
Polymer-Derived Biosilicate®-like Glass-Ceramics: Engineering of Formulations and Additive Manufacturing of Three-Dimensional Scaffolds

 ,
, 
 ,
,  ,
,  ,
,  and
and 
Abstract
:1. Introduction
2. Materials and Methods
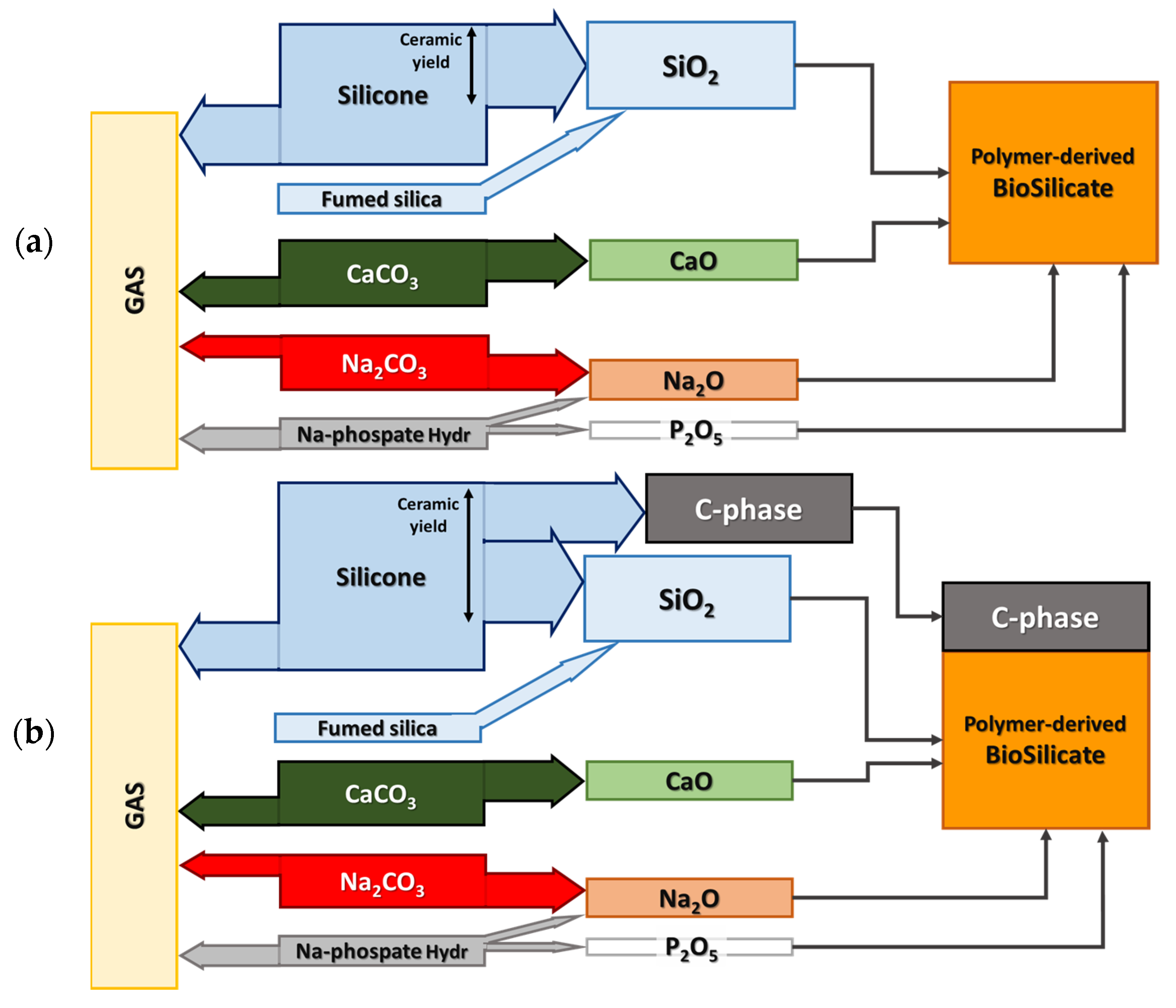
2.1. Starting Materials
2.2. Direct Ink Writing of 3D Scaffolds
2.3. Digital Light Processing of H44-Based Formulation
2.4. Microstructural Characterization
2.5. In Vitro Cytotoxicity Assay
3. Results and Discussion
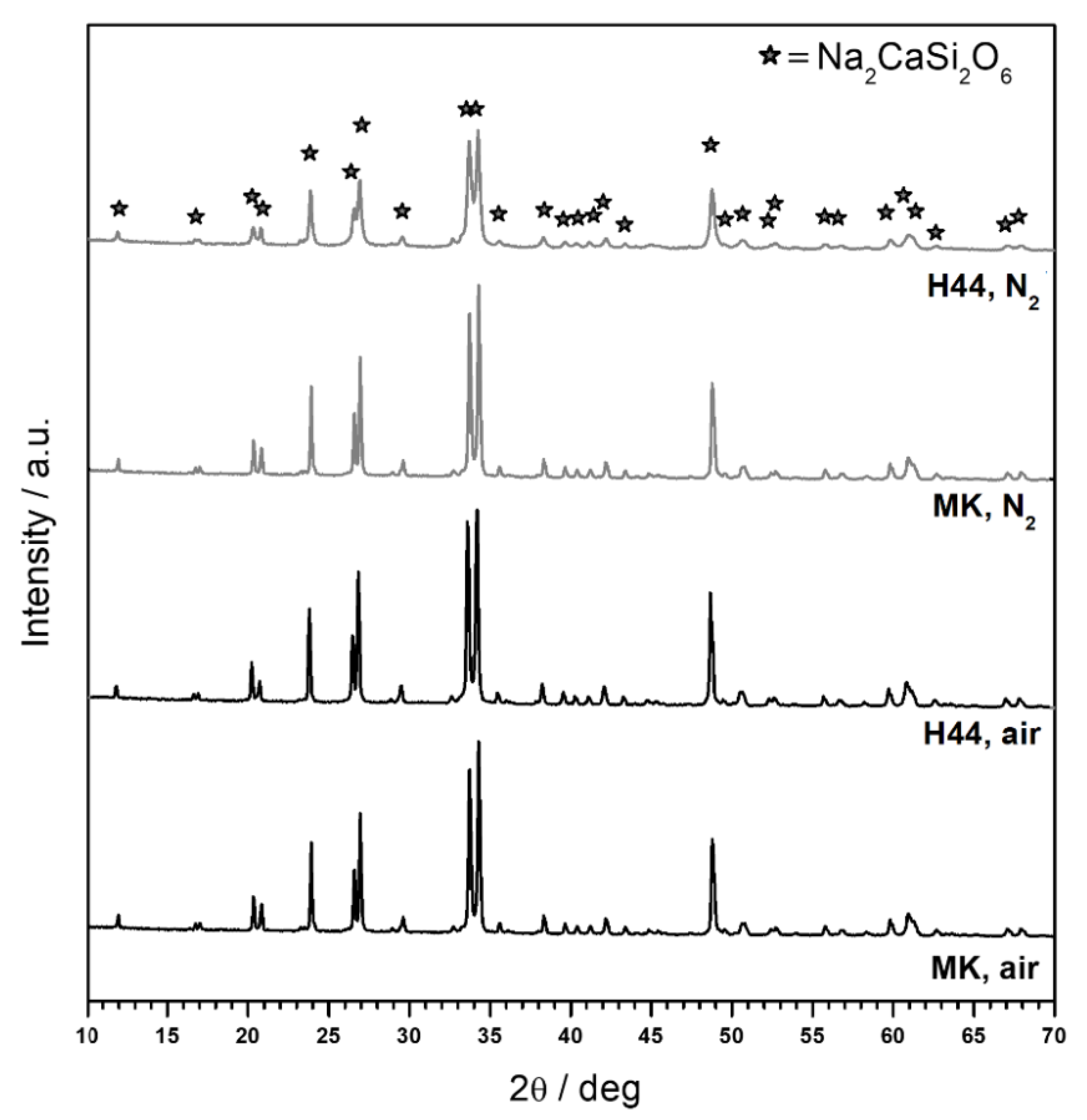
3.1. Direct Ink Writing and Firing in Air: Confirmation of Biosilicate®-like Composition
3.2. Direct Ink Writing and Firing in N2: Modulation of Biosilicate®-like/C Composites
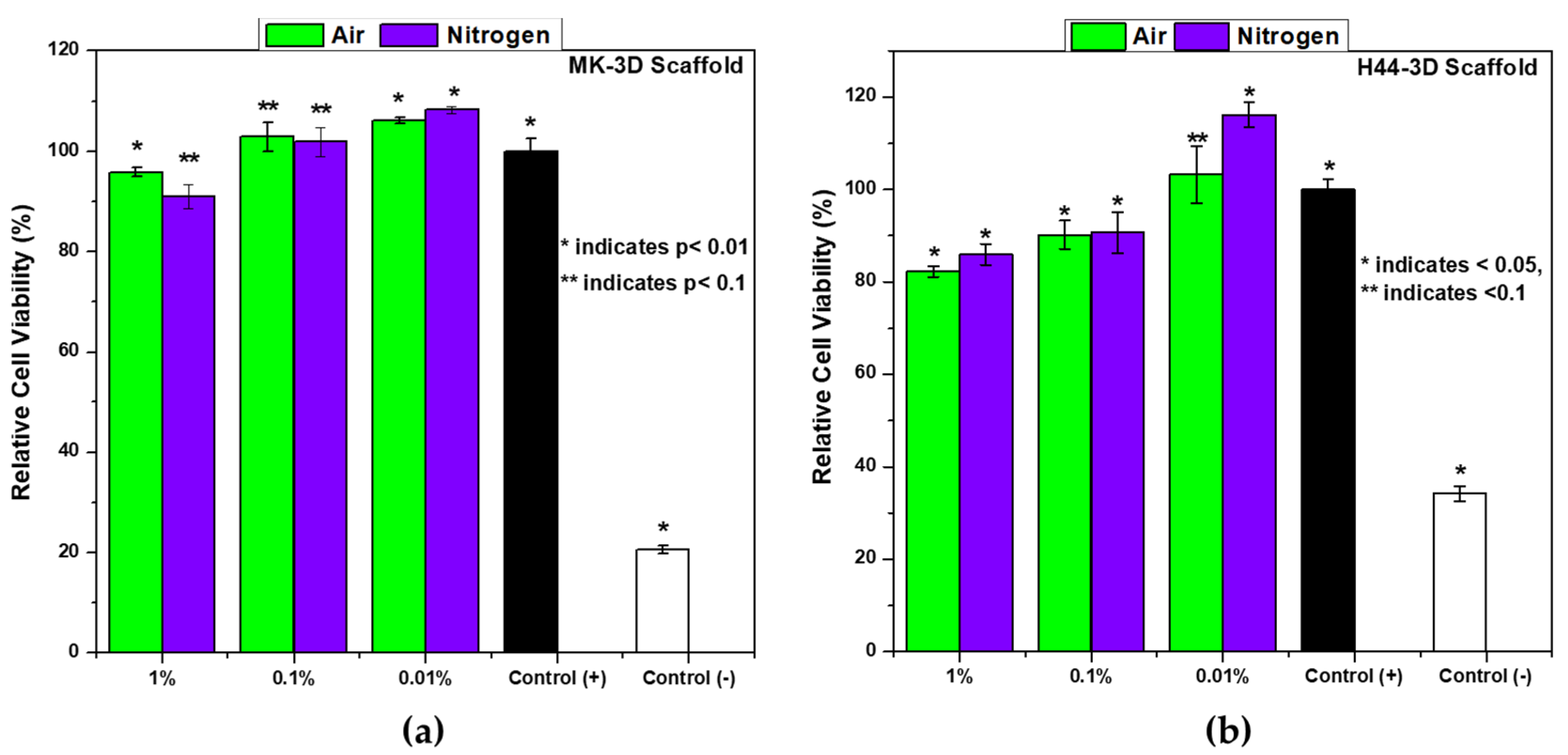
3.3. In Vitro Biocompatibility
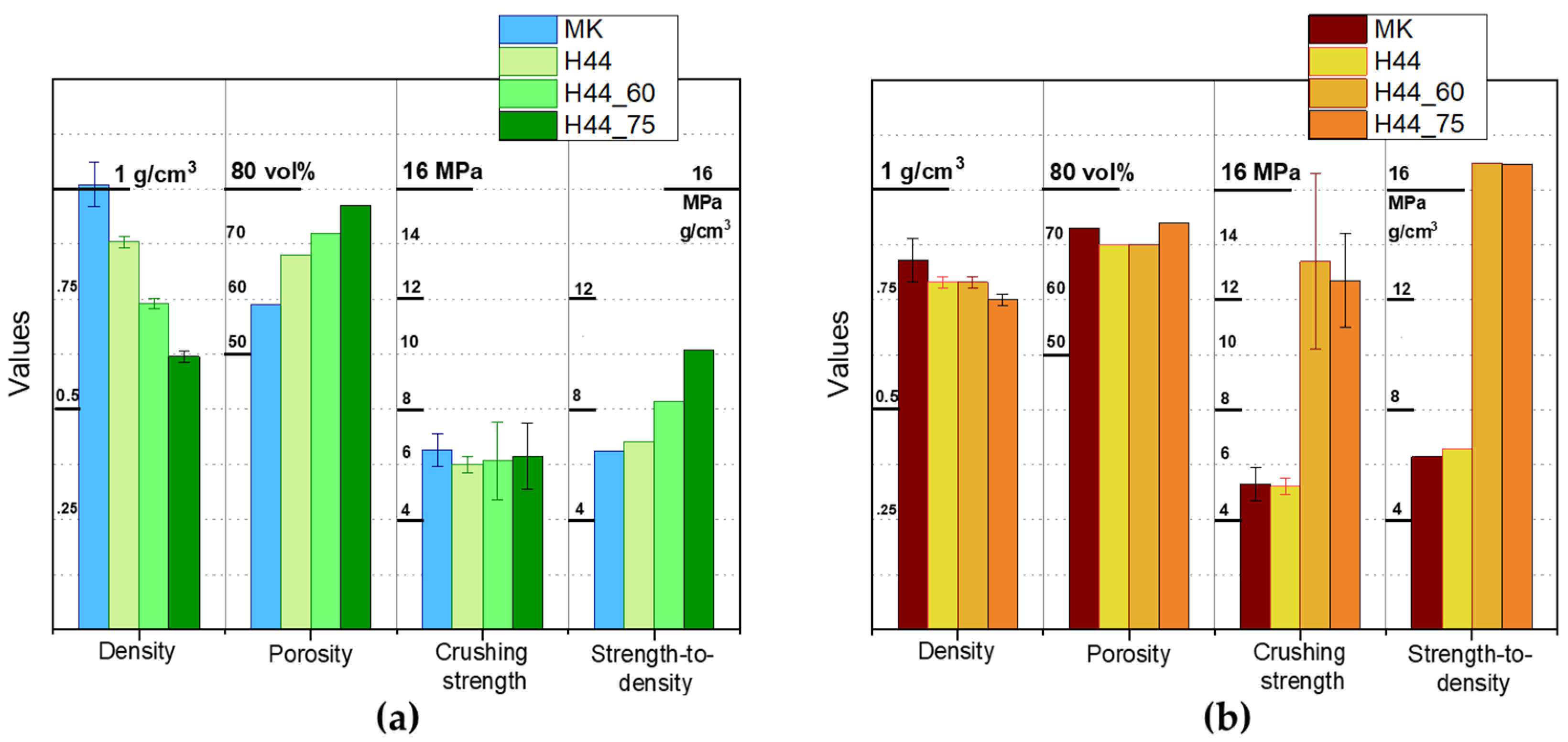
3.4. Direct Ink Writing of H44-Based Pastes: Further Microstructural Modulation
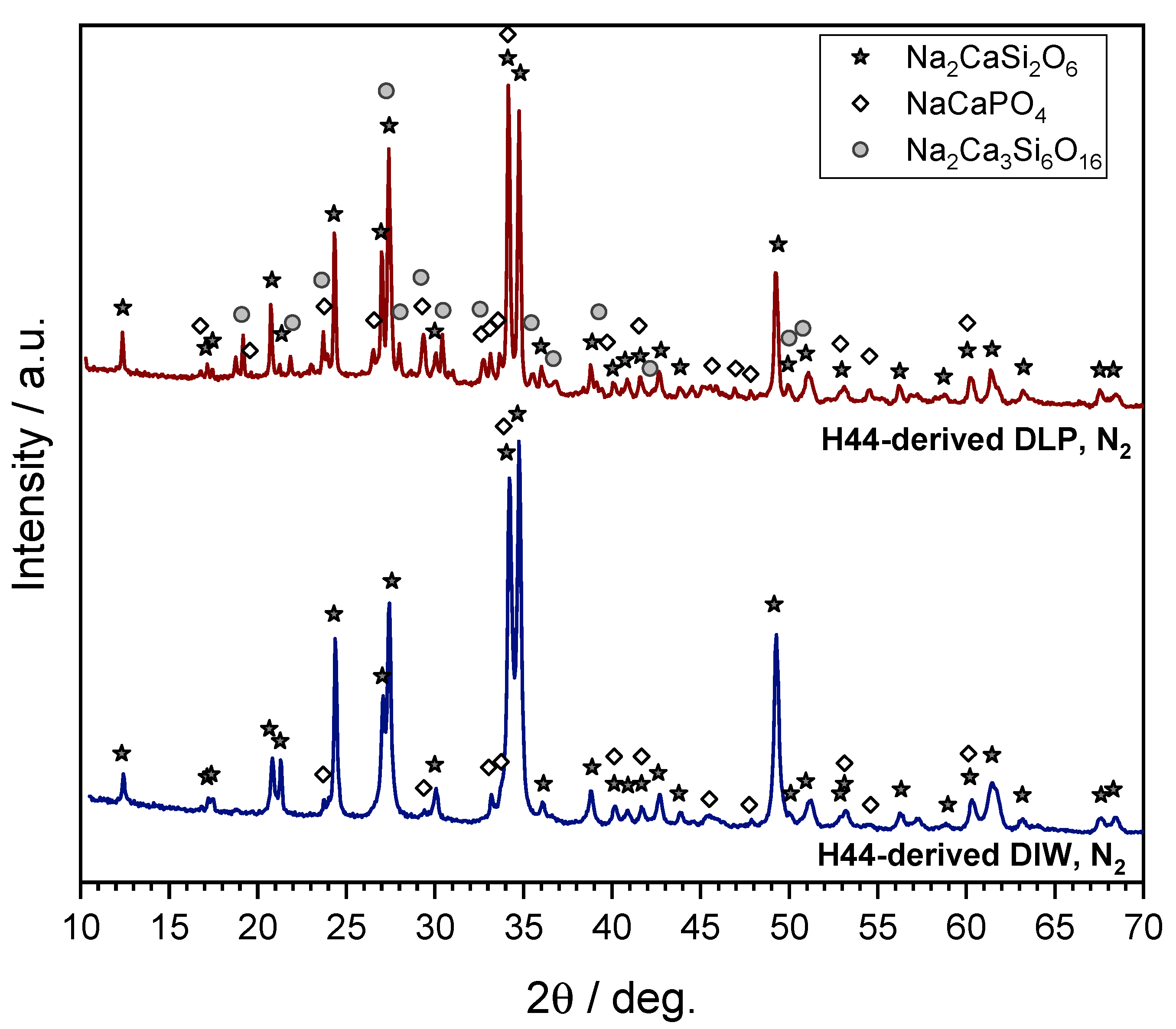
3.5. H44-Based Formulations: Extension to Digital Light Processing
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fu, S.; Zhu, M.; Zhu, Y. Organosilicon polymer-derived ceramics: An overview. J. Adv. Ceram. 2019, 8, 457–478. [Google Scholar] [CrossRef] [Green Version]
- Francis, A.; Detsch, R.; Boccaccini, A.R. Fabrication and cytotoxicity assessment of novel polysiloxane/bioactive glass films for biomedical applications. Ceram. Int. 2016, 42, 15442–15448. [Google Scholar] [CrossRef]
- Xie, F.; Gonzalo-Juan, I.; Breitzke, H.; Fasel, C.; Trapp, M.; Buntkowsky, G.; Kleebe, H.; Riedel, R.; Boccaccini, A.R.; Ionescu, E. Effect of Ca and B incorporation into silicon oxycarbide on its microstructure and phase composition. J. Am. Ceram. Soc. 2019, 102, 7645–7655. [Google Scholar] [CrossRef]
- Xie, F.; Ionescu, E.; Arango-Ospina, M.; Riedel, R.; Boccaccini, A.R.; Gonzalo-Juan, I. Facile Preparative Access to Bioactive Silicon Oxycarbides with Tunable Porosity. Materials 2019, 12, 3862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, F.; Juan, I.G.; Arango-Ospina, M.; Riedel, R.; Boccaccini, A.R.; Ionescu, E. Apatite Forming Ability and Dissolution Behavior of Boron- and Calcium-Modified Silicon Oxycarbides in Comparison to Silicate Bioactive Glass. ACS Biomater. Sci. Eng. 2019, 5, 5337–5347. [Google Scholar] [CrossRef]
- Arango-Ospina, M.; Xie, F.; Gonzalo-Juan, I.; Riedel, R.; Ionescu, E.; Boccaccini, A.R. Review: Silicon oxycarbide based materials for biomedical applications. Appl. Mater. Today 2020, 18, 100482. [Google Scholar] [CrossRef]
- Francis, A. Biological evaluation of preceramic organosilicon polymers for various healthcare and biomedical engineering applications: A review. J. Biomed. Mater. Res. Part B Appl. Biomater. 2021, 109, 744–764. [Google Scholar] [CrossRef] [PubMed]
- Bernardo, E.; Fiocco, L.; Parcianello, G.; Storti, E.; Colombo, P. Advanced Ceramics from Preceramic Polymers Modified at the Nano-Scale: A Review. Materials 2014, 7, 1927–1956. [Google Scholar] [CrossRef] [PubMed]
- Santos, J.M.; Pereira, S.; Sequeira, D.B.; Messias, A.L.; Martins, J.B.; Cunha, H.; Palma, P.J.; Santos, A.C. Biocompatibility of a bioceramic silicone-based sealer in subcutaneous tissue. J. Oral Sci. 2019, 61, 171–177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernardo, E.; Colombo, P.; Cacciotti, I.; Bianco, A.; Bedini, R.; Pecci, R.; Pardun, K.; Treccani, L.; Rezwan, K. Porous wollastonite–hydroxyapatite bioceramics from a preceramic polymer and micro- or nano-sized fillers. J. Eur. Ceram. Soc. 2012, 32, 399–408. [Google Scholar] [CrossRef]
- Fiocco, L.; Agnoli, S.; Pedron, D.; Secco, M.; Tamburini, S.; Ferroni, L.; Gardin, C.; Zavan, B.; Bernardo, E. Wollastonite-diopside-carbon composite foams from a silicone resin and inorganic fillers. Ceram. Int. 2018, 44, 931–937. [Google Scholar] [CrossRef]
- Dasan, A.; Elsayed, H.; Kraxner, J.; Galusek, D.; Bernardo, E. Hierarchically porous 3D-printed akermanite scaffolds from silicones and engineered fillers. J. Eur. Ceram. Soc. 2019, 39, 4445–4449. [Google Scholar] [CrossRef]
- Zocca, A.; Franchin, G.; Elsayed, H.; Gioffredi, E.; Bernardo, E.; Colombo, P. Direct Ink Writing of a Preceramic Polymer and Fillers to Produce Hardystonite (Ca2ZnSi2O7) Bioceramic Scaffolds. J. Am. Ceram. Soc. 2016, 99, 1960–1967. [Google Scholar] [CrossRef]
- Dasan, A.; Elsayed, H.; Kraxner, J.; Galusek, D.; Colombo, P.; Bernardo, E. Engineering of silicone-based mixtures for the digital light processing of Åkermanite scaffolds. J. Eur. Ceram. Soc. 2020, 40, 2566–2572. [Google Scholar] [CrossRef]
- Saha, A.; Raj, R.; Williamson, D.L. A Model for the Nanodomains in Polymer-Derived SiCO. J. Am. Ceram. Soc. 2006, 89, 2188–2195. [Google Scholar] [CrossRef]
- Fu, S.; Hu, H.; Chen, J.; Zhu, Y.; Zhao, S. Silicone resin derived larnite/C scaffolds via 3D printing for potential tumor therapy and bone regeneration. Chem. Eng. J. 2020, 382, 122928. [Google Scholar] [CrossRef]
- Zhu, T.; Zhu, M.; Zhu, Y. Fabrication of forsterite scaffolds with photothermal-induced antibacterial activity by 3D printing and polymer-derived ceramics strategy. Ceram. Int. 2020, 46, 13607–13614. [Google Scholar] [CrossRef]
- Shao, D.; Lu, M.; Xu, D.; Zheng, X.; Pan, Y.; Song, Y.; Xu, J.; Li, M.; Zhang, M.; Li, J.; et al. Carbon dots for tracking and promoting the osteogenic differentiation of mesenchymal stem cells. Biomater. Sci. 2017, 5, 1820–1827. [Google Scholar] [CrossRef] [PubMed]
- Saito, N.; Usui, Y.; Aoki, K.; Narita, N.; Shimizu, M.; Hara, K.; Ogiwara, N.; Nakamura, K.; Ishigaki, N.; Kato, H.; et al. Carbon nanotubes: Biomaterial applications. Chem. Soc. Rev. 2009, 38, 1897–1903. [Google Scholar] [CrossRef]
- Ogihara, N.; Usui, Y.; Aoki, K.; Shimizu, M.; Narita, N.; Hara, K.; Nakamura, K.; Ishigaki, N.; Takanashi, S.; Okamoto, M.; et al. Biocompatibility and bone tissue compatibility of alumina ceramics reinforced with carbon nanotubes. Nanomedicine 2012, 7, 981–993. [Google Scholar] [CrossRef] [Green Version]
- Shafiei, S.; Omidi, M.; Nasehi, F.; Golzar, H.; Mohammadrezaei, D.; Rad, M.R.; Khojasteh, A. Egg shell-derived calcium phosphate/carbon dot nanofibrous scaffolds for bone tissue engineering: Fabrication and characterization. Mater. Sci. Eng. C 2019, 100, 564–575. [Google Scholar] [CrossRef]
- Wu, C.; Xia, L.; Han, P.; Xu, M.; Fang, B.; Wang, J.; Chang, J.; Xiao, Y. Graphene-oxide-modified β-tricalcium phosphate bioceramics stimulate in vitro and in vivo osteogenesis. Carbon 2015, 93, 116–129. [Google Scholar] [CrossRef]
- Gao, C.; Feng, P.; Peng, S.; Shuai, C. Carbon nanotube, graphene and boron nitride nanotube reinforced bioactive ceramics for bone repair. Acta Biomater. 2017, 61, 1–20. [Google Scholar] [CrossRef]
- Hench, L.L. The story of Bioglass®. J. Mater. Sci. Mater. Med. 2006, 17, 967–978. [Google Scholar] [CrossRef] [PubMed]
- Brandão, S.M.; Schellini, S.A.; Moraes, A.D.; Padovani, C.R.; Pellizzon, C.H.; Peitl, O.; Zanotto, E.D. Biocompatibility Analysis of Bioglass®45S5 and Biosilicate®Implants in the Rabbit Eviscerated Socket. Orbit 2012, 31, 143–149. [Google Scholar] [CrossRef]
- Crovace, M.C.; Souza, M.T.; Chinaglia, C.R.; Peitl, O.; Zanotto, E.D. Biosilicate®—A multipurpose, highly bioactive glass-ceramic. In vitro, in vivo and clinical trials. J. Non-Cryst. Solids 2016, 432, 90–110. [Google Scholar] [CrossRef]
- Elsayed, H.; Rebesan, P.; Crovace, M.C.; Zanotto, E.D.; Colombo, P.; Bernardo, E. Biosilicate® scaffolds produced by 3D-printing and direct foaming using preceramic polymers. J. Am. Ceram. Soc. 2018, 102, 1010–1020. [Google Scholar] [CrossRef] [Green Version]
- Gariboldi, M.I.; Best, S.M. Effect of Ceramic Scaffold Architectural Parameters on Biological Response. Front. Bioeng. Biotechnol. 2015, 3, 151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elsayed, H.; Colombo, P.; Crovace, M.C.; Zanotto, E.D.; Bernardo, E. Suitability of Biosilicate® glass-ceramic powder for additive manufacturing of highly porous scaffolds. Ceram. Int. 2021, 47, 8200–8207. [Google Scholar] [CrossRef]
- Scheffler, M.; Takahashi, T.; Kaschta, J.; Muensted, H.; Buhler, P.; Greil, P. Pyrolytic decomposition of preceramic organo polysiloxanes. Ceram. Trans. 2000, 115, 239–250. [Google Scholar]
- Wallin, R.F.; Arscott, E.F. A practical guide to iso 10993-5: Cytotoxicity. Med. Device Diagn. Ind. 1998, 20, 96–98. [Google Scholar]
- Garriga, R.; Herrero-Continente, T.; Palos, M.; Cebolla, V.L.; Osada, J.; Muñoz, E.; Rodríguez-Yoldi, M.J. Toxicity of Carbon Nanomaterials and Their Potential Application as Drug Delivery Systems: In Vitro Studies in Caco-2 and MCF-7 Cell Lines. Nanomaterials 2020, 10, 1617. [Google Scholar] [CrossRef]
- Saha, D.; Heldt, C.L.; Gencoglu, M.F.; Vijayaragavan, K.S.; Chen, J.; Saksule, A. A study on the cytotoxicity of carbon-based materials. Mater. Sci. Eng. C 2016, 68, 101–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schelm, K.; Morales, E.A.; Scheffler, M. Mechanical and Surface-Chemical Properties of Polymer Derived Ceramic Replica Foams. Materials 2019, 12, 1870. [Google Scholar] [CrossRef] [Green Version]
- Serbena, F.C.; Zanotto, E. Internal residual stresses in glass-ceramics: A review. J. Non-Cryst. Solids 2012, 358, 975–984. [Google Scholar] [CrossRef]
- ElBatal, H.A.; El-Kheshen, A.A.; Marzouk, M.A.; Ghoneim, N.A.; ElBatal, F.H.; Ouis, M.A.; Fayad, A.M.; Abdelghany, A.M. In vitro bioactivity of silicophosphate glasses doped with ZnO, SrO or CuO. J. Theor. Appl. Phys. 2020, 14, 159–169. [Google Scholar] [CrossRef]
- Cacciotti, I.; Lombardi, M.; Bianco, A.; Ravaglioli, A.; Montanaro, L. Sol–gel derived 45S5 bioglass: Synthesis, microstructural evolution and thermal behaviour. J. Mater. Sci. Mater. Med. 2012, 23, 1849–1866. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Oxides in Biosilicate® (wt.%) | Oxide Quantities Referred to 10 g of SiO2 (g) in Biosilicate® | Source Quantities Referred to 10 g of SiO2 (g)/Source | ||
|---|---|---|---|---|
| Firing in Air | Firing in N2 | |||
| SiO2 (48.5%) | 10 | 9 | 10.71/MK | 14.09/MK (2.84 g as extra C-phase) |
| 17.00/H44 | 23.58/H44 (7.98 g as extra C-phase) | |||
| 1 | 1/Fumed silica (SiO2) | |||
| P2O5 (4%) | 0.82 | 4.13/Na2HPO4·12H2O | ||
| Na2O (23.75%) | 4.90 | 0.72 | ||
| 4.18 | 7.14/Na2CO3 | |||
| CaO (23.75%) | 4.90 | 8.74/CaCO3 | ||
| Oxides in Biosilicate® (wt.%) | Oxide Quantities Referred to 10 g of SiO2 (g) in Biosilicate® | Source Quantities Referred to 10 g of SiO2 (g)/Source | ||
|---|---|---|---|---|
| Firing in Air | Firing in N2 | |||
| SiO2 (48.5%) | 10 | 9 | 17.00/H44 | 23.58/H44 (7.98 g as extra C-phase) |
| 1 | 1/Fumed silica (SiO2) | |||
| P2O5 (4%) | 0.82 | 1.54/Na4P2O6 | ||
| Na2O (23.75%) | 4.90 | 0.72 | ||
| 4.18 | 7.14/Na2CO3 | |||
| CaO (23.75%) | 4.90 | 8.74/CaCO3 | ||
| Oxides in Biosilicate® (wt.%) | Oxide Quantities Referred to 10 g of SiO2 (g) in Biosilicate® | Source Quantities Referred to 10 g of SiO2 (g)/Source | |
|---|---|---|---|
| Firing in N2 | |||
| SiO2 (48.5%) | 10 | 26.20/H44 (8.86 g as extra C-phase) | |
| P2O5 (4%) | 0.82 | 1.54/Na4P2O6 | |
| Na2O (23.75%) | 4.90 | 0.72 | |
| 4.18 | 7.14/Na2CO3 | ||
| CaO (23.75%) | 4.90 | 8.74/CaCO3 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dogrul, F.; Ożóg, P.; Michálek, M.; Elsayed, H.; Galusek, D.; Liverani, L.; Boccaccini, A.R.; Bernardo, E. Polymer-Derived Biosilicate®-like Glass-Ceramics: Engineering of Formulations and Additive Manufacturing of Three-Dimensional Scaffolds. Materials 2021, 14, 5170. https://doi.org/10.3390/ma14185170
Dogrul F, Ożóg P, Michálek M, Elsayed H, Galusek D, Liverani L, Boccaccini AR, Bernardo E. Polymer-Derived Biosilicate®-like Glass-Ceramics: Engineering of Formulations and Additive Manufacturing of Three-Dimensional Scaffolds. Materials. 2021; 14(18):5170. https://doi.org/10.3390/ma14185170
Chicago/Turabian StyleDogrul, Fulden, Paulina Ożóg, Martin Michálek, Hamada Elsayed, Dušan Galusek, Liliana Liverani, Aldo R. Boccaccini, and Enrico Bernardo. 2021. "Polymer-Derived Biosilicate®-like Glass-Ceramics: Engineering of Formulations and Additive Manufacturing of Three-Dimensional Scaffolds" Materials 14, no. 18: 5170. https://doi.org/10.3390/ma14185170
APA StyleDogrul, F., Ożóg, P., Michálek, M., Elsayed, H., Galusek, D., Liverani, L., Boccaccini, A. R., & Bernardo, E. (2021). Polymer-Derived Biosilicate®-like Glass-Ceramics: Engineering of Formulations and Additive Manufacturing of Three-Dimensional Scaffolds. Materials, 14(18), 5170. https://doi.org/10.3390/ma14185170