
Structure and Thermodynamics of Silicon Oxycarbide Polymer-Derived Ceramics with and without Mixed-Bonding †

 ,
, 
Abstract
:1. Introduction
2. Experimental Procedures
3. Results and Discussion
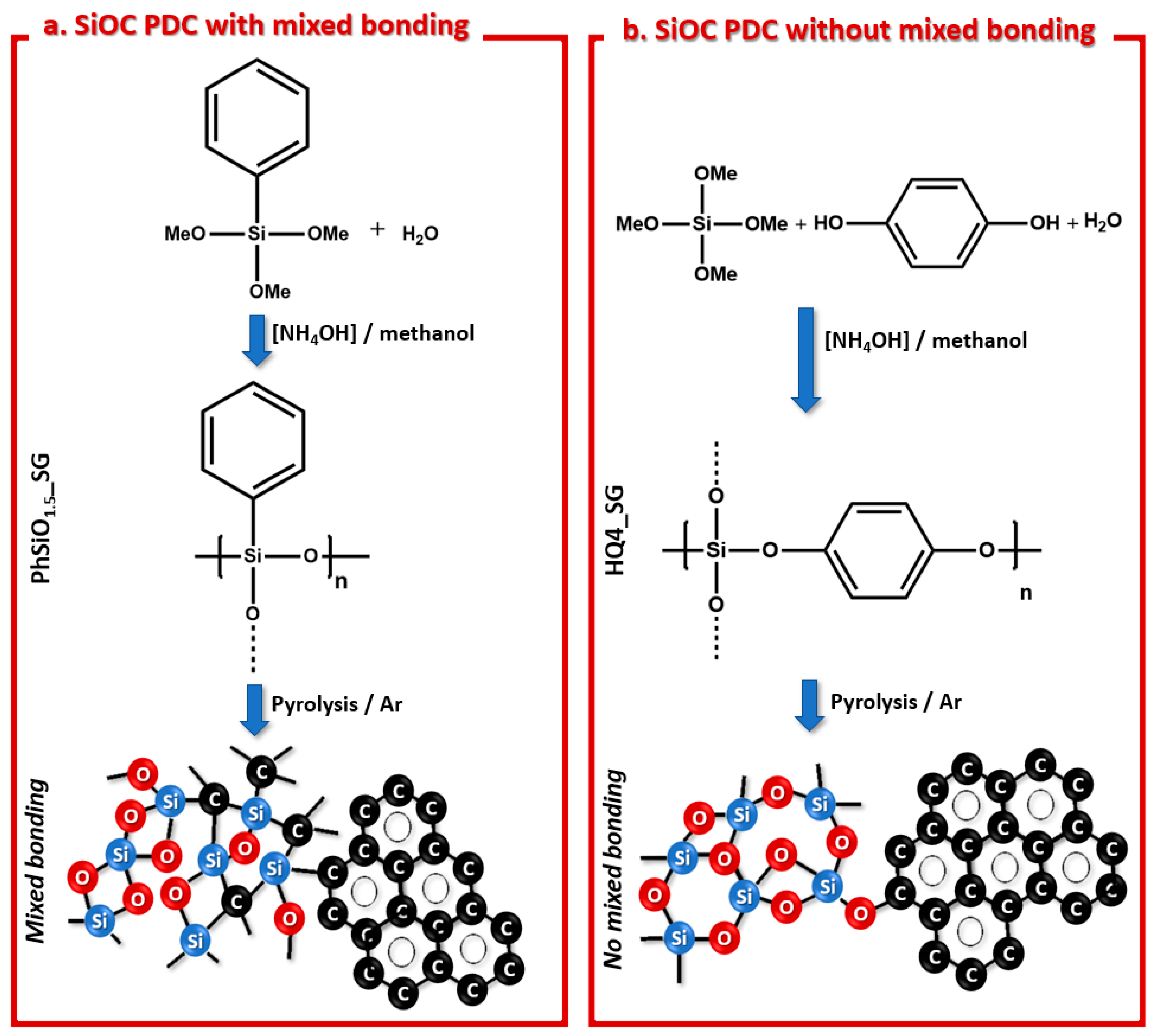
3.1. Synthesis
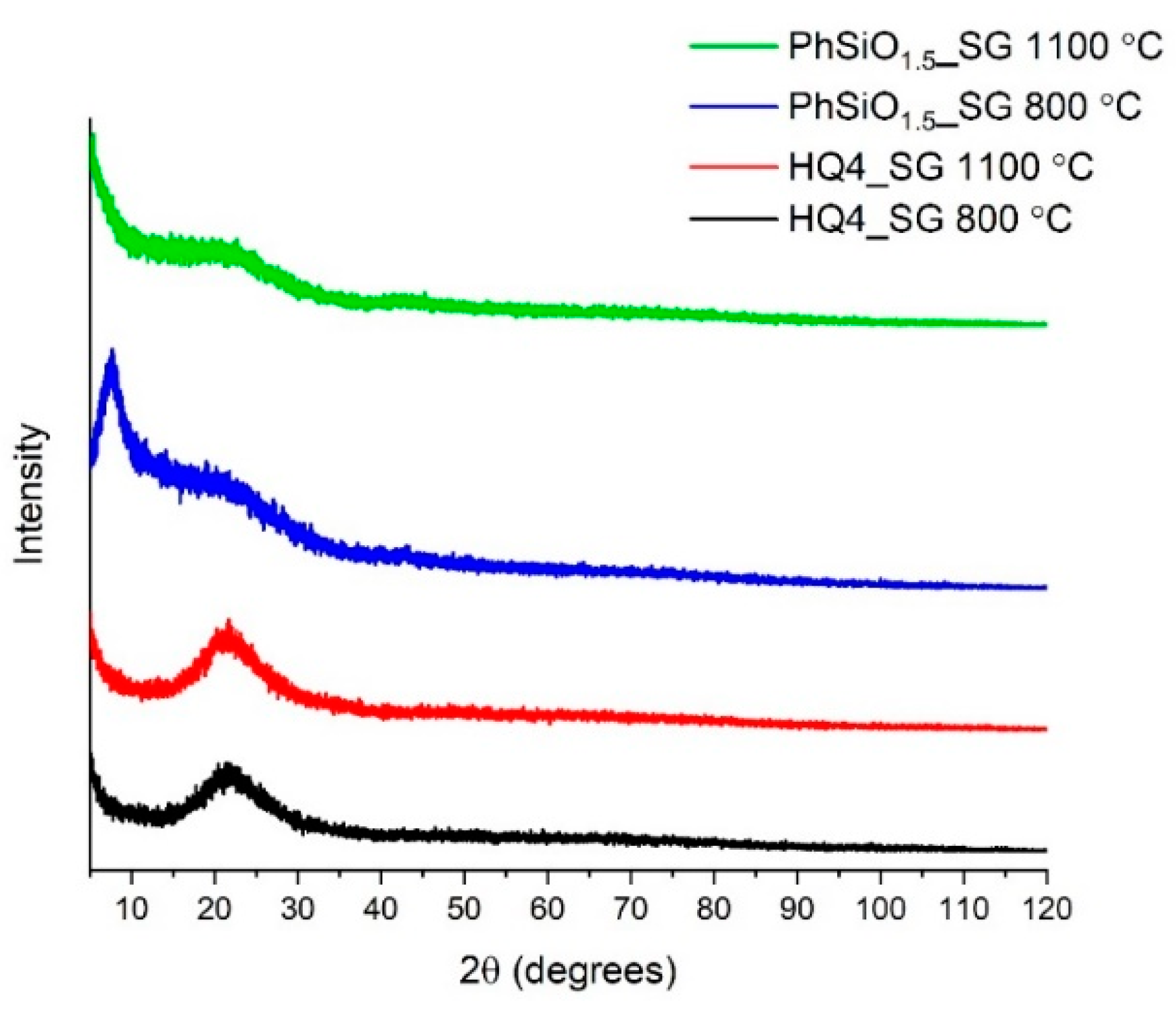
3.2. X-ray Powder Diffraction
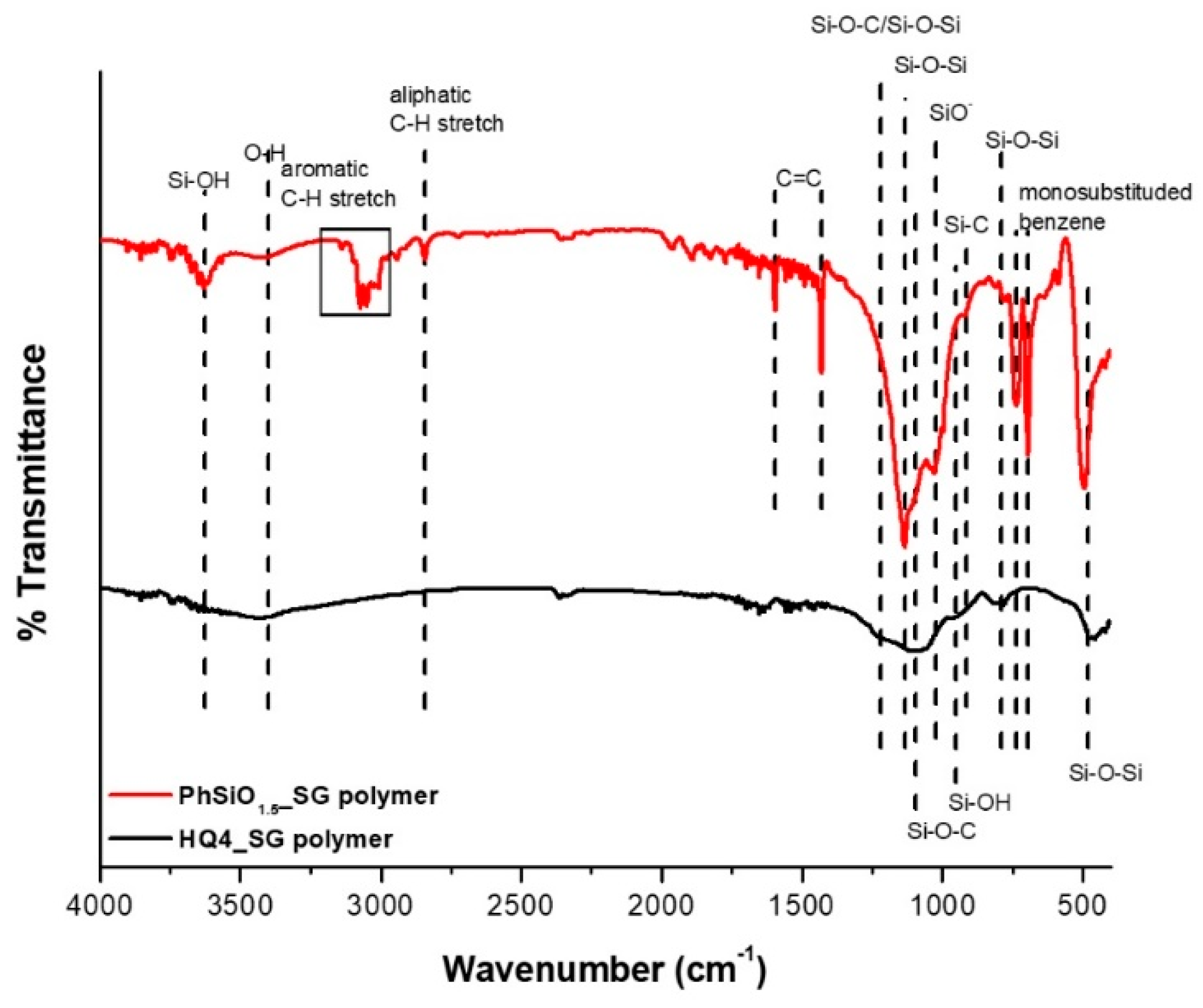
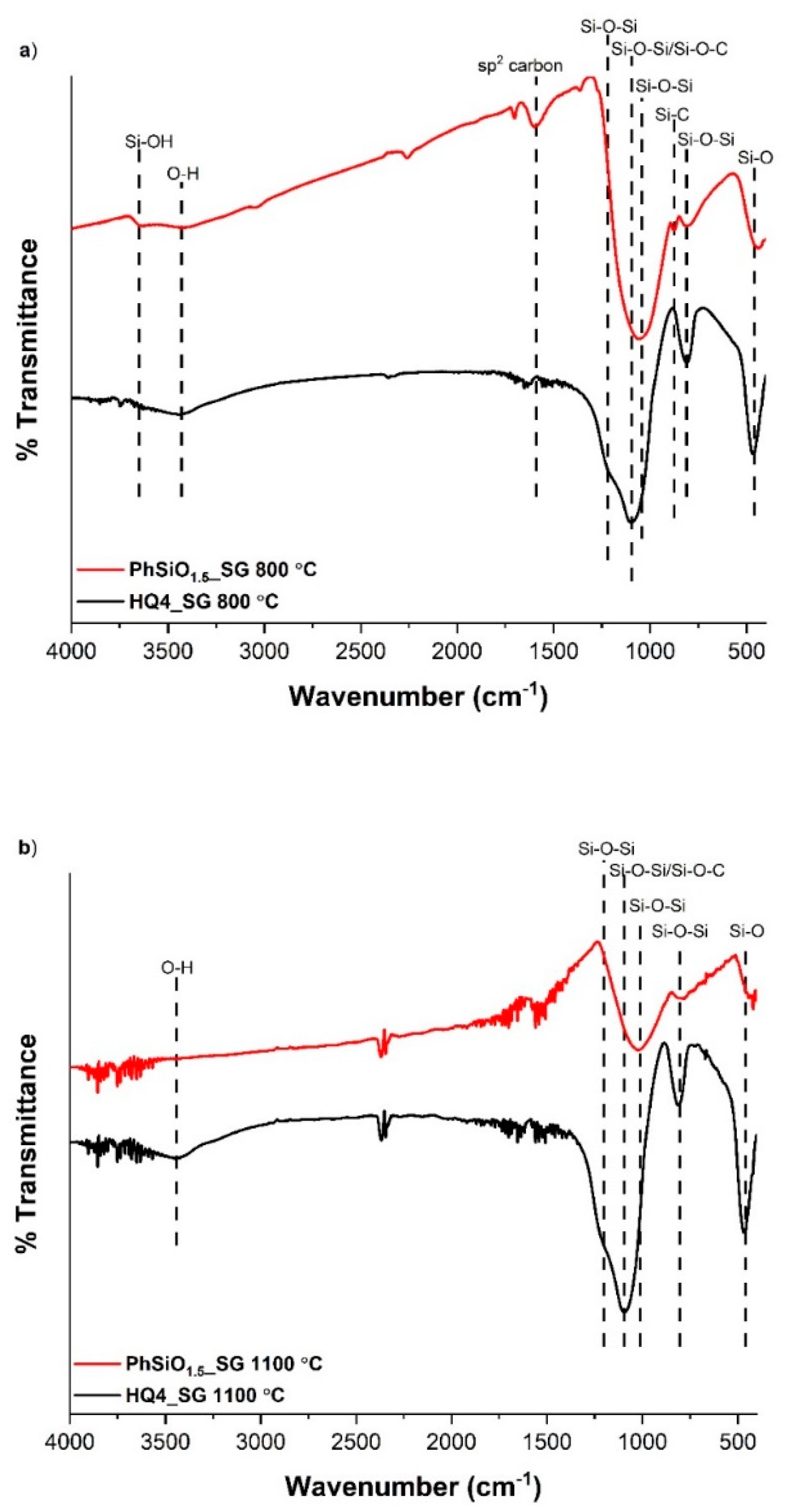
3.3. Fourier Transform Infrared Spectroscopy
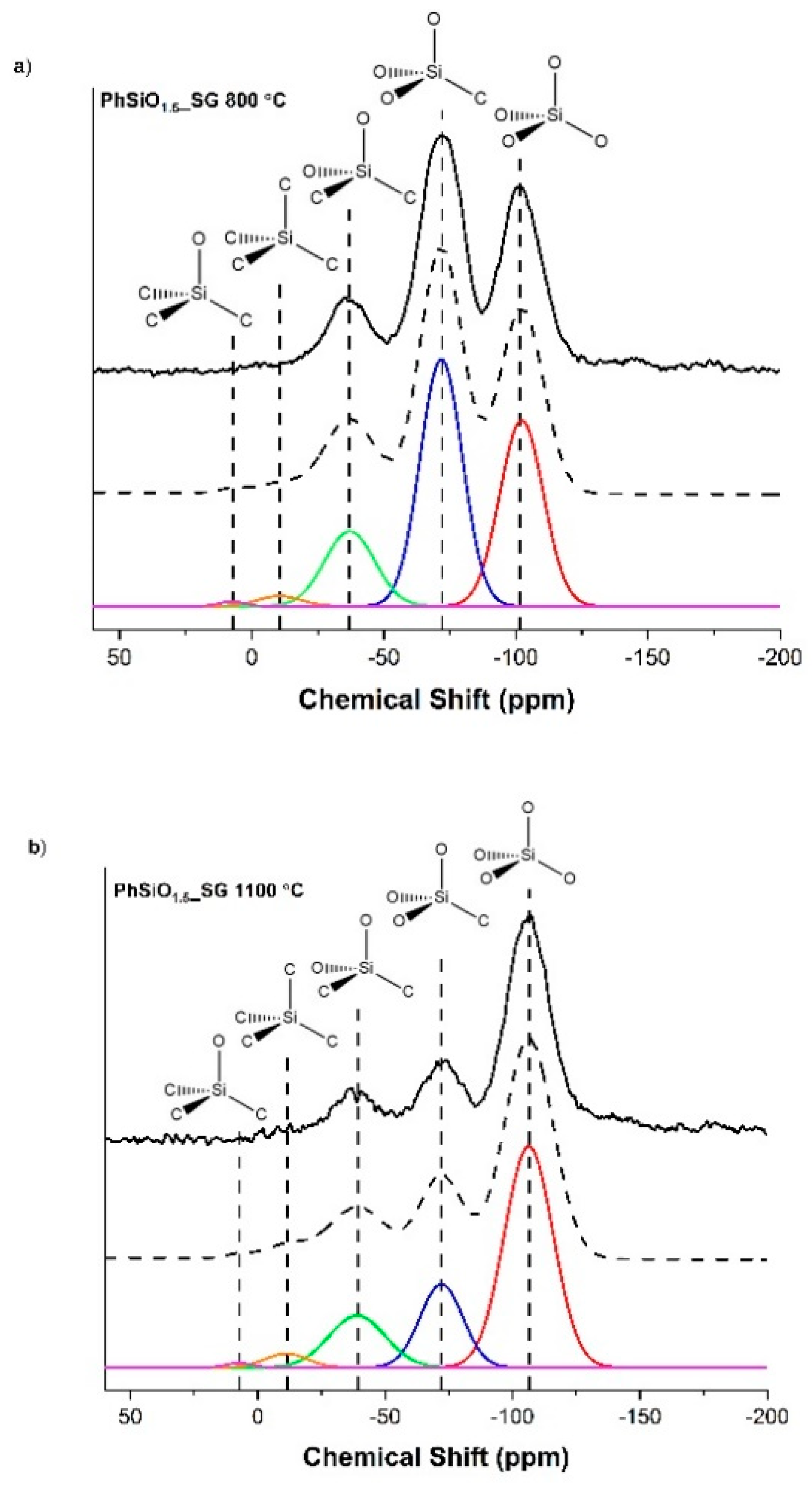
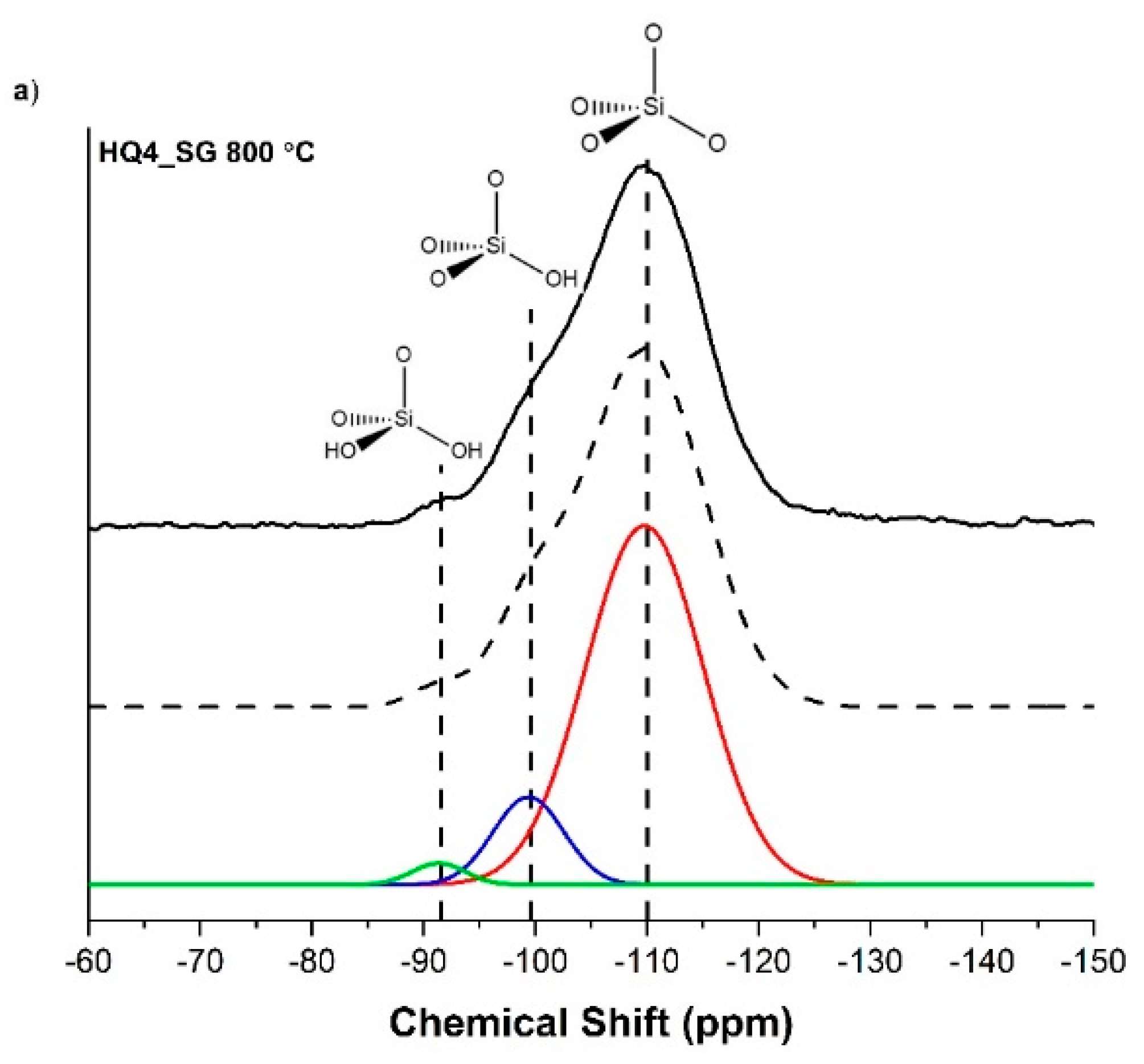
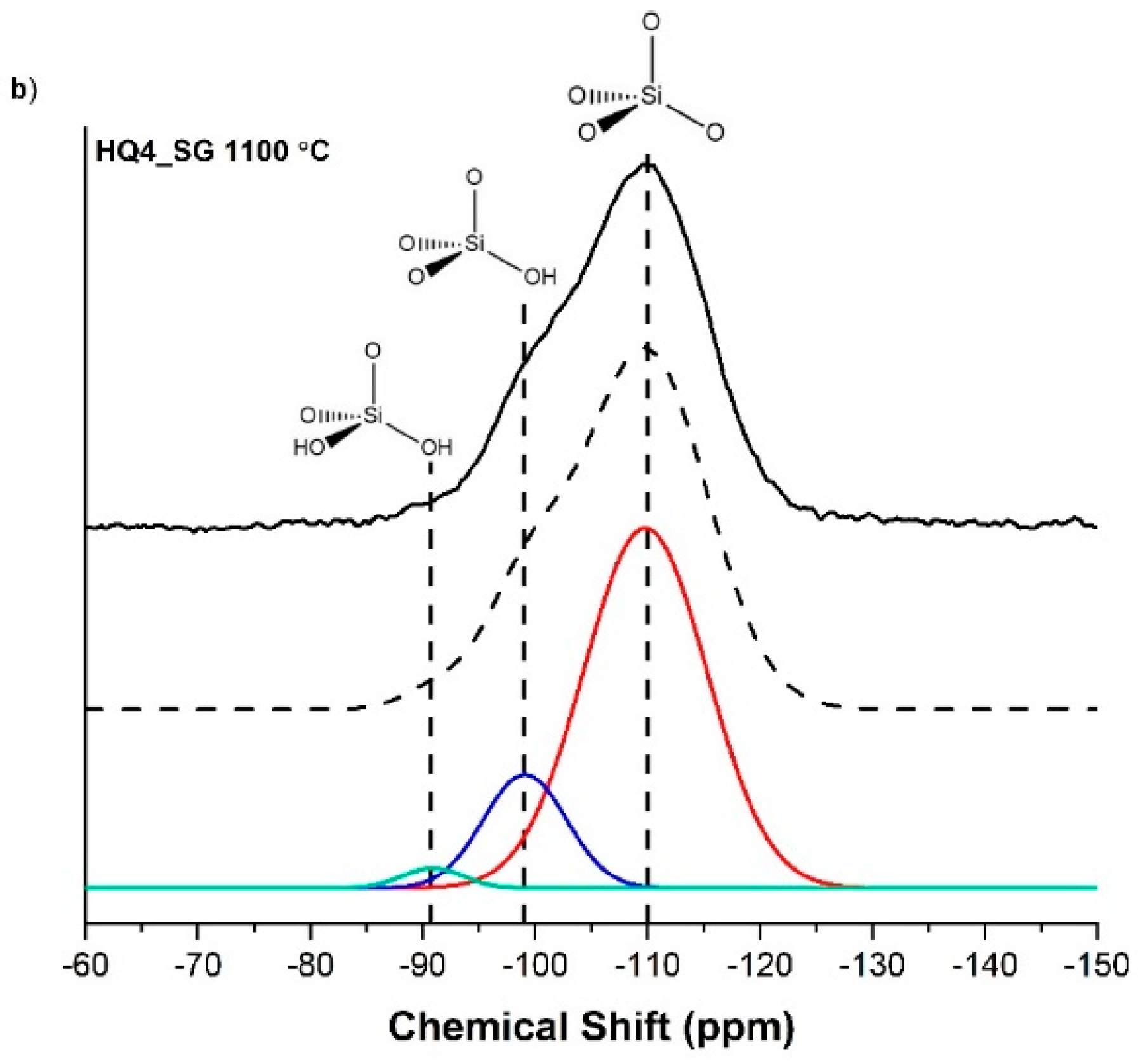
3.4. Magic Angle Spinning Nuclear Magnetic Resonance
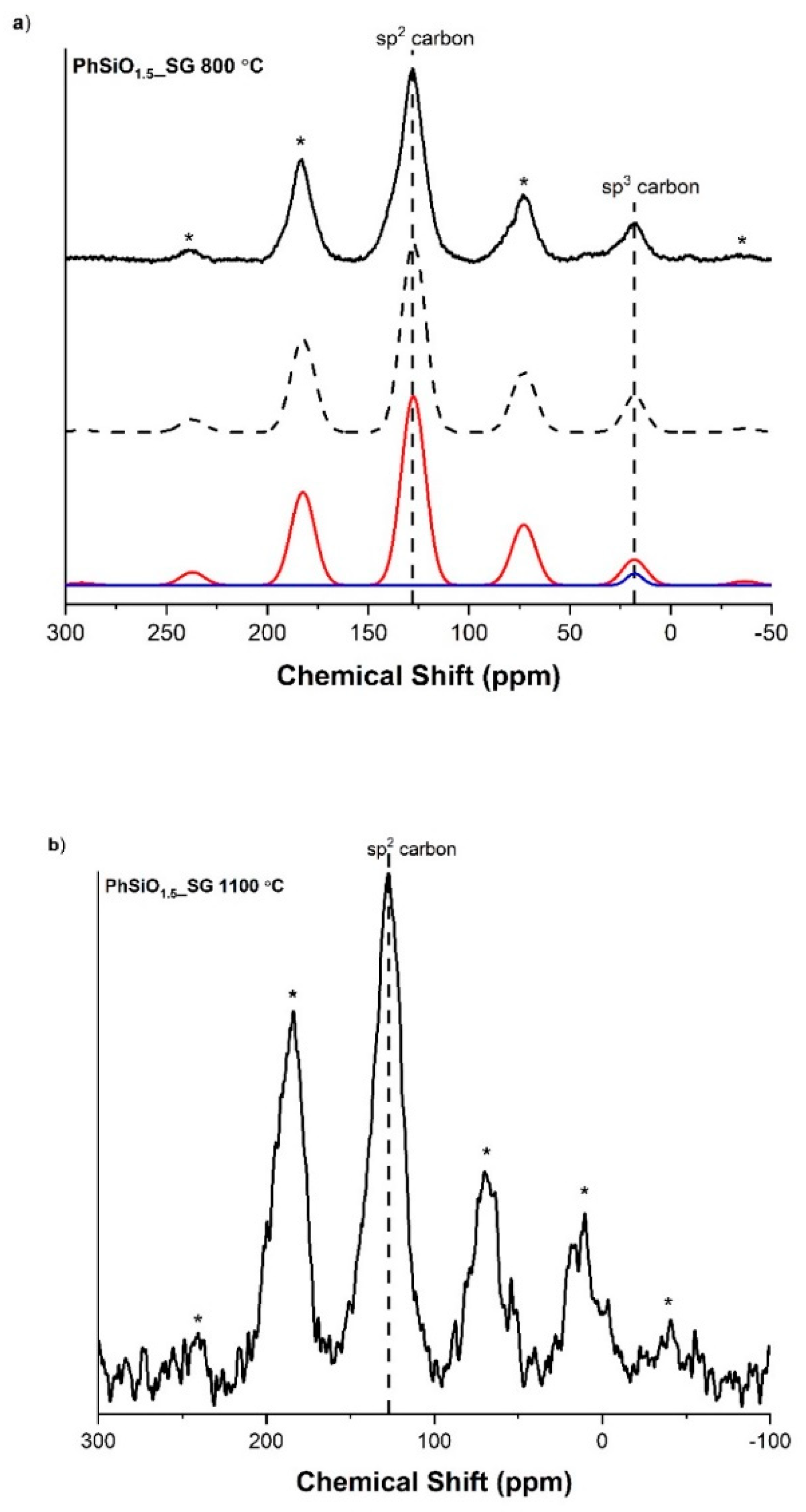
3.5. 13C(1H) Cross Polarization Magic Angle Spinning Nuclear Magnetic Resonance
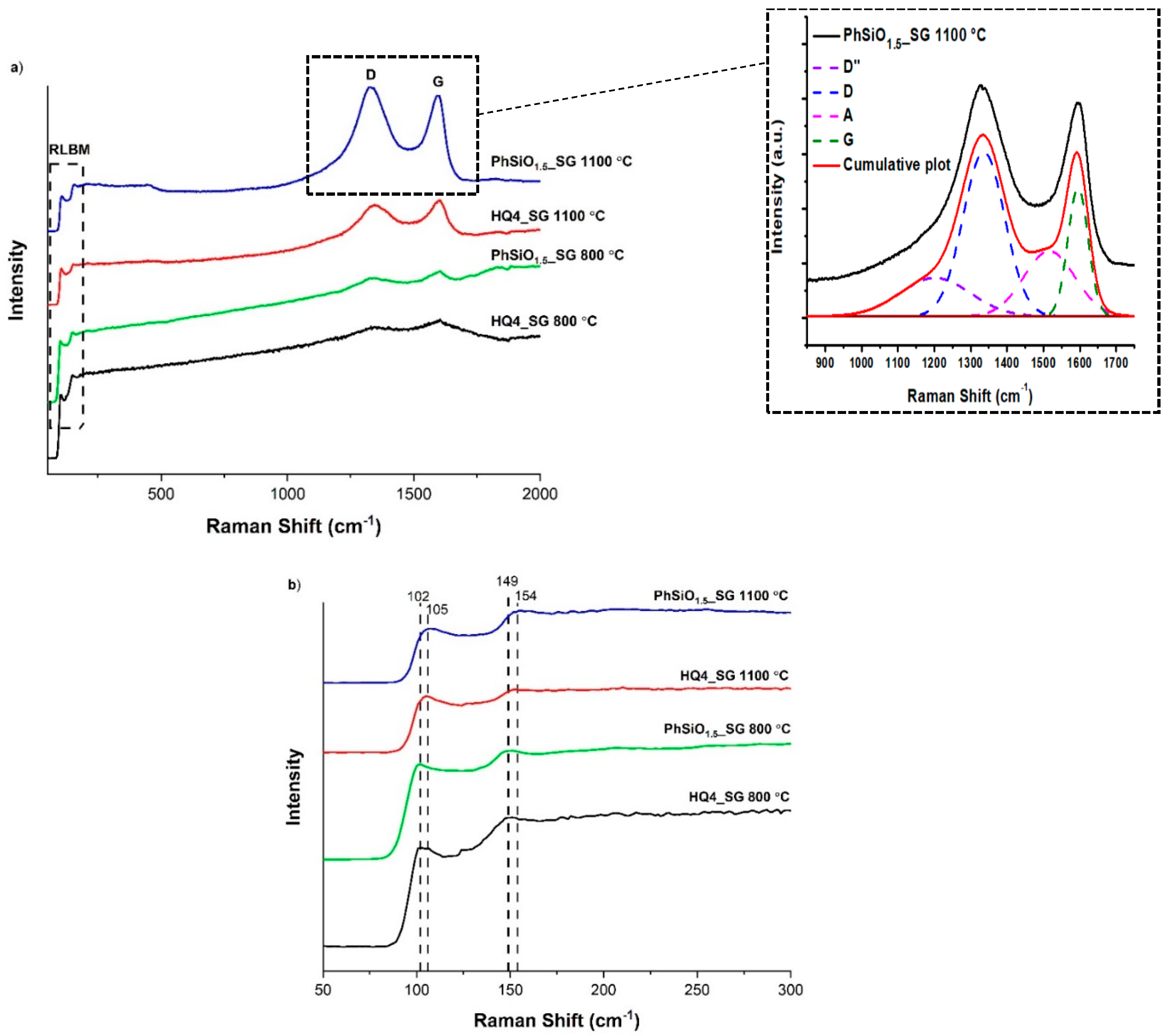
3.6. Micro-Raman Spectroscopy
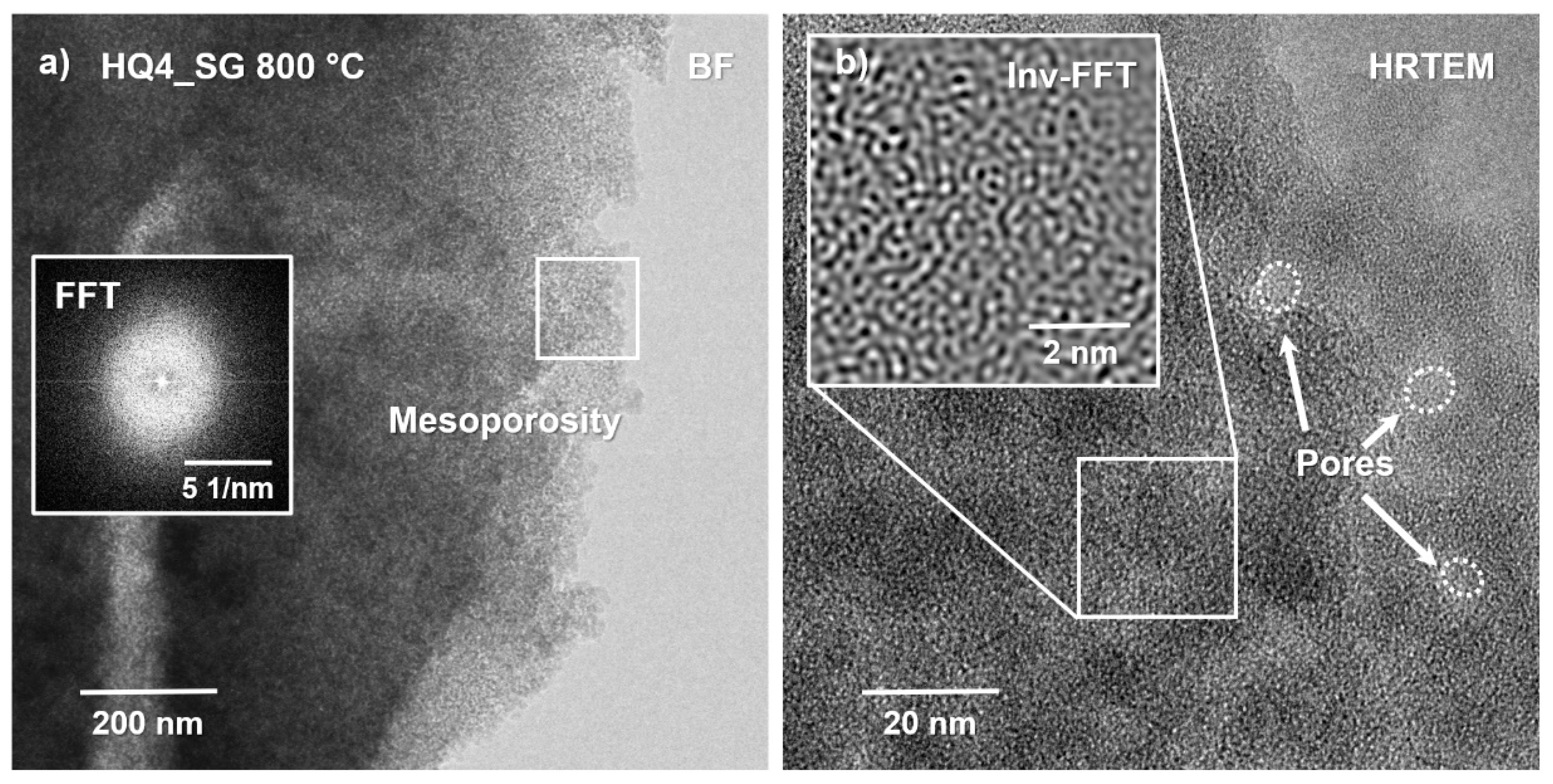
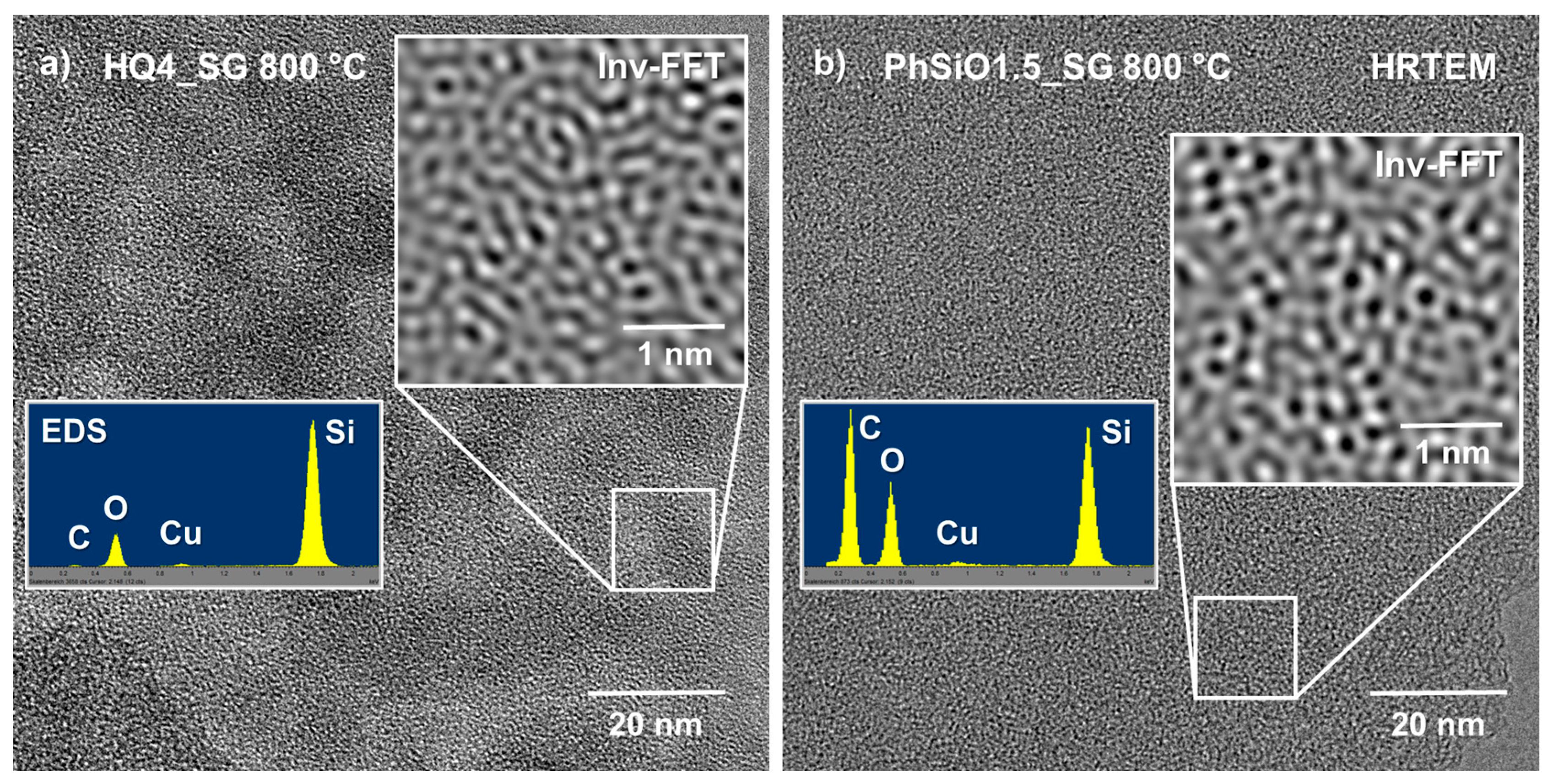
3.7. High-Resolution Transmission Electron Microscopy
4. Thermochemistry
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Colombo, P.; Mera, G.; Riedel, R.; Sorarù, G.D. Polymer-Derived Ceramics: 40 Years of Research and Innovation in Advanced Ceramics. J. Am. Ceram. Soc. 2010, 93, 1805–1837. [Google Scholar] [CrossRef]
- Mera, G.; Riedel, R. Organosilicon-Based Polymers as Precursors for Ceramics. In Polymer Derived Ceramics: From Nano-structure to Applications; Colombo, P., Riedel, R., Soraru, G.D., Kleebe, H.-J., Eds.; DEStech Publications, Inc.: Lancaster, PA, USA, 2009. [Google Scholar]
- Riedel, R.; Mera, G.; Hauser, R.; Klonczynski, A. Silicon-Based Polymer-Derived Ceramics: Synthesis Properties and Applications-A Review. J. Ceram. Soc. Jpn. 2006, 114, 425–444. [Google Scholar] [CrossRef] [Green Version]
- Shah, S.R.; Raj, R. Mechanical Properties of a Fully Dense Polymer Derived Ceramic Made by a Novel Pressure Casting Process. Acta Mater. 2002, 50, 4093–4103. [Google Scholar] [CrossRef]
- Fiocco, L.; Li, S.; Stevens, M.M.; Bernardo, E.; Jones, J.R. Biocompatibility and Bioactivity of Porous Polymer-Derived Ca-Mg Silicate Ceramics. Acta Biomater. 2017, 50, 56–67. [Google Scholar] [CrossRef]
- Ionescu, E.; Sen, S.; Mera, G.; Navrotsky, A. Structure, Energetics and Bioactivity of Silicon Oxycarbide-Based Amorphous Ceramics with Highly Connected Networks. J. Eur. Ceram. Soc. 2018, 38, 1311–1319. [Google Scholar] [CrossRef]
- Mera, G.; Navrotsky, A.; Sen, S.; Kleebe, H.-J.; Riedel, R. Polymer-Derived SiCN and SiOC Ceramics—Structure and Energetics at the Nanoscale. J. Mater. Chem. A 2013, 1, 3826–3836. [Google Scholar] [CrossRef]
- Stabler, C.; Ionescu, E.; Graczyk-Zajac, M.; Gonzalo-Juan, I.; Riedel, R. Silicon Oxycarbide Glasses and Glass-Ceramics: “All-Rounder” Materials for Advanced Structural and Functional Applications. J. Am. Ceram. Soc. 2018, 101, 4817–4856. [Google Scholar] [CrossRef]
- Pantano, C.G.; Singh, A.K.; Zhang, H. Silicon Oxycarbide Glasses. J. Sol-Gel Sci. Technol. 1999, 14, 7–25. [Google Scholar] [CrossRef]
- Mera, G.; Gallei, M.; Bernard, S.; Ionescu, E. Ceramic Nanocomposites from Tailor-Made Preceramic Polymers. Nanomaterials 2015, 5, 468–540. [Google Scholar] [CrossRef]
- Widgeon, S.J.; Sen, S.; Mera, G.; Ionescu, E.; Riedel, R.; Navrotsky, A. 29Si and 13C Solid-State NMR Spectroscopic Study of Nanometer-Scale Structure and Mass Fractal Characteristics of Amorphous Polymer Derived Silicon Oxycarbide Ceramics. Chem. Mater. 2010, 22, 6221–6228. [Google Scholar] [CrossRef]
- Morcos, R.M.; Mera, G.; Navrotsky, A.; Varga, T.; Riedel, R.; Poli, F.; Müller, K. Enthalpy of Formation of Carbon-Rich Polymer-Derived Amorphous SiCN Ceramics. J. Am. Ceram. Soc. 2008, 91, 3349–3354. [Google Scholar] [CrossRef]
- Niu, M.; Wang, H.; Chen, J.; Su, L.; Wu, D.; Navrotsky, A. Structure and Energetics of SiOC and SiOC-Modified Carbon-Bonded Carbon Fiber Composites. J. Am. Ceram. Soc. 2017, 100, 3693–3702. [Google Scholar] [CrossRef]
- Tavakoli, A.H.; Campostrini, R.; Gervais, C.; Babonneau, F.; Bill, J.; Sorarù, G.D.; Navrotsky, A. Energetics and Structure of Polymer-Derived Si–(B–)O–C Glasses: Effect of the Boron Content and Pyrolysis Temperature. J. Am. Ceram. Soc. 2014, 97, 303–309. [Google Scholar] [CrossRef]
- Varga, T.; Navrotsky, A.; Moats, J.L.; Morcos, R.M.; Poli, F.; Müller, K.; Saha, A.; Raj, R. Thermodynamically Stable SixOyCz Polymer-Like Amorphous Ceramics. J. Am. Ceram. Soc. 2007, 90, 3213–3219. [Google Scholar] [CrossRef]
- Widgeon, S.; Mera, G.; Gao, Y.; Stoyanov, E.; Sen, S.; Navrotsky, A.; Riedel, R. Nanostructure and Energetics of Carbon-Rich SiCN Ceramics Derived from Polysilylcarbodiimides: Role of the Nanodomain Interfaces. Chem. Mater. 2012, 24, 1181–1191. [Google Scholar] [CrossRef]
- Navrotsky, A. Thermochemical Studies of Nitrides and Oxynitrides by Oxidative Oxide Melt Calorimetry. J. Alloys Compd. 2001, 321, 300–306. [Google Scholar] [CrossRef]
- Navrotsky, A. Progress and New Directions in High Temperature Calorimetry Revisited. Phys. Chem. Miner. 1997, 24, 222–241. [Google Scholar] [CrossRef]
- McHale, J.M.; Navrotsky, A.; Kowach, G.R.; Disalvo, F.J. Thermochemistry of Metal Nitrides in the Ca/Zn/N System. Chem. A Eur. J. 1996, 2, 1514–1517. [Google Scholar] [CrossRef]
- Navrotsky, A. Progress and New Directions in High Temperature Calorimetry. Phys. Chem. Miner. 1977, 2, 89–104. [Google Scholar] [CrossRef]
- Morcos, R.M.; Navrotsky, A.; Varga, T.; Blum, Y.; Ahn, D.; Poli, F.; Müller, K.; Raj, R. Energetics of SixOyCz Polymer-Derived Ceramics Prepared Under Varying Conditions. J. Am. Ceram. Soc. 2008, 91, 2969–2974. [Google Scholar] [CrossRef]
- Mera, G.; Kroll, P.; Ponomarev, I.; Chen, J.; Morita, K.; Liesegang, M.; Ionescu, E.; Navrotsky, A. Metal-Catalyst-Free Access to Multiwalled Carbon Nanotubes/Silica Nanocomposites (MWCNT/SiO2) from a Single-Source Precursor. Dalt. Trans. 2019, 48, 11018–11033. [Google Scholar] [CrossRef] [PubMed]
- Narisawa, M.; Funabiki, F.; Iwase, A.; Wakai, F.; Hosono, H. Effects of Atmospheric Composition on the Molecular Structure of Synthesized Silicon Oxycarbides. J. Am. Ceram. Soc. 2015, 98, 3373–3380. [Google Scholar] [CrossRef] [Green Version]
- Graczyk-Zajac, M.; Vrankovic, D.; Waleska, P.; Hess, C.; Sasikumar, P.V.; Lauterbach, S.; Kleebe, H.-J.; Sorarù, G.D. The Li-Storage Capacity of SiOC Glasses with and without Mixed Silicon Oxycarbide Bonds. J. Mater. Chem. A 2018, 6, 93–103. [Google Scholar] [CrossRef]
- Ionescu, E.; Linck, C.; Fasel, C.; Müller, M.; Kleebe, H.; Riedel, R. Polymer-Derived SiOC/ZrO2 Ceramic Nanocomposites with Excellent High-Temperature Stability. J. Am. Ceram. Soc. 2010, 93, 241–250. [Google Scholar] [CrossRef]
- Navrotsky, A. Progress and New Directions in Calorimetry: A 2014 Perspective. J. Am. Ceram. Soc. 2014, 97, 3349–3359. [Google Scholar] [CrossRef]
- Chen, J.; King, S.W.; Muthuswamy, E.; Koryttseva, A.; Wu, D.; Navrotsky, A. Thermodynamic Stability of Low-k Amorphous SiOCH Dielectric Films. J. Am. Ceram. Soc. 2016, 99, 2752–2759. [Google Scholar] [CrossRef]
- Lee, D.W.; Kawakami, Y. Incompletely Condensed Silsesquioxanes: Formation and Reactivity. Polym. J. 2007, 39, 230–238. [Google Scholar] [CrossRef] [Green Version]
- Morimoto, S.; Imoto, H.; Naka, K. POSS Solid Solutions Exhibiting Orientationally Disordered Phase Transitions. Chem. Commun. 2017, 53, 9273–9276. [Google Scholar] [CrossRef]
- Zemnukhova, L.; Panasenko, A.; Artem’yanov, A.; Tsoy, E. Dependence of Porosity of Amorphous Silicon Dioxide Prepared from Rice Straw on Plant Variety. BioResources 2015, 10, 3713–3723. [Google Scholar] [CrossRef] [Green Version]
- Pandurangi, R.S.; Seehra, M.S.; Razzaboni, B.L.; Bolsaitis, P. Surface and Bulk Infrared Modes of Crystalline and Amorphous Silica Particles: A Study of the Relation of Surface Structure to Cytotoxicity of Respirable Silica. Environ. Health Perspect. 1990, 86, 327–336. [Google Scholar] [CrossRef]
- Morrow, B.A.; McFarlan, A.J. Surface Vibrational Modes of Silanol Groups on Silica. J. Phys. Chem. 1992, 96, 1395–1400. [Google Scholar] [CrossRef]
- Jeleń, P.; Szumera, M.; Gawęda, M.; Długoń, E.; Sitarz, M. Thermal Evolution of Ladder-like Silsesquioxanes during Formation of Black Glasses. J. Therm. Anal. Calorim. 2017, 130, 103–111. [Google Scholar] [CrossRef] [Green Version]
- Jeleń, P.; Bik, M.; Nocuń, M.; Gawęda, M.; Długoń, E.; Sitarz, M. Free Carbon Phase in SiOC Glasses Derived from Ladder-like Silsesquioxanes. J. Mol. Struct. 2016, 1126, 172–176. [Google Scholar] [CrossRef]
- Lee, K.-M.; Chang, Y.; Chi, K.; Navamathavan, R. Characteristics of SiOC(-H) Thin Films Prepared by Using Plasma-Enhanced Atomic Layer Deposition. J. Korean Phys. Soc. 2011, 59, 3074–3079. [Google Scholar] [CrossRef]
- Sitarz, M. The Structure of Simple Silicate Glasses in the Light of Middle Infrared Spectroscopy Studies. J. Non-Cryst. Solids 2011, 357, 1603–1608. [Google Scholar] [CrossRef]
- Niemiec, W.; Szczygieł, P.; Jeleń, P.; Handke, M. IR Investigation on Silicon Oxycarbide Structure Obtained from Precursors with 1:1 Silicon to Carbon Atoms Ratio and Various Carbon Atoms Distribution. J. Mol. Struct. 2018, 1164, 217–226. [Google Scholar] [CrossRef]
- Chen, Q.; Xue, F.; Ding, E. Preparation of POSS-Triol/Wollastonite Composite Particles by Liquid Phase Mechanochemical Method and Its Application in UV Curable Coatings. Sci. Eng. Compos. Mater. 2019, 26, 183–196. [Google Scholar] [CrossRef] [Green Version]
- Penso, I.; Cechinatto, E.A.; Machado, G.; Luvison, C.; Wanke, C.H.; Bianchi, O.; Soares, M.R.F. Preparation and Characterization of Polyhedral Oligomeric Silsesquioxane (POSS) Using Domestic Microwave Oven. J. Non-Cryst. Solids 2015, 428, 82–89. [Google Scholar] [CrossRef]
- Sitarz, M.; Jastrzębski, W.; Jeleń, P.; Długoń, E.; Gawęda, M. Preparation and Structural Studies of Black Glasses Based on Ladder-like Silsesquioxanes. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2014, 132, 884–888. [Google Scholar] [CrossRef] [PubMed]
- Handke, M.; Handke, B.; Kowalewska, A.; Jastrzębski, W. New Polysilsesquioxane Materials of Ladder-like Structure. J. Mol. Struct. 2009, 924–926, 254–263. [Google Scholar] [CrossRef]
- Segatelli, M.G.; Pires, A.T.N.; Yoshida, I.V.P. Synthesis and Structural Characterization of Carbon-Rich SiCxOy Derived from a Ni-Containing Hybrid Polymer. J. Eur. Ceram. Soc. 2008, 28, 2247–2257. [Google Scholar] [CrossRef]
- Handke, M.; Jastrzębski, W.; Kowalewska, A.; Mozgawa, W. Spectroscopic Study of Preceramic Polymers (Xerogels) Obtained by Hydrolytic Condensation of Ethoxycyclosiloxanes. J. Mol. Struct. 2009, 924–926, 248–253. [Google Scholar] [CrossRef]
- Grill, A.; Neumayer, D.A. Structure of Low Dielectric Constant to Extreme Low Dielectric Constant SiCOH Films: Fourier Transform Infrared Spectroscopy Characterization. J. Appl. Phys. 2003, 94, 6697–6707. [Google Scholar] [CrossRef]
- Hair, M.L. Hydroxyl Groups on Silica Surface. J. Non-Cryst. Solids 1975, 19, 299–309. [Google Scholar] [CrossRef]
- Davydov, V.Y.; Kiselev, A.V.; Zhuravlev, L.T. Study of the Surface and Bulk Hydroxyl Groups of Silica by Infra-Red Spectra and D2O-Exchange. Trans. Faraday Soc. 1964, 60, 2254–2264. [Google Scholar] [CrossRef]
- Zhuravlev, L.T. Concentration of Hydroxyl Groups on the Surface of Amorphous Silicas. Langmuir 1987, 3, 316–318. [Google Scholar] [CrossRef]
- Sen, S.; Widgeon, S.J.; Navrotsky, A.; Mera, G.; Tavakoli, A.; Ionescu, E.; Riedel, R. Carbon Substitution for Oxygen in Silicates in Planetary Interiors. Proc. Natl. Acad. Sci. USA 2013, 110, 15904–15907. [Google Scholar] [CrossRef] [Green Version]
- Liang, T.; Li, Y.-L.; Su, D.; Du, H.-B. Silicon Oxycarbide Ceramics with Reduced Carbon by Pyrolysis of Polysiloxanes in Water Vapor. J. Eur. Ceram. Soc. 2010, 30, 2677–2682. [Google Scholar] [CrossRef]
- Léonardelli, S.; Facchini, L.; Fretigny, C.; Tougne, P.; Legrand, A.P. Silicon-29 Nuclear Magnetic Resonance Study of Silica. J. Am. Chem. Soc. 1992. [Google Scholar] [CrossRef]
- Sindorf, D.W.; Maciel, G.E. Silicon-29 Nuclear Magnetic Resonance Study of Hydroxyl Sites on Dehydrated Silica Gel Surfaces, Using Silylation as a Probe. J. Phys. Chem. 1983, 87, 5516–5521. [Google Scholar] [CrossRef]
- Luhmer, M.; d’Espinose, J.B.; Hommel, H.; Legrand, A.P. High-Resolution 29Si Solid-State NMR Study of Silicon Functionality Distribution on the Surface of Silicas. Magn. Reson. Imaging 1996, 14, 911–913. [Google Scholar] [CrossRef]
- Walther, K.L.; Wokaun, A.; Baiker, A. Characterization of Porous Silica Gels Prepared via the Sol-Gel Process by 29Si CP/MAS Solid-State NMR Spectroscopy. Mol. Phys. 1990, 71, 769–780. [Google Scholar] [CrossRef]
- Freitas, J.C.C.; Cipriano, D.F.; Zucolotto, C.G.; Cunha, A.G.; Emmerich, F.G. Solid-State 13C NMR Spectroscopy Applied to the Study of Carbon Blacks and Carbon Deposits Obtained by Plasma Pyrolysis of Natural Gas. J. Spectrosc. 2016, 2016, 1543273. [Google Scholar] [CrossRef] [Green Version]
- Freitas, J.; Cunha, A.; Emmerich, F. Solid-State Nuclear Magnetic Resonance (NMR) Methods Applied to the Study of Carbon Materials. In Chemistry & Physics of Carbon. Series: Chemistry and Physics of Carbon; Ljubisa, R., Ed.; CRC Press: Boca Raton, FL, USA, 2012; pp. 85–170. ISBN 978-1-4398-2094-0. [Google Scholar] [CrossRef]
- Ruz, P.; Banerjee, S.; Pandey, M.; Sudarsan, V.; Sastry, P.U.; Kshirsagar, R.J. Structural Evolution of Turbostratic Carbon: Implications in H2 Storage. Solid State Sci. 2016, 62, 105–111. [Google Scholar] [CrossRef]
- De Souza, F.A.L.; Ambrozio, A.R.; Souza, E.S.; Cipriano, D.F.; Scopel, W.L.; Freitas, J.C.C. NMR Spectral Parameters in Graphene, Graphite, and Related Materials: Ab Initio Calculations and Experimental Results. J. Phys. Chem. C 2016, 120, 27707–27716. [Google Scholar] [CrossRef]
- Dybowski, C.; Gaffney, E.J.; Sayir, A.; Rabinowitz, M.J. Solid-State 13C and 29Si MAS NMR Spectroscopy of Silicon Carbide. Colloids Surfaces A Physicochem. Eng. Asp. 1996, 118, 171–181. [Google Scholar] [CrossRef]
- Kitaygorodskiy, A.; Wang, W.; Xie, S.-Y.; Lin, Y.; Shiral Fernando, K.A.; Wang, X.; Qu, L.; Chen, B.; Sun, Y.-P. NMR Detection of Single-Walled Carbon Nanotubes in Solution. J. Am. Chem. Soc. 2005, 127, 7517–7520. [Google Scholar] [CrossRef]
- Goze-Bac, C.; Latil, S.; Lauginie, P.; Jourdain, V.; Conard, J.; Duclaux, L.; Rubio, A.; Bernier, P. Magnetic Interactions in Carbon Nanostructures. Carbon N. Y. 2002, 40, 1825–1842. [Google Scholar] [CrossRef]
- Latil, S.; Henrard, L.; Bac, C.G.; Bernier, P.; Rubio, A. NMR Chemical Shift of Single-Wall Carbon Nanotubes. Phys. Rev. Lett. 2001, 86, 3160–3163. [Google Scholar] [CrossRef] [Green Version]
- Zurek, E.; Autschbach, J. Density Functional Calculations of the 13C NMR Chemical Shifts in (9,0) Single-Walled Carbon Nanotubes. J. Am. Chem. Soc. 2004, 126, 13079–13088. [Google Scholar] [CrossRef]
- Marques, M.A.L.; d’Avezac, M.; Mauri, F. Magnetic Response and NMR Spectra of Carbon Nanotubes from Ab Initio Calculations. Phys. Rev. B 2006, 73, 125433. [Google Scholar] [CrossRef] [Green Version]
- Abou-Hamad, E.; Kim, Y.; Talyzin, A.V.; Goze-Bac, C.; Luzzi, D.E.; Rubio, A.; Wågberg, T. Hydrogenation of C60 in Peapods: Physical Chemistry in Nano Vessels. J. Phys. Chem. C 2009, 113, 8583–8587. [Google Scholar] [CrossRef] [Green Version]
- Abou-Hamad, E.; Babaa, M.-R.; Bouhrara, M.; Kim, Y.; Saih, Y.; Dennler, S.; Mauri, F.; Basset, J.-M.; Goze-Bac, C.; Wågberg, T. Structural Properties of Carbon Nanotubes Derived from NMR. Phys. Rev. B 2011, 84, 165417. [Google Scholar] [CrossRef]
- Hodkiewicz, J.; Scientific, T.F. Characterizing Carbon Materials with Raman Spectroscopy. Prog. Mater. Sci. 2005, 17, 10. [Google Scholar] [CrossRef]
- Lei, X.-W.; Ni, Q.-Q.; Shi, J.-X.; Natsuki, T. Radial Breathing Mode of Carbon Nanotubes Subjected to Axial Pressure. Nanoscale Res. Lett. 2011, 6, 492. [Google Scholar] [CrossRef] [Green Version]
- Batra, R.C.; Gupta, S.S. Wall Thickness and Radial Breathing Modes of Single-Walled Carbon Nanotubes. J. Appl. Mech. 2008, 75. [Google Scholar] [CrossRef]
- Maultzsch, J.; Telg, H.; Reich, S.; Thomsen, C. Radial Breathing Mode of Single-Walled Carbon Nanotubes: Optical Transition Energies and Chiral-Index Assignment. Phys. Rev. B Condens. Matter Mater. Phys. 2005, 72, 205438. [Google Scholar] [CrossRef] [Green Version]
- Caņado, L.G.; Takai, K.; Enoki, T.; Endo, M.; Kim, Y.A.; Mizusaki, H.; Jorio, A.; Coelho, L.N.; Magalhães-Paniago, R.; Pimenta, M.A. General Equation for the Determination of the Crystallite Size La of Nanographite by Raman Spectroscopy. Appl. Phys. Lett. 2006, 88, 163106. [Google Scholar] [CrossRef]
- Cançado, L.G.; Jorio, A.; Ferreira, E.H.M.; Stavale, F.; Achete, C.A.; Capaz, R.B.; Moutinho, M.V.O.; Lombardo, A.; Kulmala, T.S.; Ferrari, A.C. Quantifying Defects in Graphene via Raman Spectroscopy at Different Excitation Energies. Nano Lett. 2011, 11, 3190–3196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kroll, P. Modeling the ‘Free Carbon’ Phase in Amorphous Silicon Oxycarbide. J. Non-Cryst. Solids 2005, 351, 1121–1126. [Google Scholar] [CrossRef]
- Tavakoli, A.H.; Armentrout, M.M.; Sen, S.; Navrotsky, A. Hydrogenated Si–O–C Nanoparticles: Synthesis, Structure, and Thermodynamic Stability. J. Mater. Res. 2015, 30, 295–303. [Google Scholar] [CrossRef]
- Cherkasov, N.B.; Savilov, S.V.; Ivanov, A.S.; Lunin, V.V. Bomb Calorimetry as a Bulk Characterization Tool for Carbon Nanostructures. Carbon N. Y. 2013, 63, 324–329. [Google Scholar] [CrossRef]
- Levchenko, A.A.; Kolesnikov, A.I.; Trofymluk, O.; Navrotsky, A. Energetics of Single-Wall Carbon Nanotubes as Revealed by Calorimetry and Neutron Scattering. Carbon N. Y. 2011, 49, 949–954. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Si (wt.% (at.%)) | O (wt.% (at.%)) | C (wt.% (at.%)) | H (wt.% (at.%)) |
|---|---|---|---|---|
| HQ4_SG 800 °C | 43.3 (28.6) | 53.4 (61.9) | 3.05 (4.71) | 0.26 (4.8) |
| HQ4_SG 1100 °C | 42.7 (28.3) | 50.5 (58.8) | 5.02 (7.78) | 0.28 (5.2) |
| PhSiO1.5_SG 800 °C | 24.8 (11.2) | 28.6 (22.7) | 44.36 (46.98) | 1.51 (19.1) |
| PhSiO1.5_SG 1100 °C | 27.2 (14.2) | 25.1 (23.0) | 46.51 (56.81) | 0.41 (6.0) |
| Sample | Si Species (%) | ||||
|---|---|---|---|---|---|
| SiO4 | SiO3C | SiO2C2 | SiOC4 | SiC4 | |
| PhSiO1.5_SG 800 °C | 35.55 | 45.57 | 16.44 | 0.51 | 1.93 |
| PhSiO1.5_SG 1100 °C | 60.20 | 19.77 | 16.23 | 0.61 | 3.19 |
| Sample | Qn Si Species (%) | ||
|---|---|---|---|
| SiO4 (Q4) | SiO3(OH) (Q3) | SiO2(OH)2 (Q2) | |
| HQ4_SG 800 | 85.2 | 12.5 | 2.3 |
| HQ4_SG 1100 | 80.6 | 17.2 | 2.2 |
| Sample | ωD (cm−1) | ωG (cm−1) | AD/AG | La (nm) | LD (nm) | ο (1/nm2) |
|---|---|---|---|---|---|---|
| HQ4_SG 800 °C | 1345 | 1599 | 0.9 | 44.2 | 18.4 | 0.0029 |
| HQ4_SG 1100 °C | 1350 | 1601 | 2.9 | 13.1 | 10.0 | 0.0099 |
| PhSiO1.5_SG 800 °C | 1335 | 1595 | 2.3 | 16.2 | 11.1 | 0.0080 |
| PhSiO1.5_SG 1100 °C | 1333 | 1597 | 5.5 | 6.8 | 7.2 | 0.0190 |
| Reaction | Enthalpy (∆H) |
|---|---|
| Enthalpy of oxidation (∆H0ox) at 25 °C | - |
| (1) SiaOb-d/2Cc + H2O (solid, 25 °C) + O2 (gas, 802 °C)→a SiO2 (cristobalite, 802 °C) + c CO2 (gas, 802 °C) + H2O (gas, 802 °C) | ∆H1 = ∆Hds (kJ/g at.) |
| (2) SiO2 (cristobalite, 25 °C)→SiO2 (cristobalite, 802 °C) | ∆H2 = 50.1 kJ/mol |
| (3) H2O (liquid, 25 °C)→H2O (gas, 802 °C) | ∆H3 = 73.1 kJ/mol |
| (4) O2 (gas, 25 °C)→O2 (gas, 802 °C) | ∆H4 = 25.3 kJ/mol |
| (5) CO2 (gas, 25 °C)→CO2 (gas, 802 °C) | ∆H5 = 37.5 kJ/mol |
| SiaOb-d/2Cc + H2O (solid, 25 °C) + O2 (gas, 25 °C)→a SiO2(cristobalite, 25 °C) +c CO2 (gas, 25 °C) + H2O (liquid, 25 °C) | ∆H0ox (kJ/g at.) = ∆H1 − a ∆H2 − ∆H3 + ∆H4 − c ∆H5 |
| Enthalpy of formation from the elements ∆H0f,elem at 25 °C | - |
| (1) SiaOb-d/2Cc + H2O (solid, 25 °C) + O2 (gas, 25 °C)→a SiO2(cristobalite, 25 °C) +c CO2 (gas, 25 °C) + H2O (liquid, 25 °C) | ∆H1 = ∆H0ox (kJ/g·at.) |
| (2) Si (solid, 25 °C) + O2 (gas, 25 °C)→SiO2 (cristobalite, 25 °C) | ∆H2 = −908.4 ± 2.1 kJ/mol |
| (3) H2 (gas, 25 °C) + 1/2 O2 (gas, 25 °C)→H2O (liquid, 25 °C) | ∆H3 = −285.8 ± 0.1 kJ/mol |
| (4) C (solid, 25 °C) + O2 (gas, 25 °C)→CO2 (gas, 25 °C) | ∆H4 = −393.5 ± 0.1 kJ/mol |
| a Si (solid, 25 °C) + O2 (gas, 25 °C + c C (solid, 25 °C) + H2 (gas, 25 °C)→SiaOb-d/2Cc + H2O (solid, 25 °C) | ∆H0f,elem (kJ/g-at.) = −∆H1 + a ∆H2 + ∆H3 + c ∆H4 |
| Enthalpy of formation from the components ∆H0f,comp at 25 °C | |
| (1) a Si (solid, 25 °C) + O2 (gas, 25 °C) + c C (solid, 25 °C) + H2 (gas, 25 °C)→SiaOb-d/2Cc + H2O (solid, 25 °C) | ∆H1 = ∆H0f,elem (kJ/g·at.) |
| (2) Si (solid, 25 °C) + C (solid, 25 °C)→SiC (solid, 25 °C) | ∆H2 = −73.2 ± 6.3 kJ/mol |
| (3) Si (solid, 25 °C) + O2 (gas, 25 °C)→SiO2 (cristobalite, 25 °C) | ∆H3 = −908.4 ± 2.1 kJ/mol |
| (4) H2(gas, 25 °C) + O2 (gas, 25 °C)→H2O (liquid, 25 °C) | ∆H4 = −285.83 ± 0.04 kJ/mol |
| SiC (solid, 25 °C) + SiO2 (cristobalite, 25 °C) + C (solid, 25 °C) + H2O (liquid, 25 °C)→SiaOb-d/2Cc + H2O (solid, 25 °C) | ∆H0f,comp (kJ/g-at.) = ∆H1 − ∆H2 − ∆H3 – ∆H4 |
| Sample | SiaObCcHd (a + b + c + d = 1) | ΔHds (kJ/g·at) | ΔHox (kJ/g·at) | ΔHf,ele. (kJ/g·at) | ΔHf,comp. (kJ/g·at) | |||
|---|---|---|---|---|---|---|---|---|
| a | b | c | d | |||||
| HQ4_SG 800 °C | 0.286 | 0.619 | 0.047 | 0.048 | −11.0 ±0.2 | −28.0 ±0.2 | −257.2 ±0.6 | 20.0 ±0.9 |
| HQ4_SG 1100 °C | 0.283 | 0.588 | 0.078 | 0.052 | −12.3 ±0.2 | −29.2 ±0.2 | −265.8 ±0.6 | −3.2 ±0.9 |
| PhSiO1.5_SG 800 °C | 0.112 | 0.227 | 0.470 | 0.191 | −203.8 ±3.3 | −221.0 ±3.3 | −93.2 ±3.3 | −2.5 ±3.4 |
| PhSiO1.5_SG 1100 °C | 0.142 | 0.230 | 0.568 | 0.060 | −229.5 ±3.9 | −244.7 ±3.9 | −116.4 ±3.9 | −13.9 ±3.9 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sugie, C.; Navrotsky, A.; Lauterbach, S.; Kleebe, H.-J.; Mera, G. Structure and Thermodynamics of Silicon Oxycarbide Polymer-Derived Ceramics with and without Mixed-Bonding . Materials 2021, 14, 4075. https://doi.org/10.3390/ma14154075
Sugie C, Navrotsky A, Lauterbach S, Kleebe H-J, Mera G. Structure and Thermodynamics of Silicon Oxycarbide Polymer-Derived Ceramics with and without Mixed-Bonding . Materials. 2021; 14(15):4075. https://doi.org/10.3390/ma14154075
Chicago/Turabian StyleSugie, Casey, Alexandra Navrotsky, Stefan Lauterbach, Hans-Joachim Kleebe, and Gabriela Mera. 2021. "Structure and Thermodynamics of Silicon Oxycarbide Polymer-Derived Ceramics with and without Mixed-Bonding " Materials 14, no. 15: 4075. https://doi.org/10.3390/ma14154075
APA StyleSugie, C., Navrotsky, A., Lauterbach, S., Kleebe, H. -J., & Mera, G. (2021). Structure and Thermodynamics of Silicon Oxycarbide Polymer-Derived Ceramics with and without Mixed-Bonding . Materials, 14(15), 4075. https://doi.org/10.3390/ma14154075