Chitosan-Functionalized Graphene Nanocomposite Films: Interfacial Interplay and Biological Activity


 , , , , ,
, , , , ,  , , and
, , and
Abstract
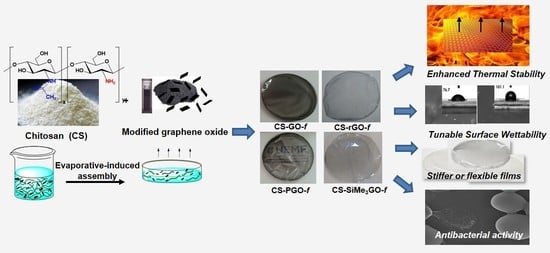
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Characterization
2.3. Preparation of Modified Graphene Fillers
2.4. Preparation of Chitosan–Graphene Films
2.5. Determination of Antimicrobial Activity
2.6. Permeability of Bacterial Cell Membranes
Confocal Microscopy
2.7. Morphological Changes of S. Aureus Cells Visualized by Scanning Electron Microscopy (SEM)
2.8. Hemolysis Assay
2.9. The Adsorption of Hemoglobin (Hb)
2.10. Methemoglobin (Met-Hb)
2.11. Catalase Activity
2.12. Statistical Analysis
3. Results
3.1. Synthesis of Functionalized Graphene Fillers
3.2. Preparation and Characterization of Chitosan-Modified Graphene Films
3.3. Biological Activity of Chitosan-Modified Graphene Films
3.3.1. Antimicrobial Activity
3.3.2. Hemolysis
3.3.3. Intracellular Catalase (CAT) Activity and Hemoglobin Oxidation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Siracusa, V.; Rocculi, P.; Romani, S.; Rosa, M.D. Biodegradable polymers for food packaging: A review. Trends Food Sci. Technol. 2008, 19, 634–643. [Google Scholar] [CrossRef]
- Colvin, V.L. The potential environmental impact of engineered nanomaterials. Nat. Biotechnol. 2003, 21, 1166–1170. [Google Scholar] [CrossRef]
- Swain, S.K.; Mohanty, F. Bionanocomposites for Packaging Applications; Jawaid, M., Swain, S.K., Eds.; Springer International Publishing: Cham, Switzerland, 2018; pp. 191–208. [Google Scholar]
- Sahariah, P.; Másson, M. Antimicrobial chitosan and chitosan derivatives: A review of the structure-activity relationship. Biomacromolecules 2017, 18, 3846–3868. [Google Scholar] [CrossRef]
- El Kadib, A. Chitosan as a sustainable organocatalyst: A concise overview. ChemSusChem 2015, 8, 217–244. [Google Scholar] [CrossRef] [PubMed]
- Varma, A.J.; Deshpande, S.V.; Kennedy, J.F. Metal complexation by chitosan and its derivatives: A Review. Carbohydr. Polym. 2004, 55, 77–93. [Google Scholar] [CrossRef]
- El Kadib, A.; Bousmina, M. Chitosan bio-based organic-inorganic hybrid aerogel microspheres. Chem. Eur. J. 2012, 18, 8264–8277. [Google Scholar] [CrossRef] [PubMed]
- Lou, M.M.; Zhu, B.; Muhammad, I.; Li, B.; Xie, G.L.; Wang, Y.L.; Li, H.Y.; Sun, G.C. Antibacterial activity and mechanism of action of chitosan solutions against apricot fruit rot pathogen Burkholderia seminalis. Carbohydr. Res. 2011, 346, 1294–1301. [Google Scholar] [CrossRef]
- Frindy, S.; Primo, A.; Qaiss Ael, K.; Bouhfid, R.; Lahcini, M.; Garcia, H.; Bousmina, M.; El Kadib, A. Insightful understanding of the role of clay topology on the stability of biomimetic hybrid chitosan-clay thin films and CO2 dried porous aerogel microspheres. Carbohydr. Polym. 2016, 146, 353–361. [Google Scholar] [CrossRef]
- Frindy, S.; Primo, A.; Ennajih, H.; Qaiss Ael, K.; Bouhfid, R.; Lahcini, M.; Essassi, E.M.; Garcia, H.; El Kadib, A. Chitosan-graphene oxide films and CO2-dried porous aerogel microspheres: Interfacial interplay and stability. Carbohydr. Polym. 2017, 167, 297–305. [Google Scholar] [CrossRef]
- Pighinelli, L.; Kucharska, M. Chitosan-hydroxyapatite composites. Carbohydr. Carbohydr. Polym. 2013, 93, 256–262. [Google Scholar]
- Nazeer, M.A.; Yilgör, E.; Yilgör, I. Intercalated chitosan/hydroxyapatite nanocomposites: Promising materials for bone tissue engineering applications. Carbohydr. Polym. 2017, 175, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Pistone, A.; Iannazzo, D.; Celesti, C.; Piperopoulos, E.; Ashok, D.; Cembran, A.; Tricoli, A.; Nisbet, D. Engineering of chitosan-hydroxyapatite-magnetite hierarchical scaffolds for guided bone growth. Materials 2019, 12, 2321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.F.; Shen, L.; Zhang, W.D.; Tong, Y.J. Preparation and mechanical properties of chitosan/carbon nanotubes composites. Biomacromolecules 2005, 6, 3067–3072. [Google Scholar] [CrossRef] [PubMed]
- Zabihi, E.; Babaei, A.; Shahrampour, D.; Arab-Bafrani, Z.; Mirshahidi, K.S.; Joz Majidi, H. Facile and rapid in-situ synthesis of chitosan-ZnO nano-hybrids applicable in medical purposes; a novel combination of morphology-conducting agent. Int. J. Biol. Macromol. 2019, 131, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Jaworski, S.; Hinzmann, M.; Sawosz, E.; Grodzik, M.; Kutwin, M.; Wierzbicki, M.; Strojny, B.; Vadalasetty, K.P.; Lipińska, L.; Chwalibog, A. Interaction of different forms of graphene with chicken embryo red blood cells. Environ. Sci. Pollut. Res. 2017, 24, 21671–21679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, K.H.; Lin, Y.S.; Macosko, C.W.; Haynes, C.L. Cytotoxicity of graphene oxide and graphene in human erythrocytes and skin fibroblast. ACS Appl. Mater. Interfaces 2011, 3, 2607–2615. [Google Scholar] [CrossRef]
- Sasidharan, A.; Panchakarla., L.S.; Sadanandan, A.R.; Ashokan, A.; Chandran, P.; Girish, C.M.; Menon, D.; Nair, S.V.; Rao, C.N.R.; Kovakutty, M. Hemocompatibility and macrophage response of pristine and functionalized graphene. Small 2012, 8, 1251–1263. [Google Scholar] [CrossRef]
- Hummers, W.S.; Offeman, R.E. Preparation of graphitic oxide. J. Am. Chem. Soc. 1958, 80, 1339. [Google Scholar] [CrossRef]
- Szunerits, S.; Boukherroub, R. Antibacterial activity of graphene-based materials. J. Mater. Chem. B 2016, 4, 6892–6912. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Zeng, T.H.; Hofmann, M.; Burcombe, E.; Wei, J.; Jiang, R.; Kong, J.; Chen, Y. Antibacterial activity of graphite, graphite oxide, graphene oxide, reduced graphene oxide: Membrane and oxidative stress. ACS Nano 2011, 5, 6971–6980. [Google Scholar] [CrossRef]
- Bao, H.; Pan, Y.; Ping, Y.; Sahoo, N.G.; Wu, T.; Li, L.; Gan, L.H. Chitosan-Functionalized Graphene Oxide as a Nanocarrier for Drug and Gene Delivery. Small 2011, 7, 1569–1578. [Google Scholar] [CrossRef] [PubMed]
- Han, D.; Yan, L.; Chen, W.; Li, W. Preparation of chitosan/grapheneoxide composite film with enhanced mechanical strenght in the wet state. Carbohydr. Polym. 2011, 83, 653–658. [Google Scholar] [CrossRef]
- Dharupaneedi, S.P.; Anjanapura, R.V.; Han, J.M.; Aminabhavi, T.M. Functionalized Graphene Sheets Embedded in Chitosan Nanocomposite Membranes for Ethanol and Isopropanol Dehydration via Pervaporation. Ind. Eng. Chem. Res. 2014, 53, 14474–14484. [Google Scholar] [CrossRef]
- Kim, D.S.; Dhand, V.; Rhee, K.Y.; Park, S.-J. Study on the Effect of Silanization and Improvement in the Tensile Behavior of Graphene-Chitosan-Composite. Polymers 2015, 7, 527–551. [Google Scholar] [CrossRef] [Green Version]
- Valencia Zapata, M.E.; Mina Hernandez, J.H.; Grande Tovar, C.D.; Valencia Llano, C.H.; Diaz Escobar, J.A.; Vázquez-Lasa, B.; San Román, J.; Rojo, L. Novel bioactive and antibacterial acrylic bone cement nanocomposites modified with graphene oxide and chitosan. Int. J. Mol. Sci. 2019, 20, 2938. [Google Scholar] [CrossRef] [Green Version]
- Kosowska, K.; Domalik-Pyzik, P.; Krok-Borkowicz, M.; Chłopek, J. Synthesis and characterization of chitosan/reduced graphene oxide hybrid composites. Materials 2019, 12, 2077. [Google Scholar] [CrossRef] [Green Version]
- Fan, H.; Wang, L.; Zhao, K.; Li, N.; Shi, Z.; Ge, Z.; Jin, Z. Fabrication, mechanical properties, and biocompatibility of graphene-reinforced chitosan composites. Biomacromolecules 2010, 11, 2345–2351. [Google Scholar] [CrossRef]
- Aebi, H. Catalase in vitro. In Methods in Enzymology; Packer, L., Ed.; Academic Press: Cambridge, MA, USA, 1984; Volume 105, pp. 121–126. [Google Scholar]
- Drabkin, D.L. Spectrophotometric studies; the crystallographic and optical properties of the hemoglobin of man in comparison with those of other species. J. Biol. Chem. 1946, 164, 703–723. [Google Scholar]
- Anouar, A.; Katir, N.; Mamede, A.-S.; Aboulaich, A.; Draoui, K.; Royer, S.; El Kadib, A. Synthesis and multifaceted use of phosphorylated graphene oxide: Growth of titanium dioxide clusters, interplay with gold nanoparticles and exfoliated sheets in bioplastics. Mater. Chem. Front. 2019, 3, 242–250. [Google Scholar] [CrossRef]
- Park, S.; An, J.; Potts, J.R.; Velamakanni, A.; Murali, S.; Ruoff, R.S. Hydrazine-reduction of graphite- and graphene oxide. Carbon 2011, 49, 3019–3023. [Google Scholar] [CrossRef]
- Cobos, M.; González, B.; Jesús Fernández, M.; Dolores Fernández, M. Chitosan-graphene oxide nanocomposites: Effect of graphene oxide nanosheets and glycerol plasticizer on thermal and mechanical properties. J. Appl. Polym. Sci. 2017, 134, 45092–45106. [Google Scholar] [CrossRef]
- Lin-Vien, D.; Colthup, N.B.; Fateley, W.G.; Grasselli, J.G. Organosilicon compounds. In The Handbook of Infrared and Raman Characteristic Rrequency of Organic Molecules; Academic Press: San Diego, CA, USA, 1991; Volume 25, pp. 251–261. [Google Scholar]
- Zhang, M.; Li, X.H.; Gong, Y.D.; Zhao, N.M.; Zhang, X.F. Properties and biocompatibility of chitosan films modified by blending with PEG. Biomaterials 2002, 23, 2641–2648. [Google Scholar] [CrossRef]
- Mission, E.G.; Agutaya, J.K.C.N.; Quitain, A.T.; Sasaki, M.; Kida, T. Carbocatalysed hydrolytic cleaving of the glycosidic bond in fucoidan under microwave irradiation. RSC Adv. 2019, 9, 30325–30334. [Google Scholar] [CrossRef] [Green Version]
- Aldana, A.A.; Toselli, R.; Strumia, M.C.; Martinelli, M. Chitosan films modified selectively on one side with dendritic molecules. J. Mater. Chem. 2012, 22, 22670–22677. [Google Scholar] [CrossRef]
- Chabbi, J.; Agil, A.; Katir, N.; Vertruyen, B.; Jerome, C.; Lahcini, M.; El Kadib, A. Aldehyde-conjugated chitosan-graphene oxide glucodynamers: Ternary cooperative assembly and controlled chemical release. Carbohydr. Polym. 2020, 230, 115634. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Yang, C.; Hedrick, J.L.; Yang, Y.Y. Antimicrobial silica particles synthesized via ring-opening grafting of cationic amphiphilic cyclic carbonates: Effects of hydrophobicity and structure. Polym. Chem. 2016, 7, 2192–2201. [Google Scholar] [CrossRef]
- Mazaheri, M.; Akhavan, O.; Simchi, A. Flexible bactericidal graphene oxide-chitosan layers for stem cell proliferation. Appl. Surf. Sci. 2014, 301, 456–462. [Google Scholar] [CrossRef]
- Li, P.; Gao, Y.; Sun, Z.; Chang, D.; Gao, G.; Dong, A. Synthesis, characterization, and bactericidal evaluation of chitosan/guanidine functionalized graphene oxide composites. Molecules 2017, 22, 12. [Google Scholar] [CrossRef] [Green Version]
- Konwar, A.; Kalita, S.; Kotoky, J.; Chowdhury, D. Chitosan–Iron Oxide Coated Graphene Oxide Nanocomposite Hydrogel: A Robust and Soft Antimicrobial Biofilm. ACS Appl. Mat. Int. 2016, 8, 20625–20634. [Google Scholar] [CrossRef]
- Kumar, P.; Huo, P.; Zhang, R.; Liu, B. Antibacterial properties of graphene-based nanomaterials. Nanomaterials 2019, 9, 737. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Wang, G.; Zhu, H.; Zhang, M.; Zheng, X.; Di, Z.; Liu, X.; Wang, X. Antibacterial activity of large-area monolayer graphene film manipulated by charge transfer. Sci. Rep. 2014, 4, 4359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinto, A.M.; Goncalves, I.C.; Magalhaes, F.D. Graphene-based materials biocompatibility: A review. Colloids Surf. B Biointerfaces 2013, 11, 188–202. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Hu, C.; Shao, L. The antimicrobial activity of nanoparticles: Present situation and prospects for the future. Int. J. Nanomed. 2017, 12, 1227–1249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Deng, F.; Hu, Y.; Sun, J.; Yang, Y. Antibacterial activity of graphene-modified anode on Shewanella oneidensis MR-1 biofilm in microbial fuel cell. J. Power Sources 2015, 290, 80–86. [Google Scholar] [CrossRef]
- Pędziwiatr-Werbicka, E.; Miłowska, K.; Podlas, M.; Marcinkowska, M.; Ferenc, M.; Brahmi, Y.; Katir, N.; Majoral, J.P.; Felczak, A.; Boruszewska, A.; et al. Oleochemical-tethered-SBA-15-type mesoporous silicates with tunable nanoscopic order, carboxylic reactive-surface and hydrophobic framework: Impact on cellular toxicity, hemilysis and antibacterial activity. Chem. Eur. J. 2014, 20, 9596–9606. [Google Scholar] [CrossRef] [PubMed]
- Joz Majidi, H.; Babaei, A.; Arab Bafrani, Z.; Shahrampour, D.; Zabihi, E.; Jafari, S.M. Investigating the best strategy to diminish the toxicity and enhance the antibacterial activity of graphene oxide by chitosan addition. Carbohydr. Polym. 2019, 225, 115220. [Google Scholar] [CrossRef] [PubMed]
- Yildirim, A.; Ozgur, E.; Bayindir, M. Impact of mesoporous silica nanoparticle surface functionality on hemolytic activity, thrombogenicity and non-specific protein adsorption. J. Mater. Chem. B 2013, 1, 1909–1920. [Google Scholar] [CrossRef] [Green Version]
- Wei, X.L.; Ge, Z.Q. Effect of graphene oxide on conformation and activity of catalase. Carbon 2013, 60, 401–409. [Google Scholar] [CrossRef]
- Scibior, D.; Czeczot, H. Catalase: Structure, properties, functions. Postepy Hig. Med. Dosw. 2006, 60, 170–180. [Google Scholar]
- Maćczak, A.; Cyrkler, M.; Bukowska, B.; Michałowicz, J. Bisphenol A, Bisphenol S, bisphenol F and bisphenol AF induce different oxidative stress and damage in human red bloods cells (in vitro study). Toxicol. In Vitro 2017, 41, 143–149. [Google Scholar]








© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wrońska, N.; Anouar, A.; El Achaby, M.; Zawadzka, K.; Kędzierska, M.; Miłowska, K.; Katir, N.; Draoui, K.; Różalska, S.; Piwoński, I.; et al. Chitosan-Functionalized Graphene Nanocomposite Films: Interfacial Interplay and Biological Activity. Materials 2020, 13, 998. https://doi.org/10.3390/ma13040998
Wrońska N, Anouar A, El Achaby M, Zawadzka K, Kędzierska M, Miłowska K, Katir N, Draoui K, Różalska S, Piwoński I, et al. Chitosan-Functionalized Graphene Nanocomposite Films: Interfacial Interplay and Biological Activity. Materials. 2020; 13(4):998. https://doi.org/10.3390/ma13040998
Chicago/Turabian StyleWrońska, Natalia, Aicha Anouar, Mounir El Achaby, Katarzyna Zawadzka, Marta Kędzierska, Katarzyna Miłowska, Nadia Katir, Khalid Draoui, Sylwia Różalska, Ireneusz Piwoński, and et al. 2020. "Chitosan-Functionalized Graphene Nanocomposite Films: Interfacial Interplay and Biological Activity" Materials 13, no. 4: 998. https://doi.org/10.3390/ma13040998
APA StyleWrońska, N., Anouar, A., El Achaby, M., Zawadzka, K., Kędzierska, M., Miłowska, K., Katir, N., Draoui, K., Różalska, S., Piwoński, I., Bryszewska, M., El Kadib, A., & Lisowska, K. (2020). Chitosan-Functionalized Graphene Nanocomposite Films: Interfacial Interplay and Biological Activity. Materials, 13(4), 998. https://doi.org/10.3390/ma13040998