Å | 13.1438(3) | 8.57220(10) | 10.5197(2) | 8.751(3) |
| c, Å | 19.6376(5) | 12.43490(10) | 14.8175(4) | 9.047(3) |
| α, ° | 90 | 90 | 69.2830(10) | 78.376(6) |
| β, ° | 90 | 127.2540(10) | 80.262(2) | 63.283(5) |
| γ, ° | 90 | 90 | 78.8820(10) | 61.151(5) |
| volume, Å3 | 4163.62(18) | 1286.38(3) | 1240.99(5) | 526.5(3) |
| Z | 2 | 4 | 2 | 2 |
| ρcalc, Mg cm−3 | 1.797 | 1.905 | 1.974 | 1.762 |
| F000 | 2260 | 728 | 728 | 280 |
| μ, mm−1 | 4.729 | 5.598 | 5.802 | 3.999 |
| Radiation | Cu Kα (1.54178 Å) | Cu Kα (1.54178 Å) | Cu Kα (1.54178 Å) | Mo Kα (0.71073 Å) |
| Temp., K | 100 | 296 | 100 | 100 |
| residuals: a R; Rw | 0.0439, 0.1216 | 0.0267, 0.0763 | 0.0356, 0.0849 | 0.0168, 0.0428 |
| goodness of fit | 1.078 | 1.084 | 1.101 | 1.062 |
| Complex | Temperature | Ex λmax | Em λmax | Lifetime |
|---|---|---|---|---|
| 1 | 298 K | 334 nm | 524 nm | 63 μs |
| 78 K | 315 nm | 416 nm | 78 ns | |
| 542 nm | 0.31 μs | |||
| 2 | 298 K | 340 nm | 420 nm | 6.3 μs |
| 78 K | 332 nm | 440 nm | 33 μs |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nicholas, A.D.; Bullard, R.M.; Wheaton, A.M.; Streep, M.; Nicholas, V.A.; Pike, R.D.; Patterson, H.H. Synthesis and Luminescence of Optical Memory Active Tetramethylammonium Cyanocuprate(I) 3D Networks. Materials 2019, 12, 1211. https://doi.org/10.3390/ma12081211
Nicholas AD, Bullard RM, Wheaton AM, Streep M, Nicholas VA, Pike RD, Patterson HH. Synthesis and Luminescence of Optical Memory Active Tetramethylammonium Cyanocuprate(I) 3D Networks. Materials. 2019; 12(8):1211. https://doi.org/10.3390/ma12081211
Chicago/Turabian StyleNicholas, Aaron D., Rebeka M. Bullard, Amelia M. Wheaton, Michaela Streep, Victoria A. Nicholas, Robert D. Pike, and Howard H. Patterson. 2019. "Synthesis and Luminescence of Optical Memory Active Tetramethylammonium Cyanocuprate(I) 3D Networks" Materials 12, no. 8: 1211. https://doi.org/10.3390/ma12081211
APA StyleNicholas, A. D., Bullard, R. M., Wheaton, A. M., Streep, M., Nicholas, V. A., Pike, R. D., & Patterson, H. H. (2019). Synthesis and Luminescence of Optical Memory Active Tetramethylammonium Cyanocuprate(I) 3D Networks. Materials, 12(8), 1211. https://doi.org/10.3390/ma12081211