
Biomimetic Growth of Hydroxyapatite on SiO2 Microspheres to Improve Its Biocompatibility and Gentamicin Loading Capacity

 and
and 
Abstract
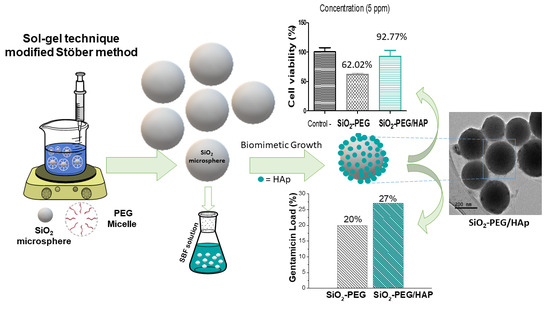
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
2.2.1. Synthesis of SiO2 Microspheres: Effect of Surfactant and Heat Treatment
2.2.2. Biomimetic Growth of Hydroxyapatite on SiO2
2.2.3. In Vitro Biocompatibility
Hemolysis Test
Cytotoxicity Using the Cell Line 3T3: MTT Test
2.2.4. Gentamicin Loading Capacity
2.2.5. Characterization
3. Results and Discussion
3.1. Synthesis of SiO2 Microspheres: Effect of Surfactant and Heat Treatment
3.2. Biomimetic Growth of Hydroxyapatite on SiO2
3.3. In Vitro Biocompatibility
3.3.1. Hemolysis Test
3.3.2. Cytotoxicity Using the Cell Line 3T3: MTT Test
3.4. Gentamicin Loading Capacity
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Farid, S.B.H. Bioceramics: For Materials Science and Engineering, 1st ed.; Elsevier Ltd.: Cambridge, MA, USA, 2019; pp. 97–117. [Google Scholar]
- Riggs, B.L.; Khosla, S.; Melton, L.J. Sex steroids and the construction and conservation of the adult skeleton. Endocr. Rev. 2002, 22, 279–302. [Google Scholar] [CrossRef] [PubMed]
- Bharadwaz, A.; Jayasuriya, A.C. Recent Trends in the application of widely used natural and synthetic polymer nanocomposites in bone tissue regeneration. Mater. Sci. Eng. C 2020, 110, 110698. [Google Scholar] [CrossRef] [PubMed]
- Rufino-Senra, M.; Barbosa de Lima, R.; Saboya-Souza, D.H.; Vieira-Marques, M.F.; Neves-Monteiro, S. Thermal characterization of hydroxyapatite or carbonated hydroxyapatite hybrid composites with distinguished collagens for a bone graft. J. Mater. Res. Technol. 2020, 9, 7190–7200. [Google Scholar] [CrossRef]
- Salimi, S. Functionally graded calcium phosphate bioceramics: An overview of preparation and properties. Ceram. Int. 2020, 46, 19664–19668. [Google Scholar] [CrossRef]
- Bystrova, A.V.; Dekhtyar, Y.D.; Popov, A.I.; Coutinho, J.; Bystrov, V.S. Modified Hydroxyapatite structure and properties: Modeling and synchrotron data analysis of modified hydroxyapatite structure. Ferroelectrics 2015, 475, 135–147. [Google Scholar] [CrossRef]
- Beck, G.R.; Ha, S.W.; Camalier, C.E.; Yamaguchi, M.; Li, Y.; Lee, J.K.; Weitzmann, M.N. Bioactive silica-based nanoparticles stimulate bone-forming osteoblasts, suppress bone-resorbing osteoclasts, and enhance bone mineral density in vivo. Nanomed. Nanotechnol. Biol. Med. 2012, 8, 793–803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santin, M.; Phillips, G. History of biomimetic, bioactive, and bioresponsive biomaterials. In Biomimetic, Bioresponsive, and Bioactive Materials: An Introduction to Integrating Materials with Tissues, 1st ed.; Santin, M., Phillips, G., Eds.; Wiley: San Francisco, CA, USA, 2012; pp. 1–30. [Google Scholar]
- Yilmaz, B.; Pazarceviren, A.E.; Tezcaner, H.; Evis, Z. Historical development of simulated body fluids used in biomedical applications: A review. Microchem. J. 2020, 155, 104713. [Google Scholar] [CrossRef]
- Vallés-Lluch, A.; Gallego-Ferrer, G.; Monleón-Pradas, M. Biomimetic apatite coating on P(EMA-co-HEA)/SiO2 hybrid nanocomposites. Polymers 2009, 50, 2874–2884. [Google Scholar] [CrossRef]
- Shi, S.; Goto, T.; Cho, S.H.; Sekino, T. Surface-morphology modification of ceramic-based composites for a photocatalytic activity via simple chemical and heat treatments. J. Ceram. Soc. Jpn 2000, 108, 118–121. [Google Scholar] [CrossRef] [Green Version]
- Catauro, M.; Bollino, F.; Papale, F.; Gallicchio, M.; Pacifico, S. Influence of the polymer amount on bioactivity and biocompatibility of SiO2/PEG hybrid materials synthesized by sol-gel technique. Mater. Sci. Eng. C 2015, 48, 548–555. [Google Scholar] [CrossRef]
- Moussa, M.; Mankoci, S.; Ustriyana, P.; Zhang, N.; Abdelmagid, S.; Molenda, J.; Murphy, W.L.; Safadi, F.F.; Sahai, N. Orthosilicic acid, Si(OH)4, stimulates osteoblast differentiation in vitro by upregulating miR-146a to antagonize NF-κB activation. Acta Biomater. 2016, 39, 192–202. [Google Scholar] [CrossRef]
- Mehra, R.R.; Tiwari, P.; Basu, A.; Duttkonar, A. Correction: In search of bioinspired hydrogels from amphiphilic peptides: A template for nanoparticle stabilization for the sustained release of anticancer drugs. New J. Chem. 2019, 43, 11666–11678. [Google Scholar] [CrossRef]
- Wu, C.; Chang, J.; Fan, W. Bioactive mesoporous calcium-silicate nanoparticles with excellent mineralization ability, osteostimulation, drug-delivery and antibacterial properties for filling. J. Mater. Chem. 2012, 22, 16801–16809. [Google Scholar] [CrossRef]
- Kurdyukov, D.A.; Eurov, D.A.; Kirilenko, D.A.; Sokolov, V.V.; Golubev, V.G. Tailoring the size and microporosity of Stöber silica particles. Microporous Mesoporous Mater. 2018, 258, 205–210. [Google Scholar] [CrossRef]
- Lei, Y.; Chen, X.; Song, H.; Hu, Z.; Cao, B. The influence of thermal treatment on the microstructure and thermal insulation performance of silica aerogels. J. Non. Cryst. Solids 2017, 470, 178–183. [Google Scholar] [CrossRef]
- Ríos, F.; Fernández-Arteaga, A.; Fernández-Serrano, M.; Jurado, E.; Lechuga, M. Silica micro- and nanoparticles reduce the toxicity of surfactant solutions. J. Hazard. Mater. 2018, 353, 436–443. [Google Scholar] [CrossRef]
- Ibarra-Alonso, M.C.; Reyna-Martínez, R.; Narro Céspedes, R.I.; Reyes Acosta, Y.K.; Martínez-Luevanos, A.; Zugasti-Cruz, A.; Neira-Velázquez, M.G.; Sánchez-Valdés, S.; Soria-Arguello, G. Effect of thermal and argon plasma treatment in SiO2 spheres, assessing the effectiveness in the elimination of organic waste. Rev. Mex. Ing. Química 2020, 19, 1071–1080. [Google Scholar] [CrossRef]
- Kokubo, T.; Takadama, H. How useful is SBF in predicting in vivo bone bioactivity. Biomaterials 2006, 27, 15. [Google Scholar] [CrossRef]
- ASTM F756-13. Standard Practice for Assessment of Hemolytic Properties of Materials. 2014. Available online: https://www.astm.org/Standards/F756.htm (accessed on 15 March 2021).
- 10993–5:2009. International Organization for Standardization. Biological Evaluation of Medical Devices—Part 5: Tests for In Vitro Cytotoxicity. 2009. Available online: https://www.iso.org/standard/36406.html (accessed on 15 March 2021).
- Travaglini, L.; Picchetti, P.; Del Giudice, A.; Galantini, L.; De Cola, L. Tuning and controlling the shape of mesoporous silica particles with CTAB/sodium deoxycholate catanionic mixtures. Microporous Mesoporous Mater. 2019, 279, 423–431. [Google Scholar] [CrossRef]
- Wiercigroch-Walkosz, K.; Cichos, J.; Karbowiak, M. Growth of silica shell on hydrophobic upconverting nanocrystals—Mechanism and control of porosity. Colloids Surf. A Physicochem. Eng. Asp. 2019, 572, 1–9. [Google Scholar] [CrossRef]
- Raju, M.; van Duin, A.C.T.; Fichthorn, K. Mechanisms of Oriented Attachment of TiO2 Nanocrystals in Vacuum and Humid Environments: Reactive Molecular Dynamics. Nano Lett. 2014, 14, 1836–1842. [Google Scholar] [CrossRef]
- Gholizadeh, R.; Wang, Y. Molecular dynamics simulation of the aggregation phenomenon in the late stages of silica materials preparation. Chem. Eng. Sci. 2018, 184, 62–71. [Google Scholar] [CrossRef]
- Ching-Hung, L.; Yi-Ming, S. Influence of the surface properties of nano-silica on the dispersion and isothermal crystallization kinetics of PHB/silica nanocomposites. Mater. Chem. Phys. 2017, 199, 88–97. [Google Scholar]
- Liu, Y.; Tourbin, M.; Lachaize, S.; Guiraud, P. Silica nanoparticles separation from water: Aggregation by cetyltrimethylammonium bromide (CTAB). Chemosphere 2013, 92, 681–687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo-Yong, H.; Sheng-Ming, X.; Lin-Yan, L.; Xue-Jun, W. Effect of surfactants on dispersion property and morphology of nano-sized nickel powders. Trans. Nonferrous Met. Soc. China 2014, 24, 3739–3746. [Google Scholar] [CrossRef]
- He, S.; Huang, Y.; Chen, G.; Feng, M.; Dai, H.; Yuan, B.; Chen, X. Effect of heat treatment on hydrophobic silica aerogel. J. Am. Chem. Soc. 2010, 132, 4834–4842. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Haynes, C.L. Impacts of Mesoporous Silica Nanoparticle Size, Pore Ordering, and Pore Integrity on Hemolytic Activity. J. Am. Chem. Soc. 2010, 132, 4834–4842. [Google Scholar] [CrossRef]
- Yu, H.; Zhang, H.; Wang, X.; Gu, Z.; Li, X.; Deng, F. Local structure of hydroxy—Peroxy apatite: A combined XRD, FT-IR, Raman, SEM, and solid-state NMR study. J. Phys. Chem. Solids 2007, 68, 1863–1871. [Google Scholar] [CrossRef]
- Londoño, M.E.; Echavarría, A.; La Calle, F.D. Características cristaloquímicas de la hidrixiapatita sintética tratada a diferentes temperaturas. Esc. Ing. Antioq. 2006, 5, 109–118. [Google Scholar]
- Siriphannon, P.; Kameshima, Y.; Yasumori, A.; Okada, K.; Hayashi, S. Formation of hydroxyapatite on CaSiO3 powders in simulated body fluid. J. Eur. Ceram. Soc. 2002, 22, 511–520. [Google Scholar] [CrossRef]
- Angelopoulou, A.; Efthimiadou, E.K.; Kordas, G.; Angelopoulou, E.K. Efthimiadou. A new approach to fabricate bioactive silica binary and ternary hybrid microspheres. Mater. Sci. Eng. C 2015, 53, 76–82. [Google Scholar] [CrossRef]
- Li, B.; Luo, W.; Wang, Y.; Wu, H.; Zhang, C. Bioactive SiO2-CaO-P2O5 hollow nanospheres for drug delivery. J. Non. Cryst. Solids 2016, 447, 98–103. [Google Scholar] [CrossRef]
- Fisichella, M.; Dabboue, H.; Bhattacharyya, S.; Saboungi, M.L.; Salvetat, J.P.; Hevor, T.; Guerin, M. Mesoporous silica nanoparticles enhance MTT formazan exocytosis in HeLa cells and astrocytes. Toxicol. Vitr. 2009, 23, 697–703. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.H.K.; Simon, C.G., Jr. Fast setting calcium phosphate-chitosan scaffold: Mechanical properties and biocompatibility. Biomaterials 2005, 26, 1337–1348. [Google Scholar] [CrossRef]
- Tamanna, T.; Bulitta, J.B.; Yu, A. Controlling antibiotic release from mesoporous silica nano-drug carriers via self-assembled polyelectrolyte coating. J. Mater. Sci. Mater. Med. 2015, 26, 117. [Google Scholar] [CrossRef] [PubMed]
- Ji, J.; Hao, S.; Wu, D.; Huang, R.; Xu, Y. Preparation, characterization and in vitro release of chitosan nanoparticles loaded with gentamicin and salicylic acid. Carbohydr. Polym. 2011, 85, 803–808. [Google Scholar] [CrossRef]
- Cebrián, V.; Yagüe, C.; Arruebo, M.; Martín-Saavedra, F.M.; Santamaría, J.; Vilaboa, N. On the role of the colloidal stability of mesoporous silica nanoparticles as gene delivery vectors. J. Nanopart. Res. 2011, 13, 4097–4108. [Google Scholar] [CrossRef]
- Varache, M.; Bezverkhyy, I.; Bouyer, F.; Chassagnon, R.; Baras, F.; Bouyer, F. Improving structural stability of water-dispersed MCM-41 silica nanoparticles through post-synthesis pH aging process. J. Nanopart. Res. 2015, 17, 1–13. [Google Scholar] [CrossRef]
- Burleigh, M.C.; Markowitz, M.A.; Jayasundera, S.; Spector, M.S.; Thomas, C.W.; Gaber, B. Mechanical and hydrothermal stabilities of aged periodic mesoporous organosilicas. J. Phys. Chem. B 2003, 107, 12628–12634. [Google Scholar] [CrossRef]
- Broyer, M.; Valange, S.; Bellat, J.P.; Bertrand, O.; Weber, G.; Gabelica, Z. Influence of aging, thermal, hydrothermal, and mechanical treatments on the porosity of MCM-41 mesoporous silica. Langmuir 2002, 18, 5083–5091. [Google Scholar] [CrossRef]
- Adeniran, B.; Mokaya, R. On the Shelf Life and Aging Stability of Mesoporous Silica: Insights on Thermodynamically Stable MCM-41 Structure from Assessment of 12-Year-Old Samples. Chem. Mater. 2012, 24, 4450–4458. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Band (cm−1) | Assignation |
|---|---|
| 3000–3500 | ν O–H |
| 2937, 2985, 1446 and 1392 | CH3 and CH2 groups |
| 1049 | ν Si–O–Si |
| 962 and 567 | ν Si–OH |
| 803 | ν O–Si–O |
| 465 | δ Si–O |
| Parameter | SiO2-PEG | SiO2-CTAB | SiO2-SDS |
|---|---|---|---|
| Specific surface area (m2/g) | 24.00 | 5.20 | 10.90 |
| Mean pore size (nm) | 7.07 | 8.09 | 8.34 |
| Pore volume (cm3/g) | 0.04 | 0.02 | 0.01 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Herrera-Alonso, A.E.; Ibarra-Alonso, M.C.; Esparza-González, S.C.; Estrada-Flores, S.; García-Cerda, L.A.; Martínez-Luévanos, A. Biomimetic Growth of Hydroxyapatite on SiO2 Microspheres to Improve Its Biocompatibility and Gentamicin Loading Capacity. Materials 2021, 14, 6941. https://doi.org/10.3390/ma14226941
Herrera-Alonso AE, Ibarra-Alonso MC, Esparza-González SC, Estrada-Flores S, García-Cerda LA, Martínez-Luévanos A. Biomimetic Growth of Hydroxyapatite on SiO2 Microspheres to Improve Its Biocompatibility and Gentamicin Loading Capacity. Materials. 2021; 14(22):6941. https://doi.org/10.3390/ma14226941
Chicago/Turabian StyleHerrera-Alonso, Alejandra E., María C. Ibarra-Alonso, Sandra C. Esparza-González, Sofía Estrada-Flores, Luis A. García-Cerda, and Antonia Martínez-Luévanos. 2021. "Biomimetic Growth of Hydroxyapatite on SiO2 Microspheres to Improve Its Biocompatibility and Gentamicin Loading Capacity" Materials 14, no. 22: 6941. https://doi.org/10.3390/ma14226941
APA StyleHerrera-Alonso, A. E., Ibarra-Alonso, M. C., Esparza-González, S. C., Estrada-Flores, S., García-Cerda, L. A., & Martínez-Luévanos, A. (2021). Biomimetic Growth of Hydroxyapatite on SiO2 Microspheres to Improve Its Biocompatibility and Gentamicin Loading Capacity. Materials, 14(22), 6941. https://doi.org/10.3390/ma14226941