
Modeling of Nonlinear Optical Phenomena in Host-Guest Systems Using Bond Fluctuation Monte Carlo Model: A Review

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
1.1. Outline
1.2. Monte Carlo Simulations of Polymer Systems
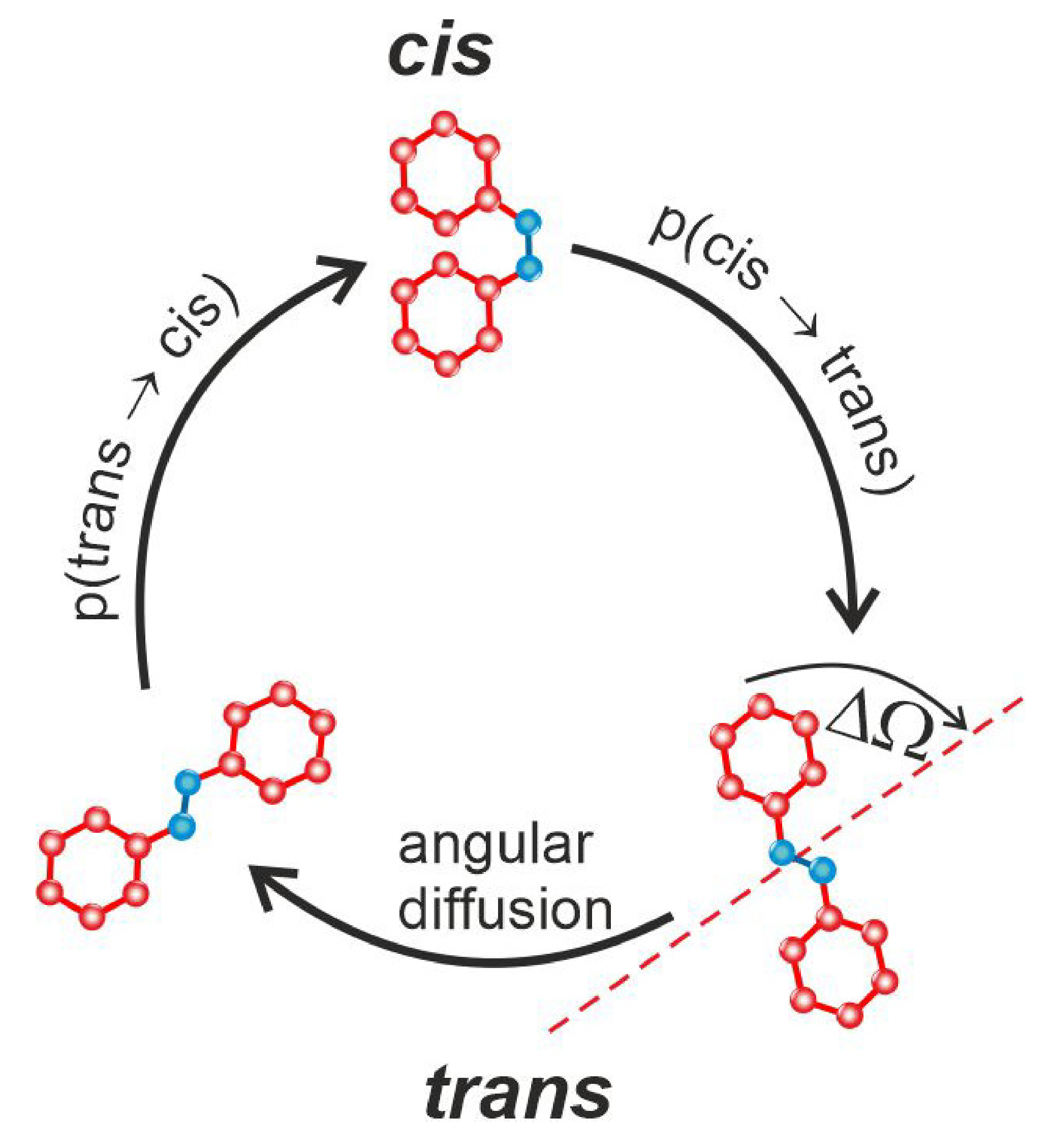
1.3. Photoinduced Mass Transport in Functionalized Azo-Polymers: Concepts
1.4. Scope of the Review
2. Materials and Methods
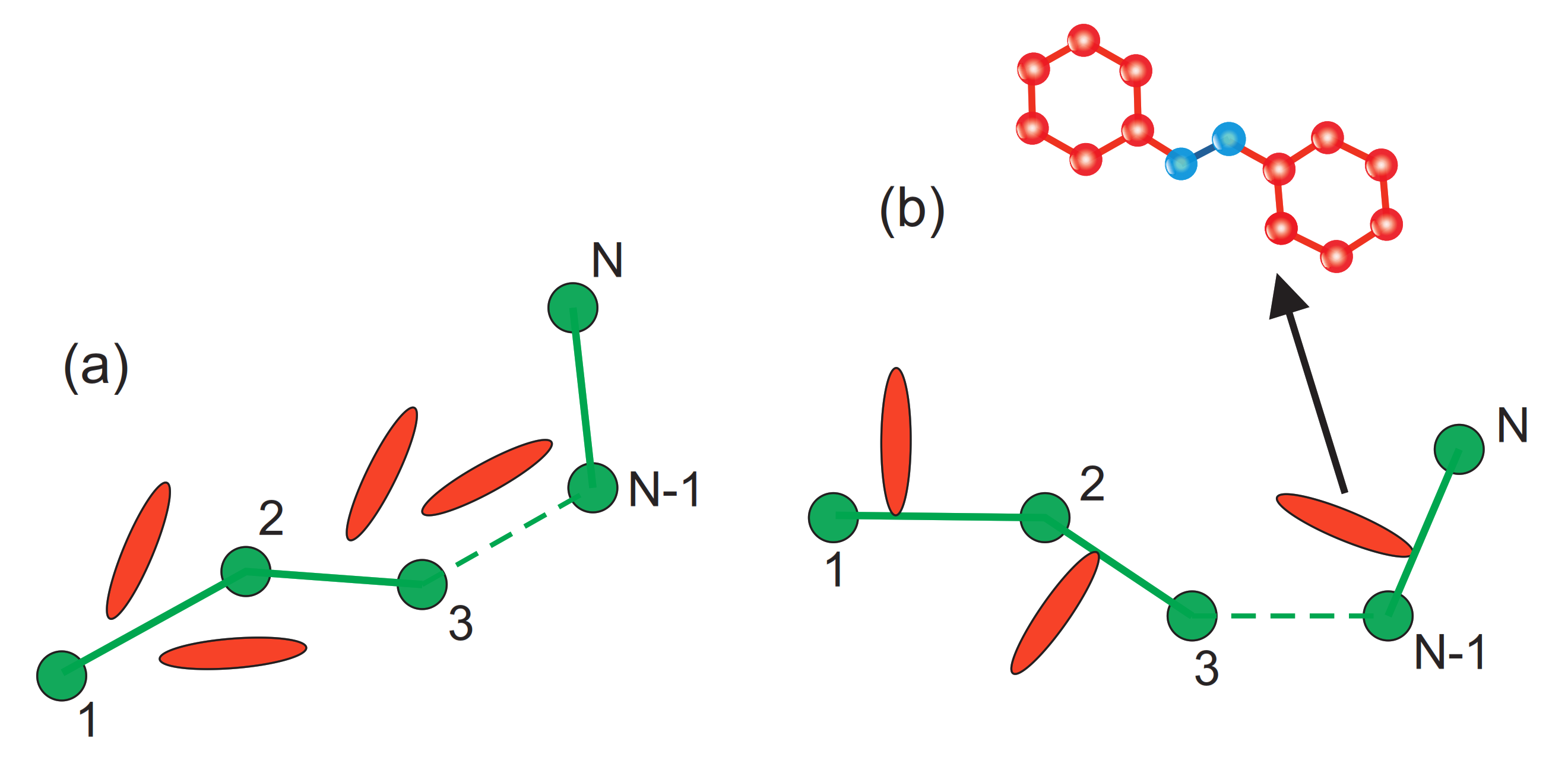
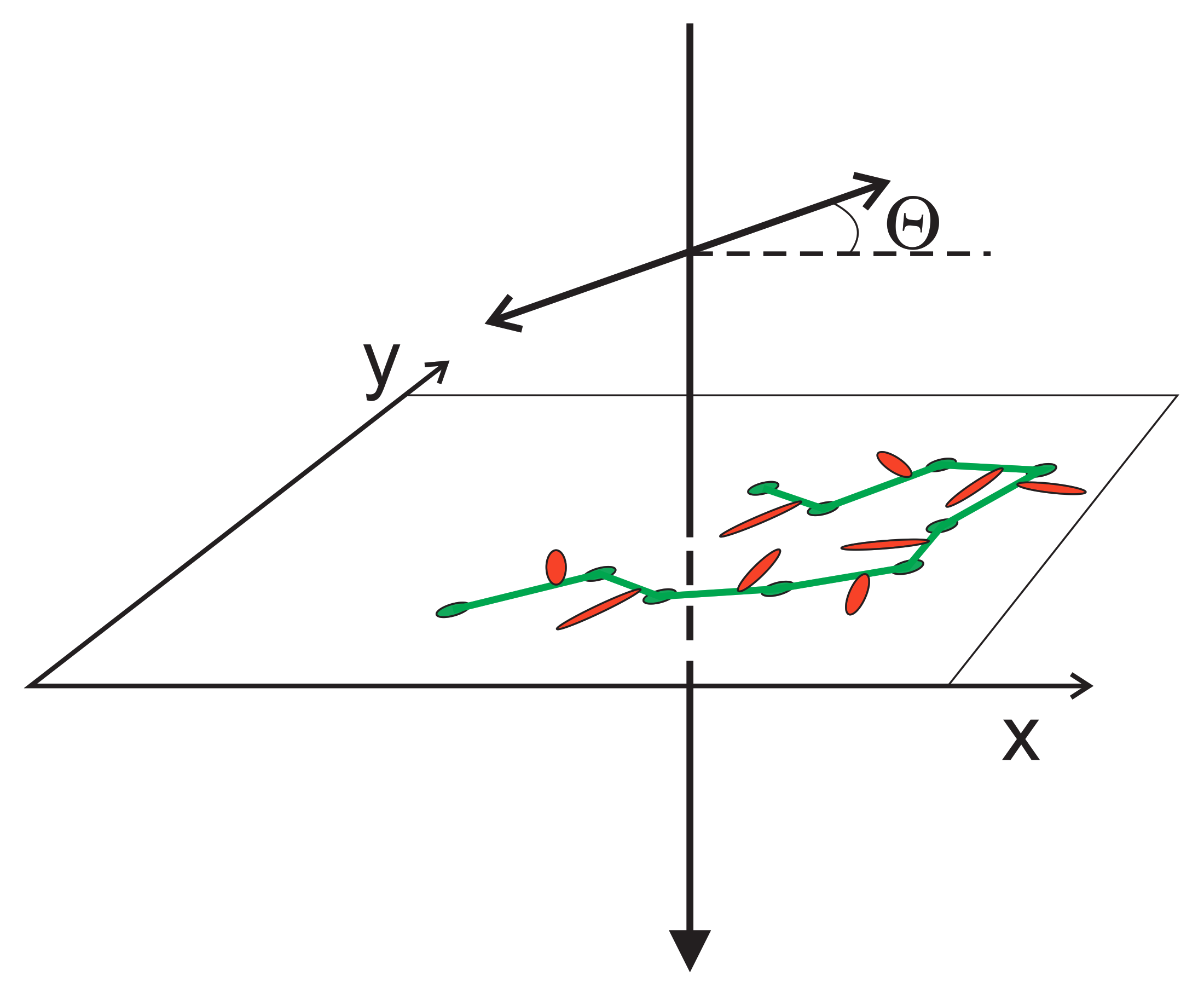
2.1. Theory: “Microscopic” Orientation Potential
2.1.1. Linearly Polarized Light
2.1.2. Elliptically Polarized Light
2.1.3. Photo-Induced Mechanical Stress
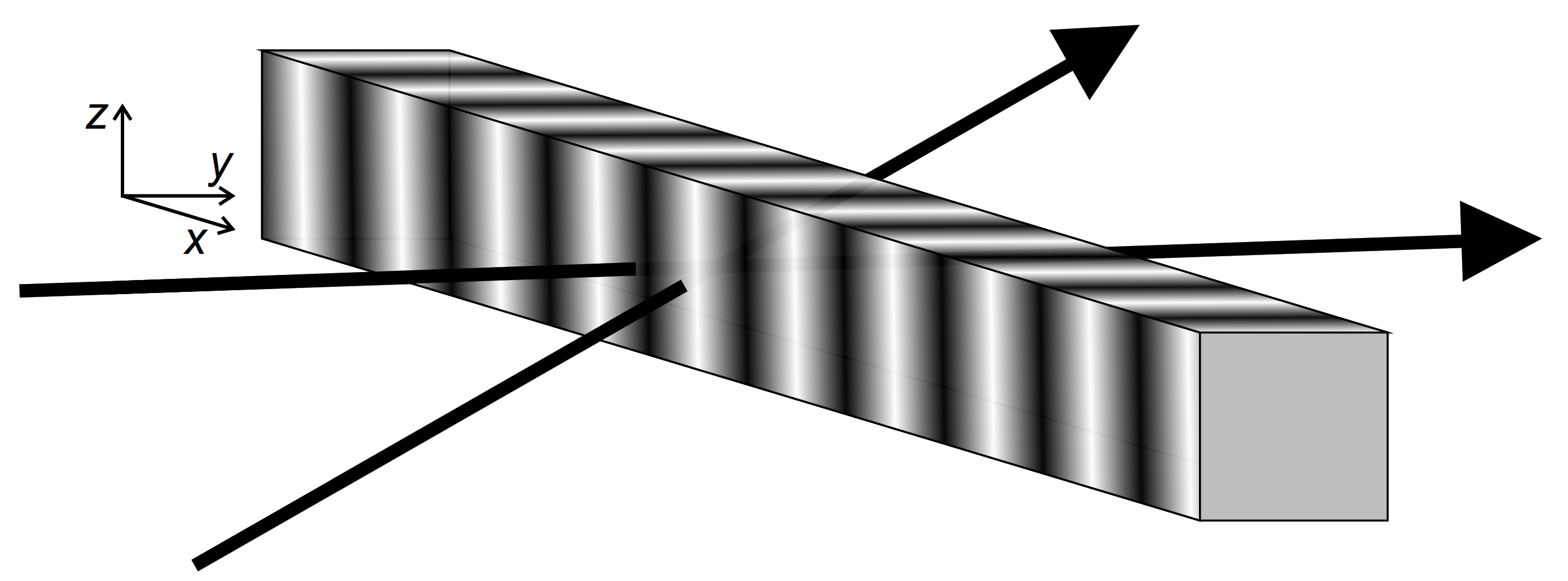
2.2. Degenerate Two Wave Mixing Experiment
2.3. Monte Carlo Simulations of Host-Guest Systems in BFM Approach
2.3.1. Metropolis Monte Carlo Method
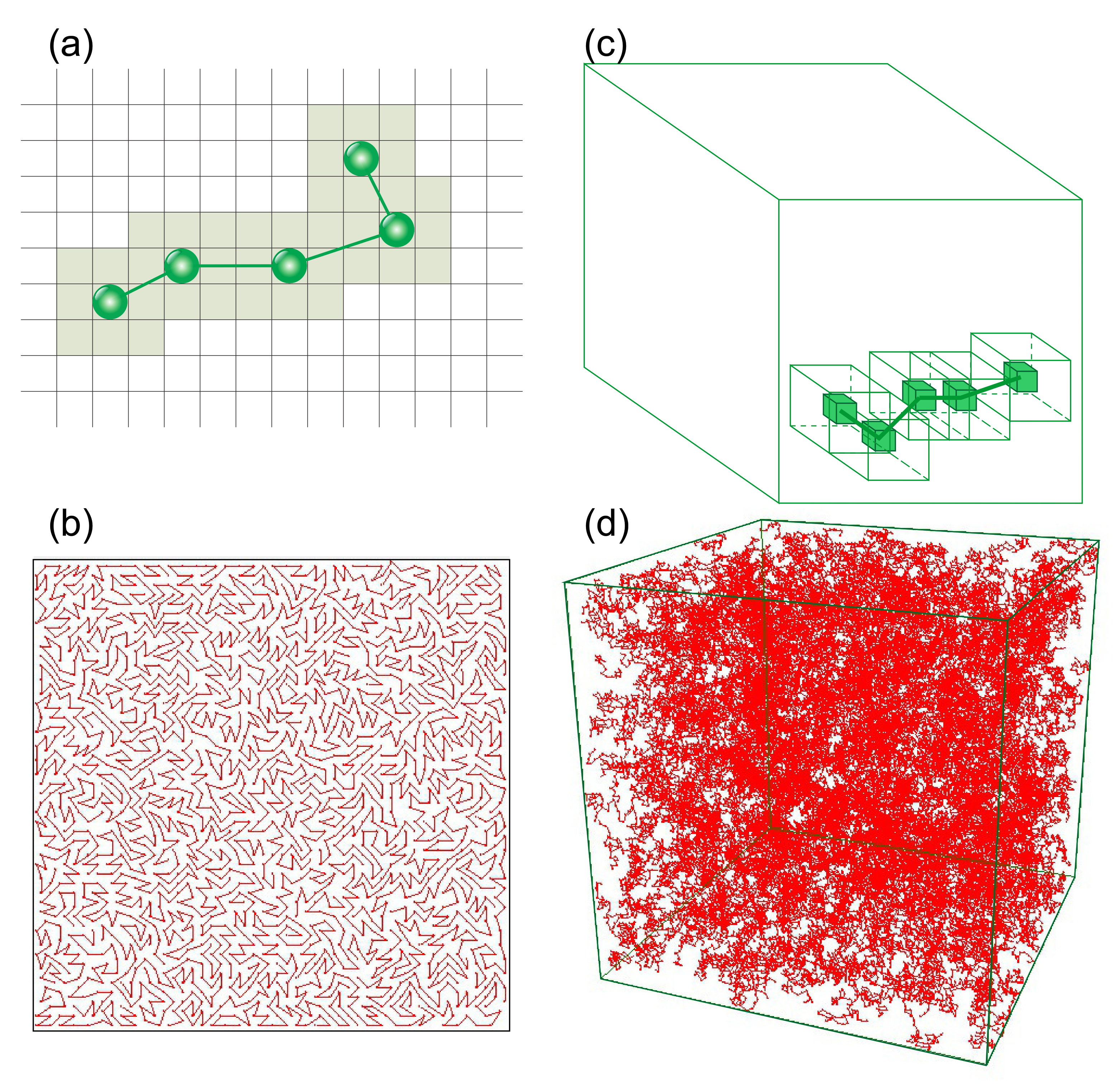
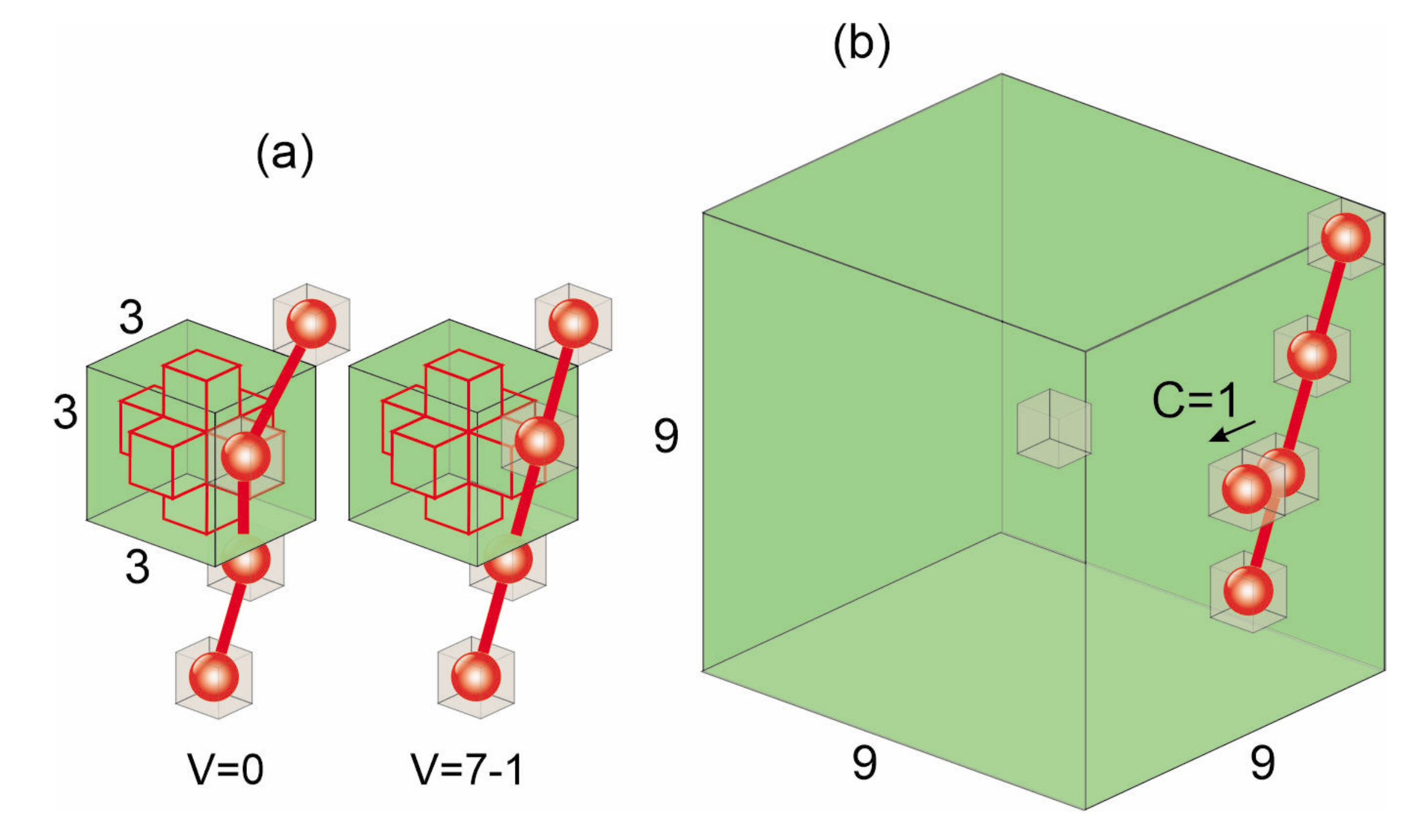
2.3.2. BFM Model in 2D and 3D
2.3.3. Local Characterization of the BFM Matrix
2.3.4. Light-Matter Interaction for Host-Guest Systems in BFM Approach Polarization Effects
2.3.5. Details of MC Simulations
2.4. Characterization of Physical Effects: Parameters
2.4.1. Diffraction Efficiency
2.4.2. SHG Efficiency
2.4.3. Displacement Complex Dynamics
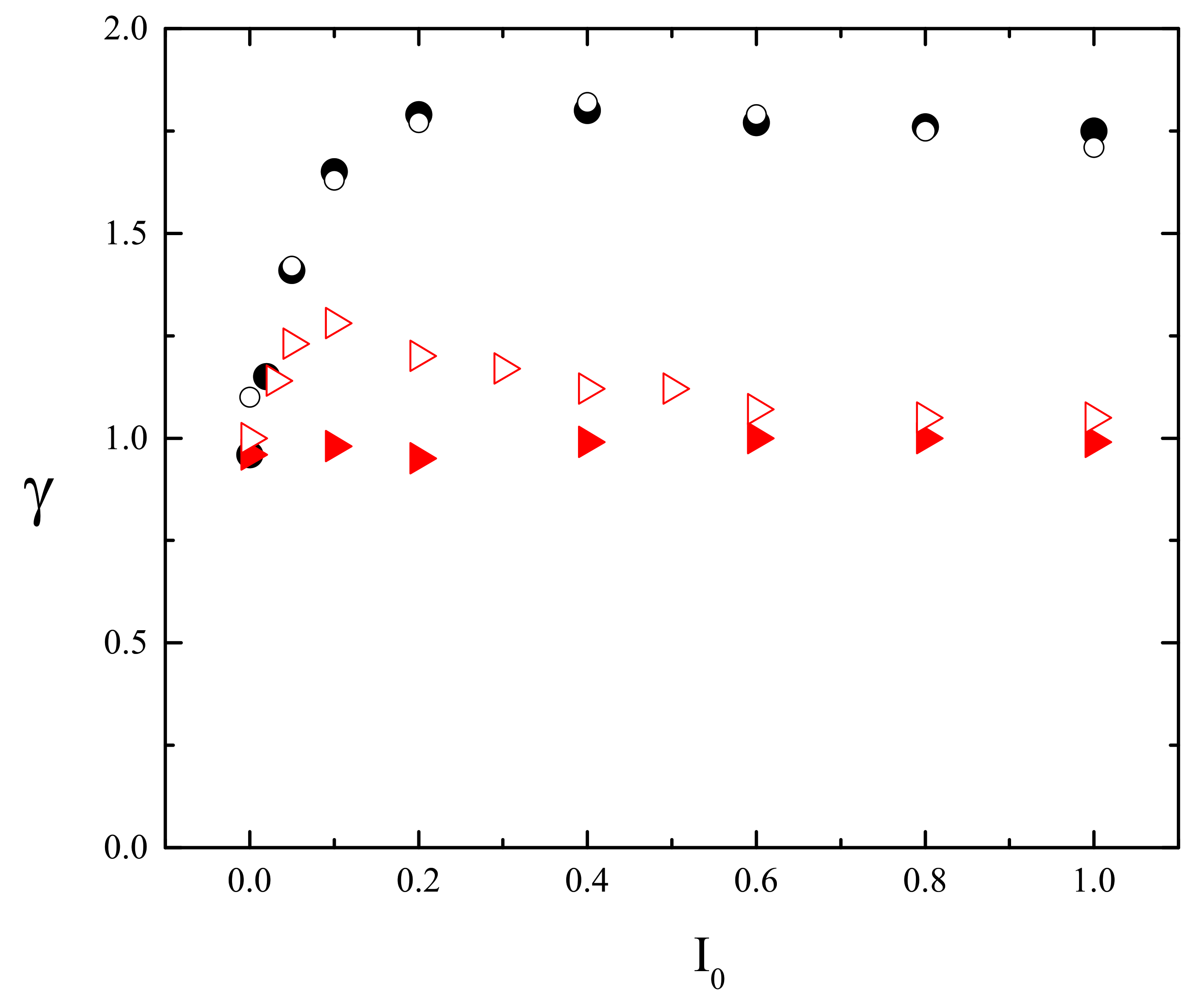
3. Results
3.1. Polymer Chain Motion in Orientation Approach
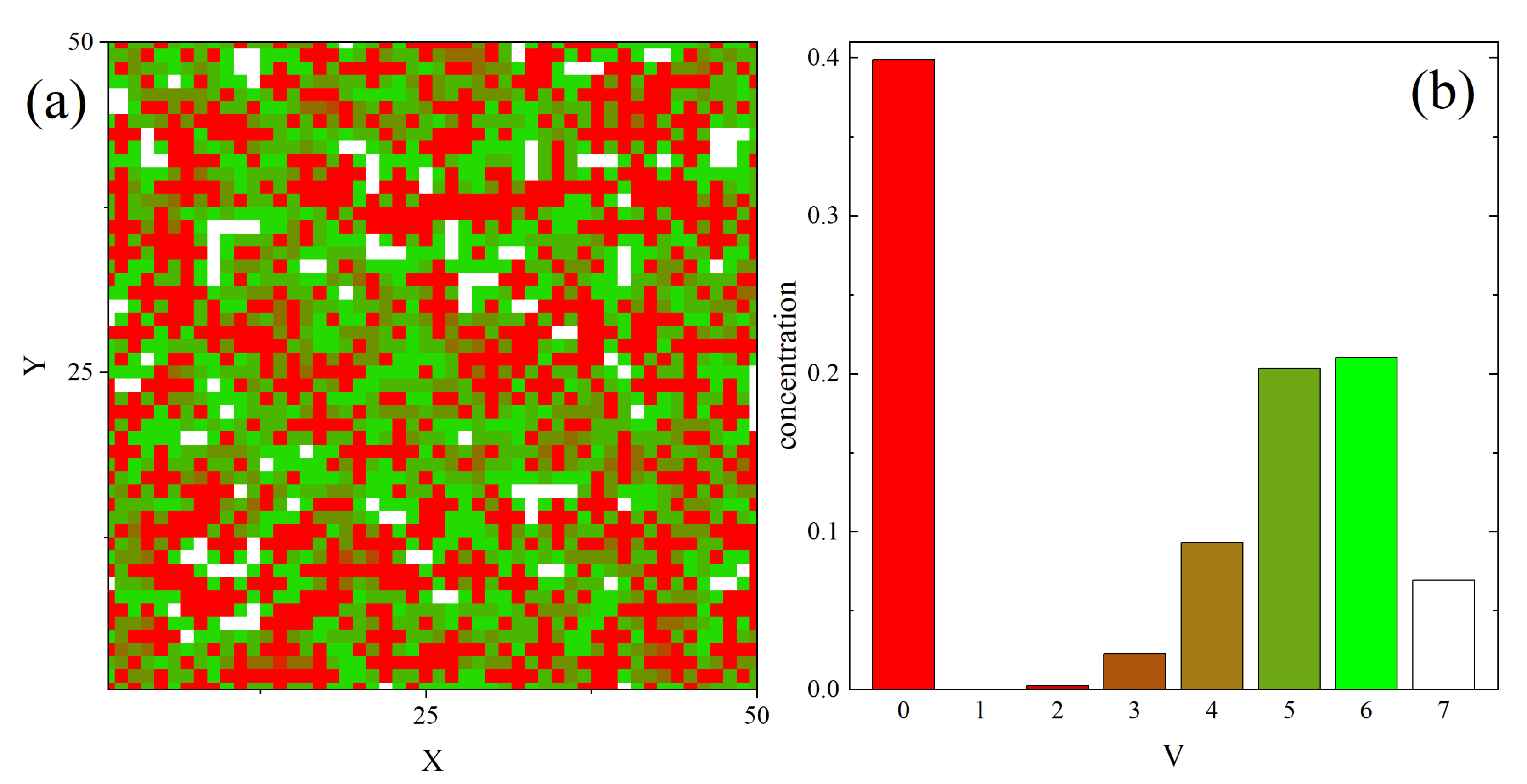
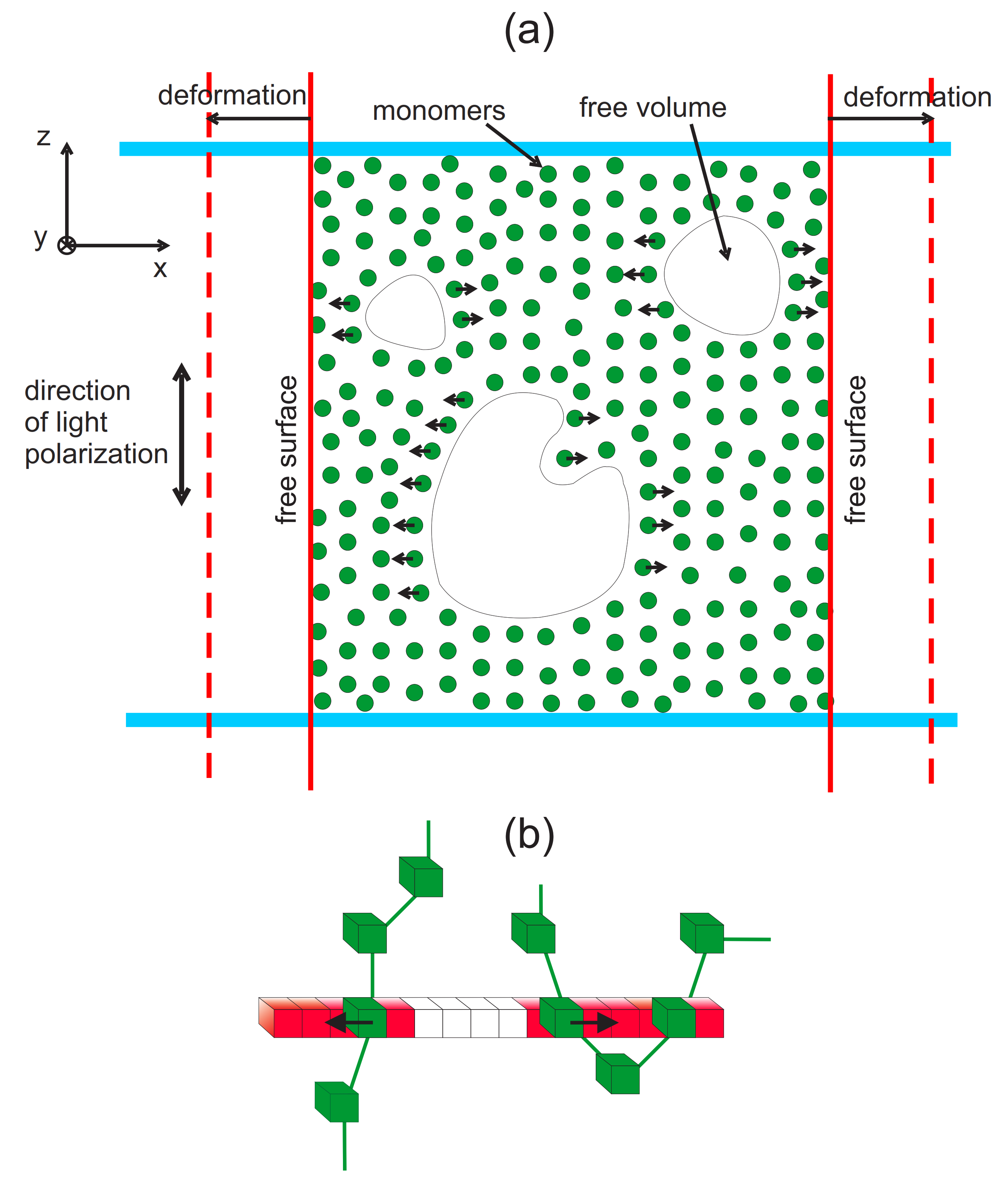
3.2. Complex Structure of Local Free Volume in Polymer Matrix
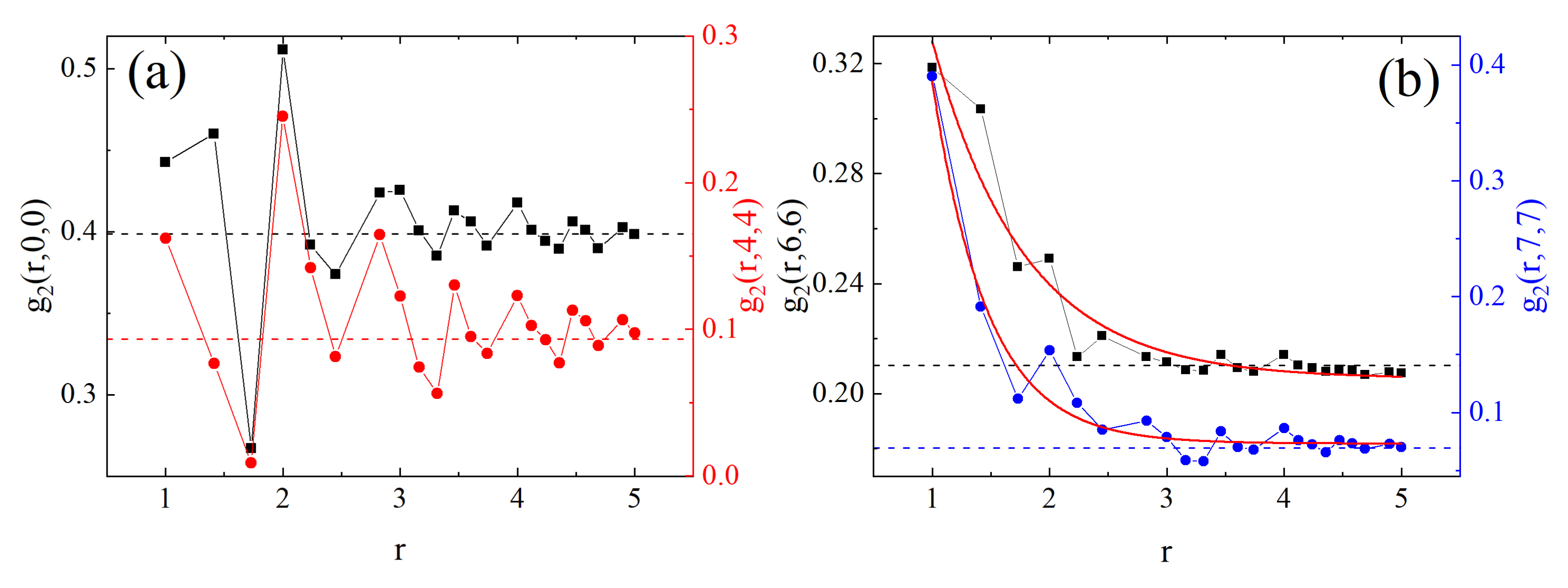
3.2.1. Mosaic-Like States: Maps of and Correlations
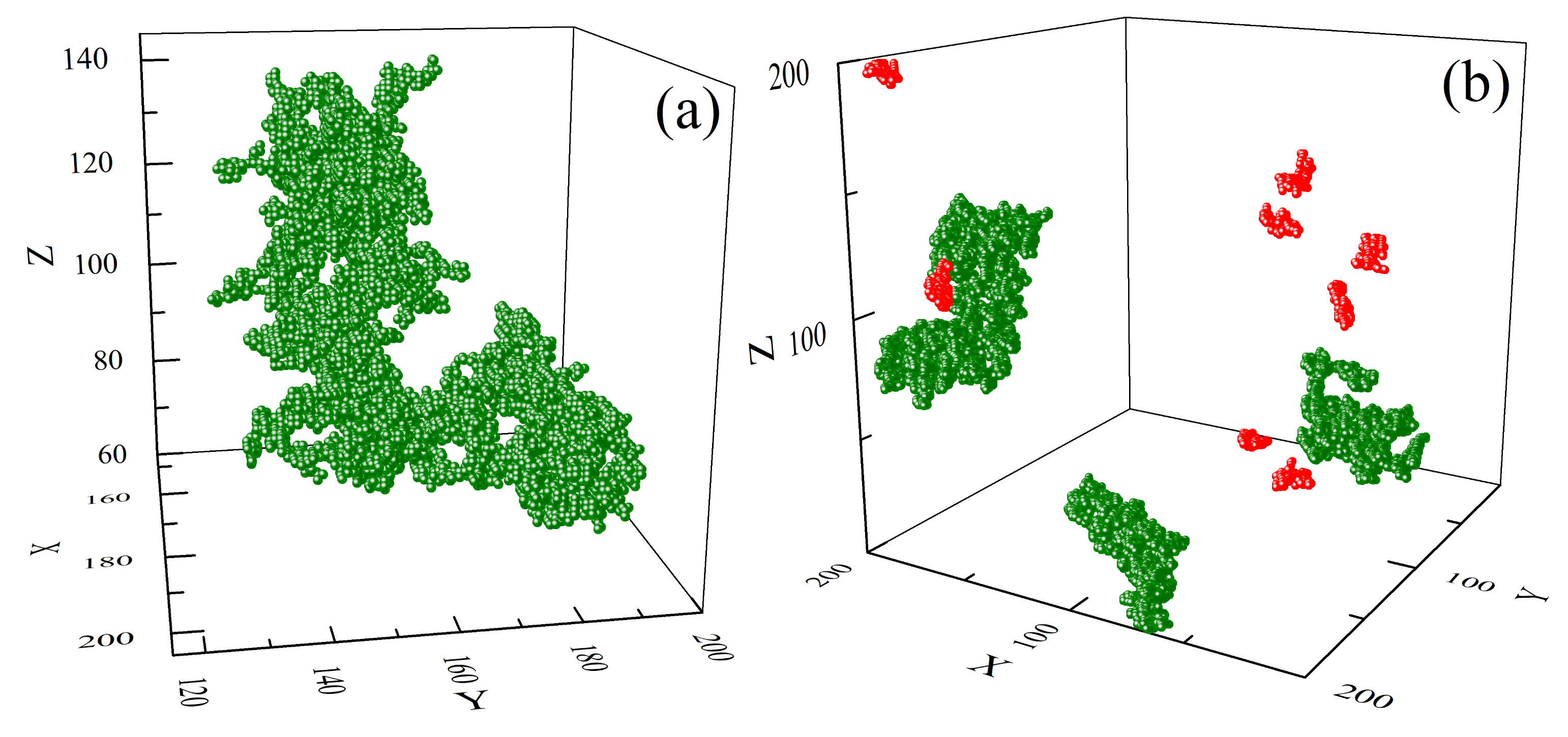
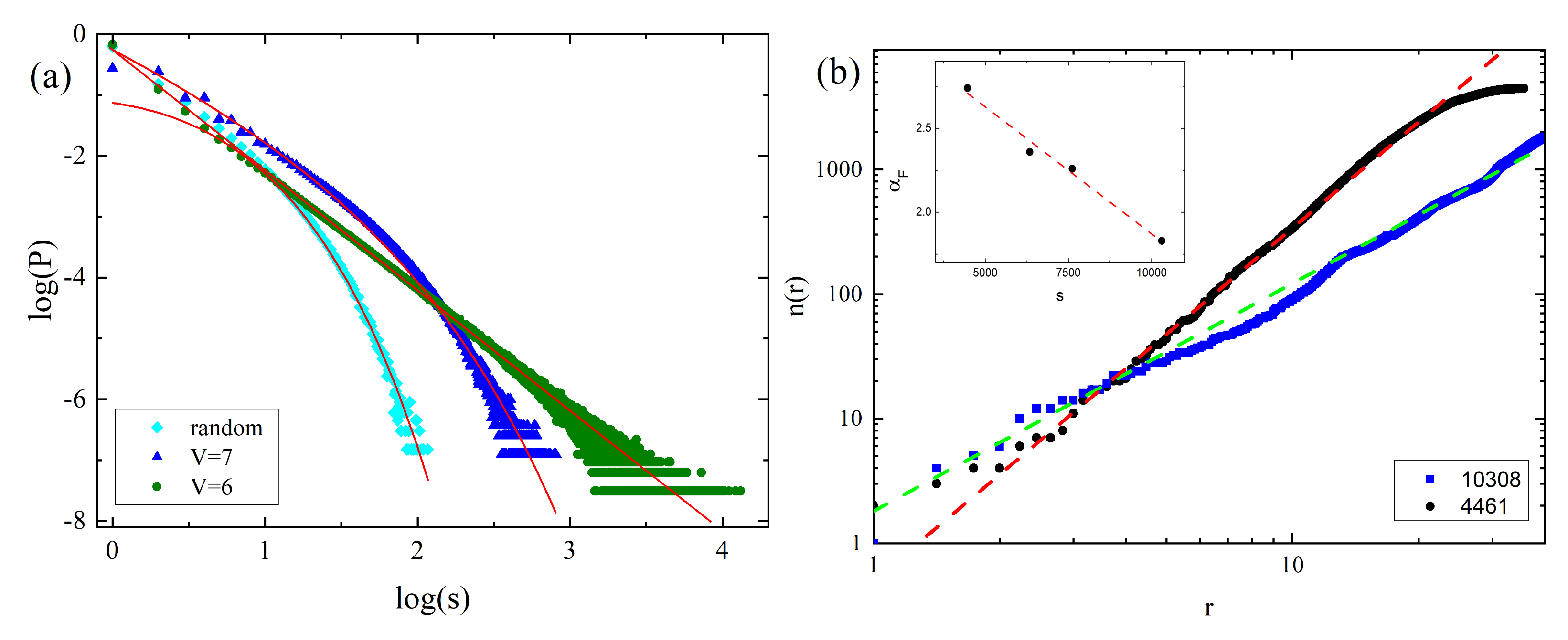
3.2.2. Clusters: Configurations, Distribution, Fractal Features
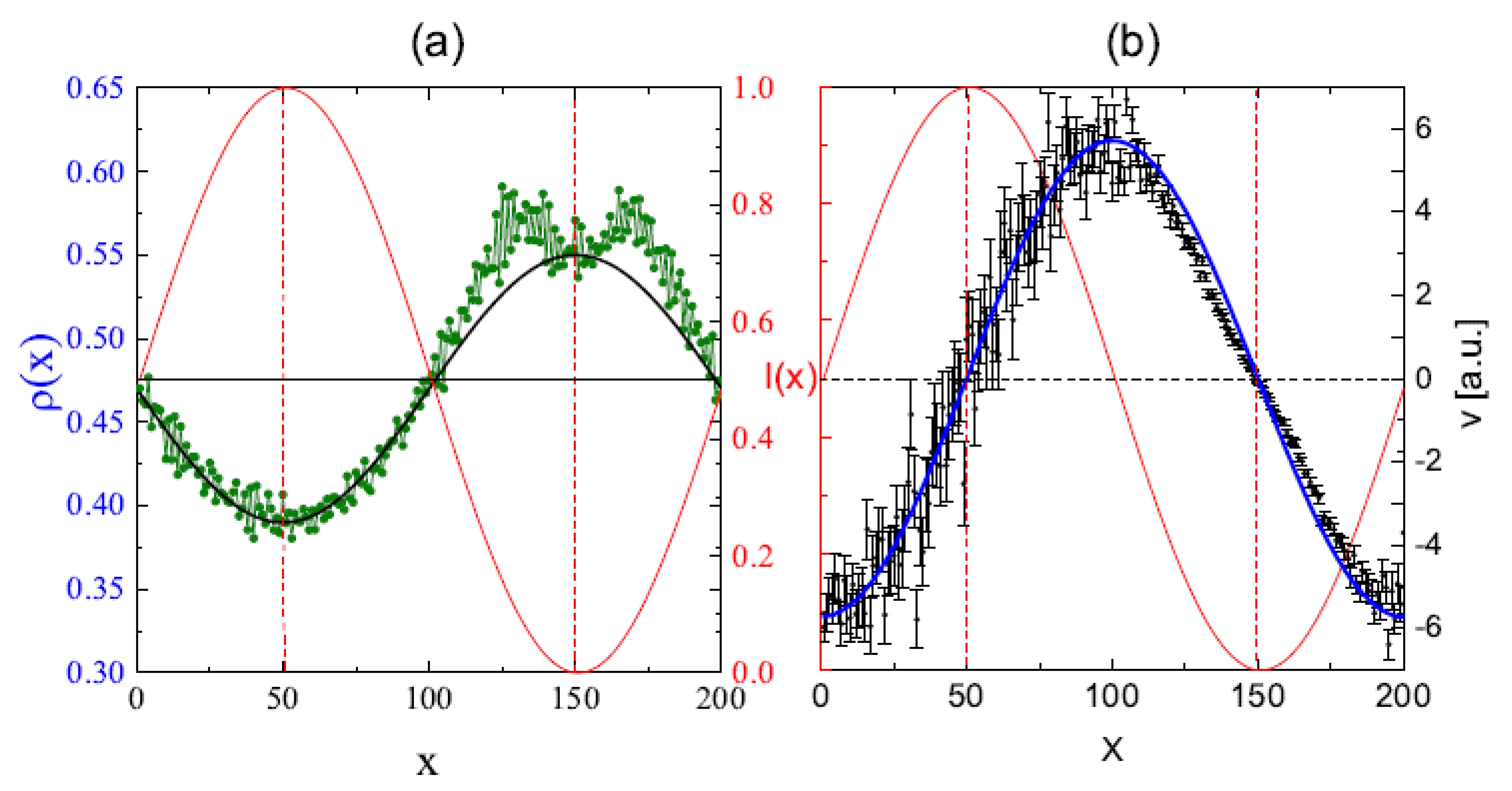
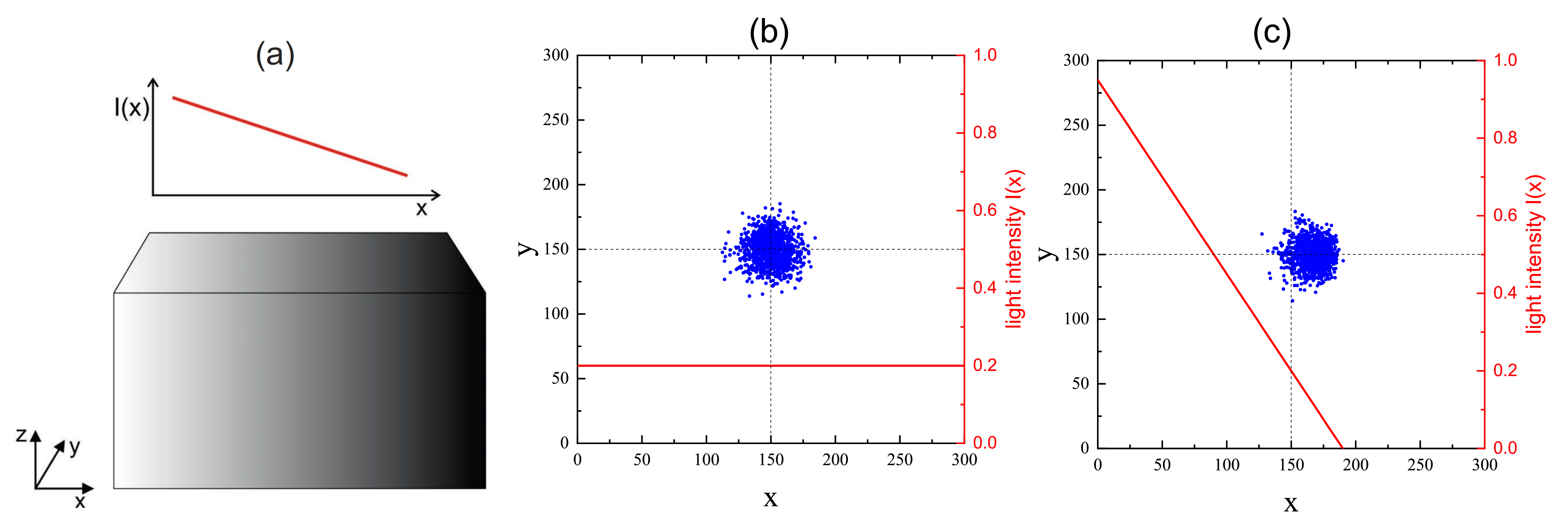
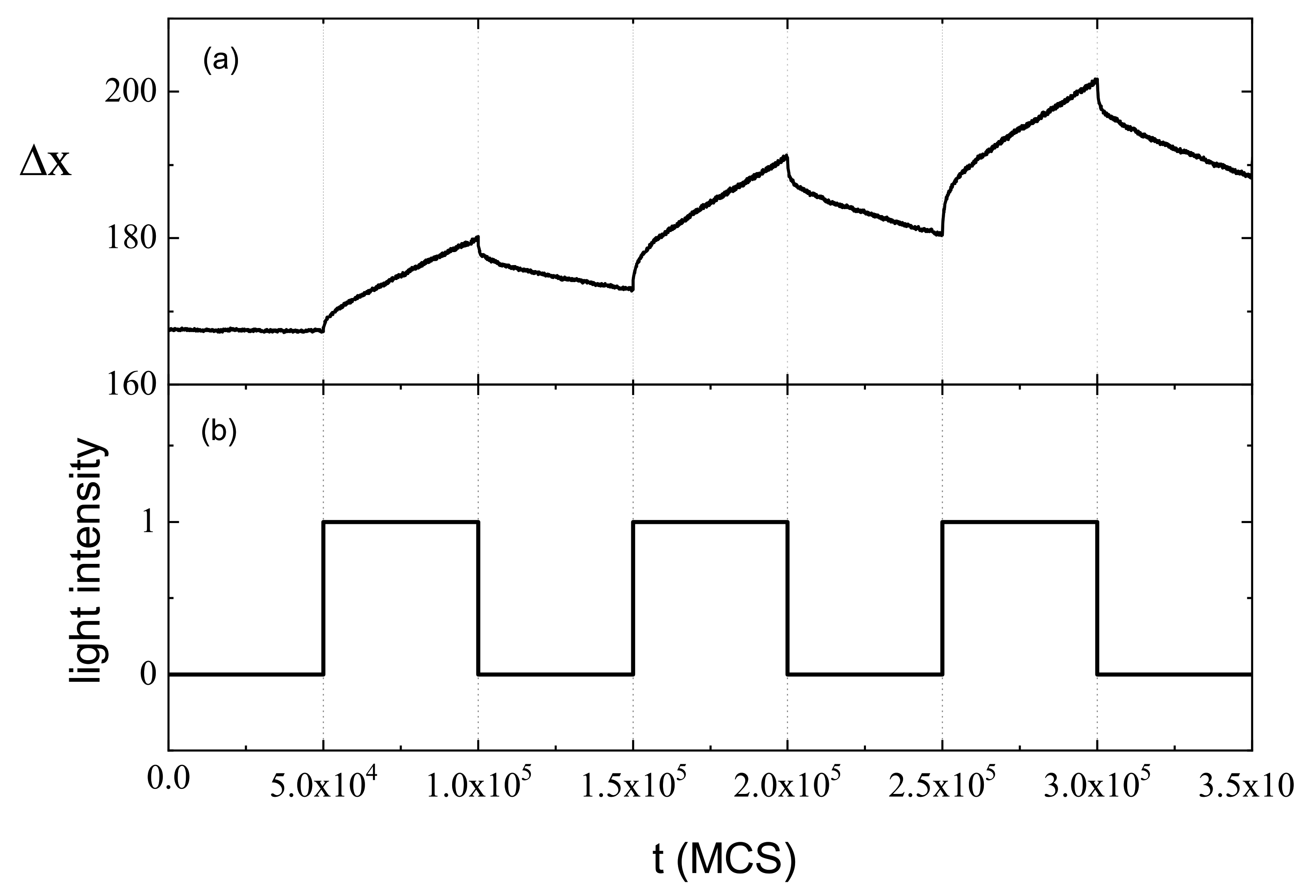
3.3. Inscription of Surface Relief Grating
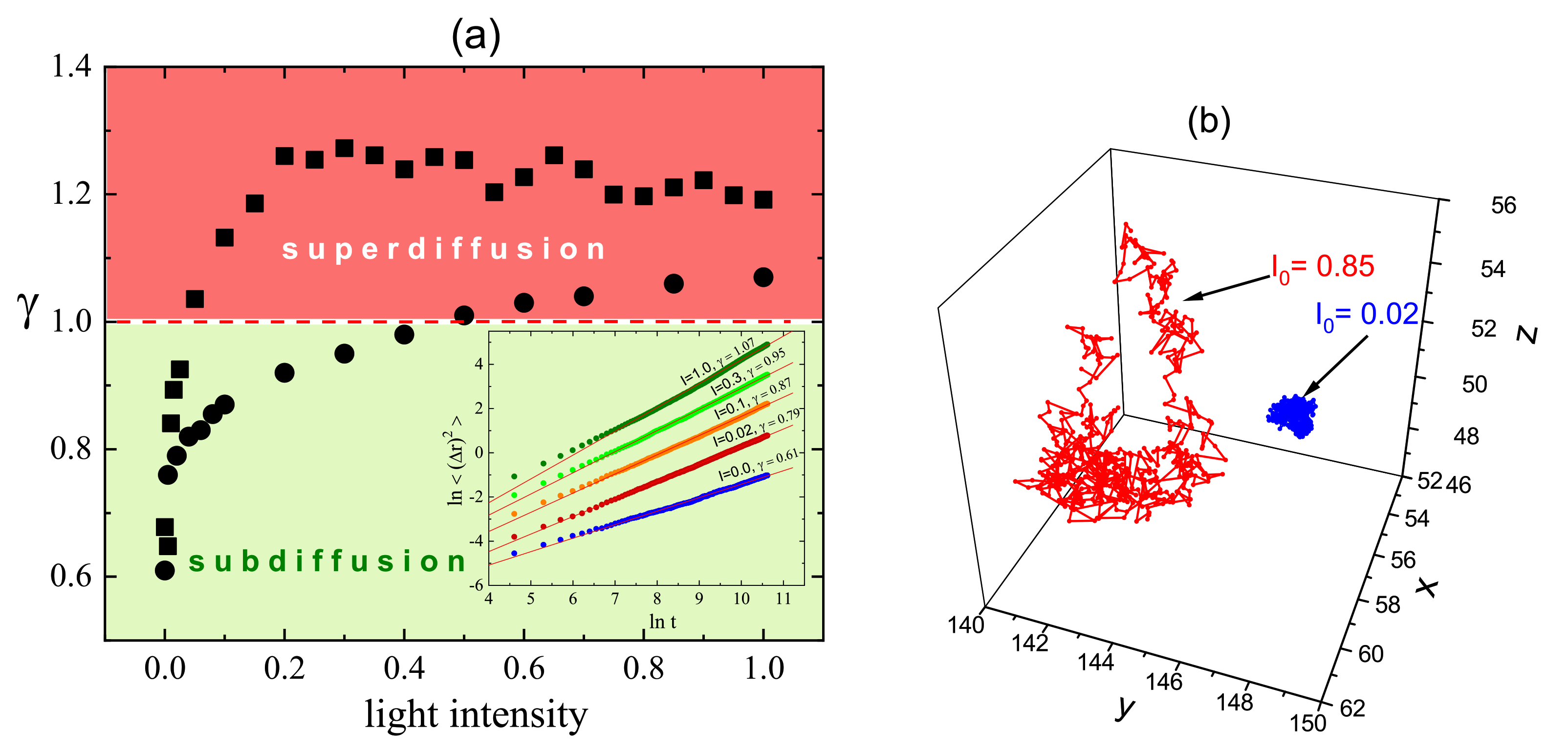
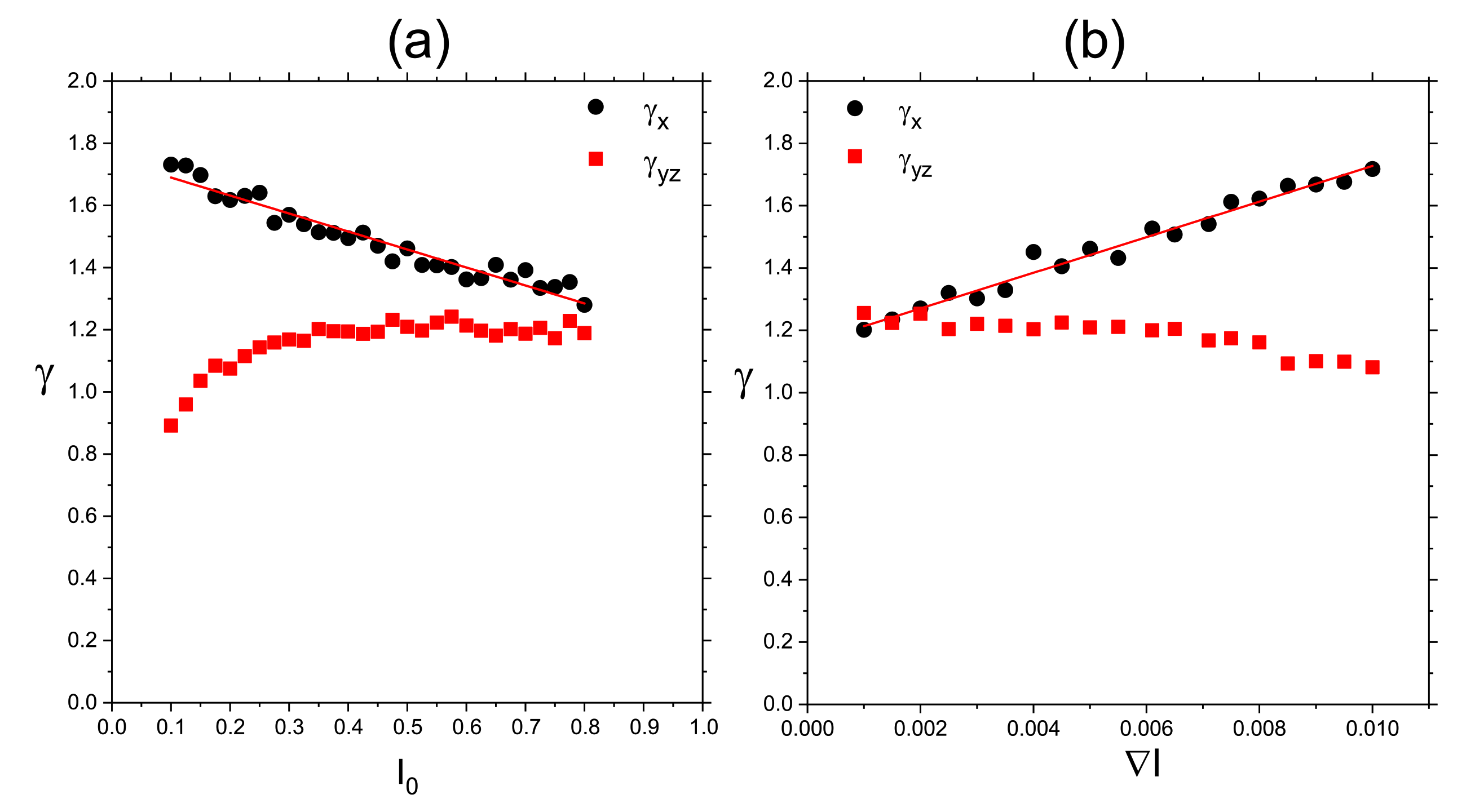
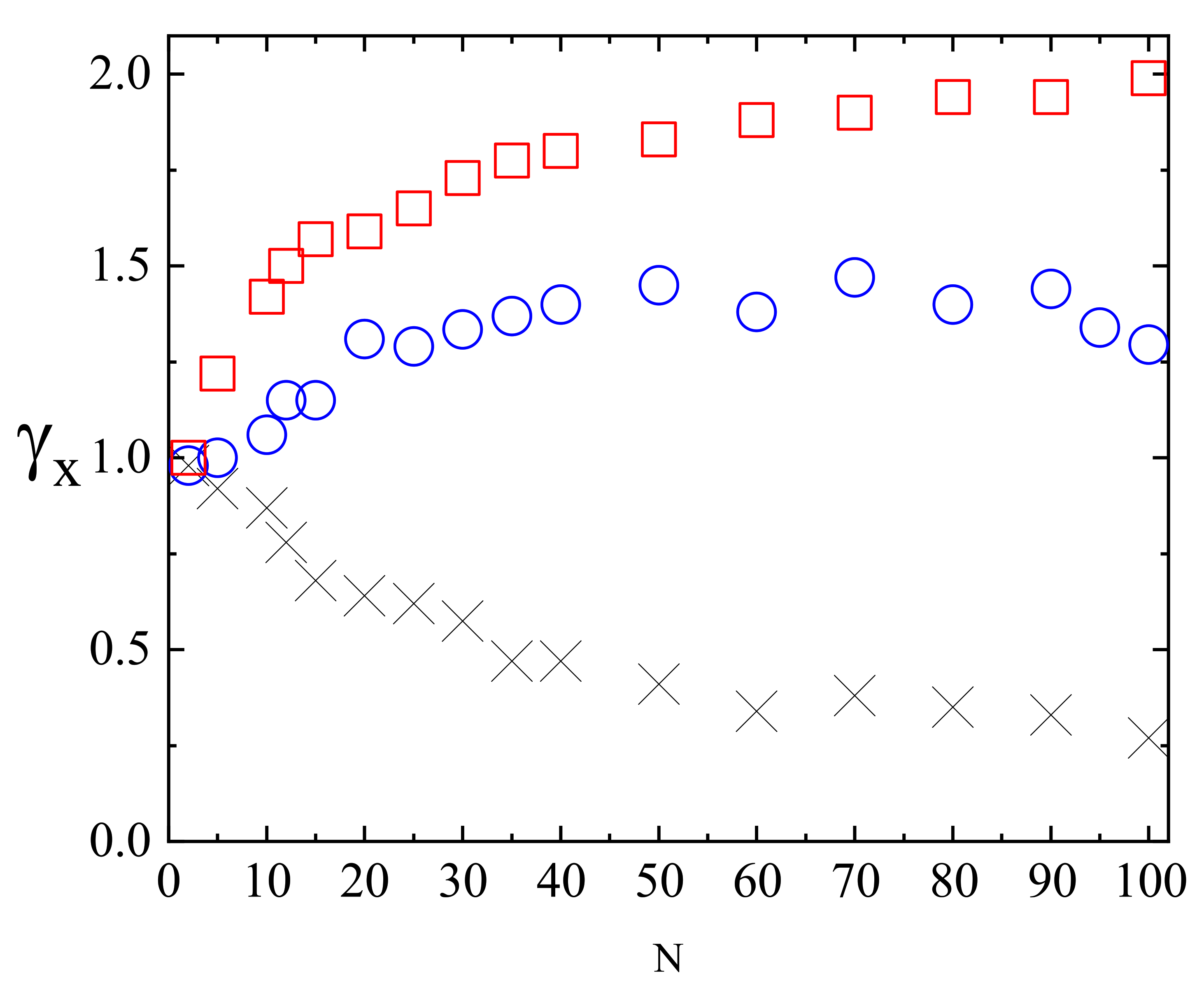
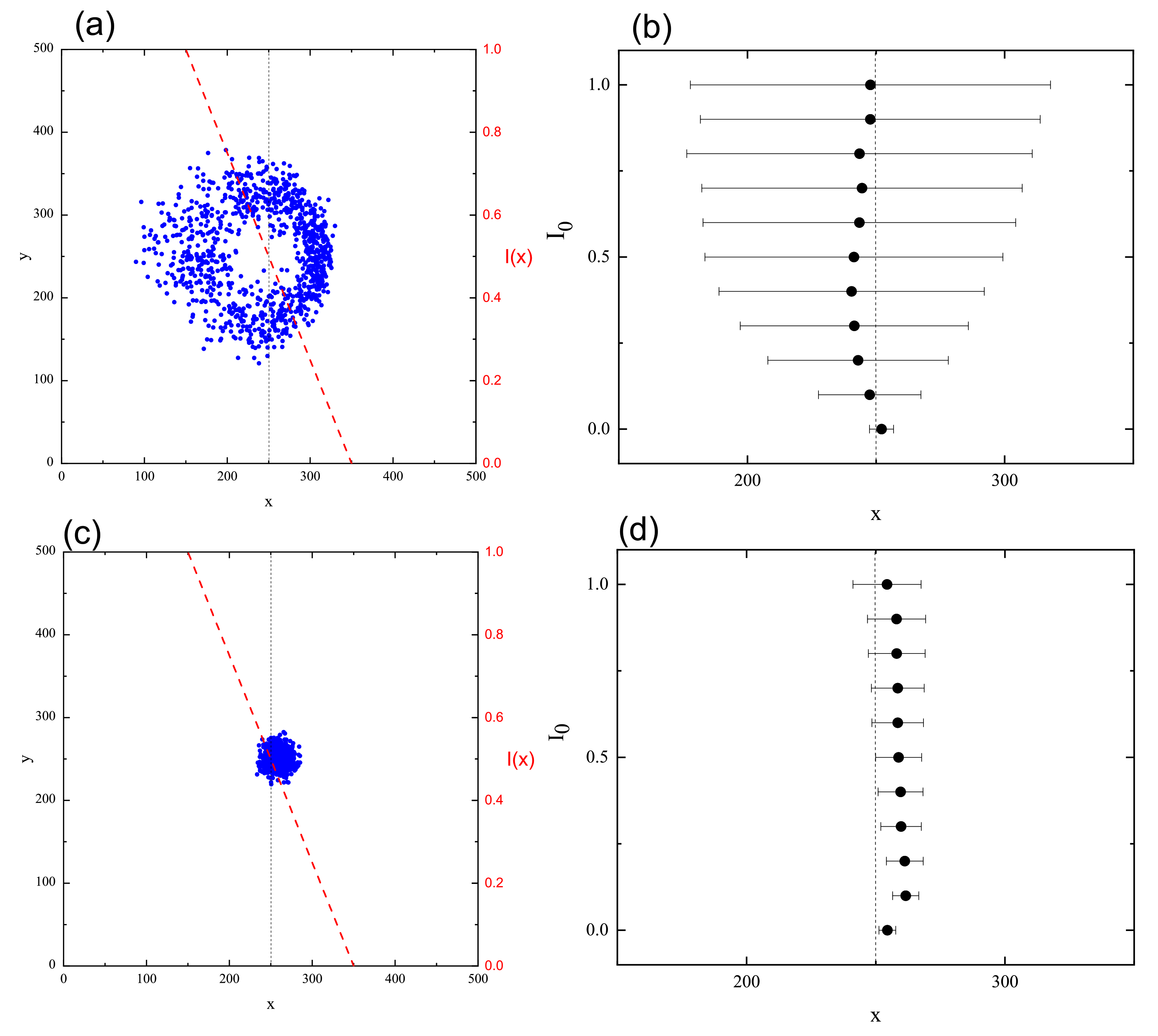
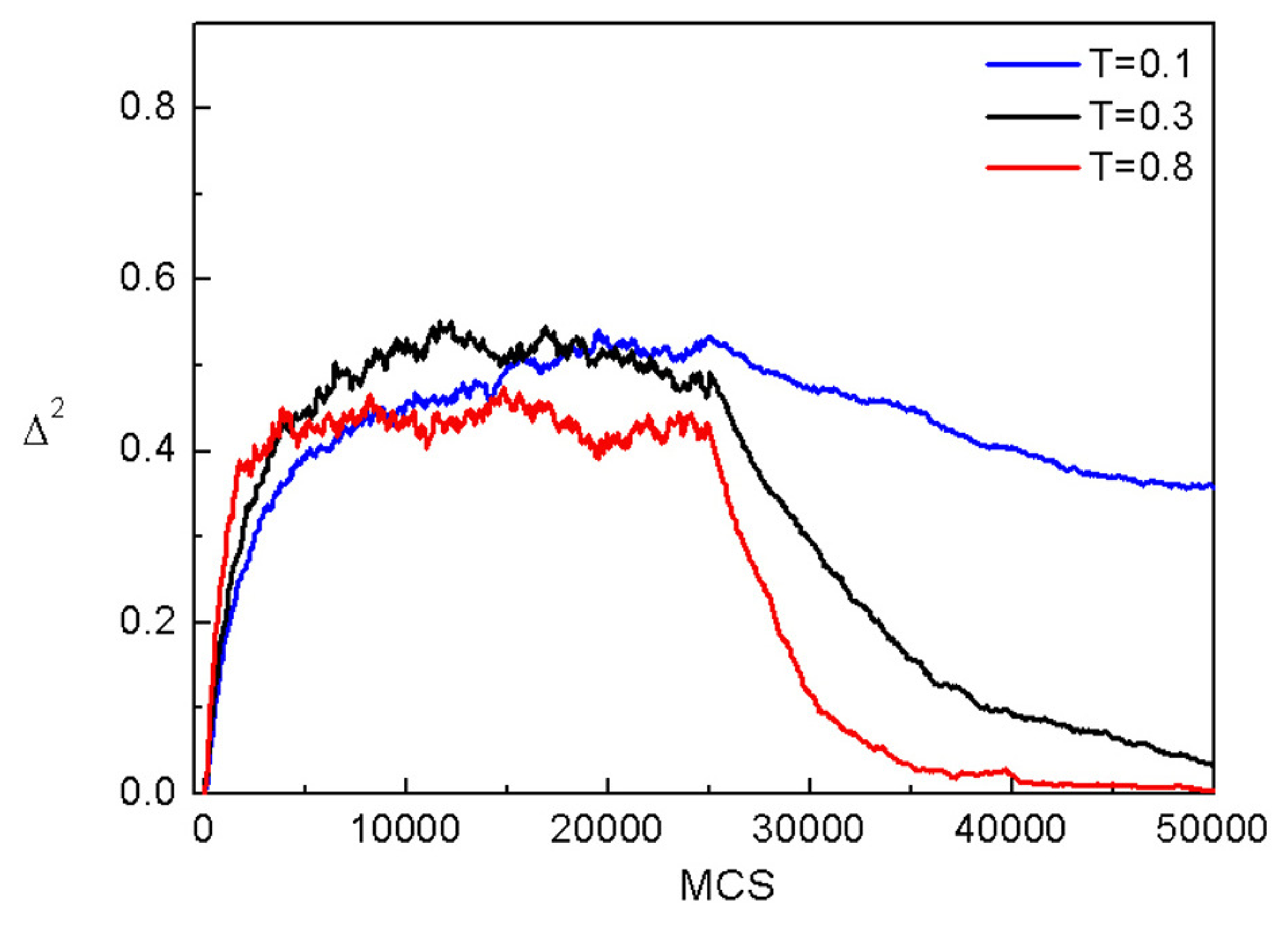
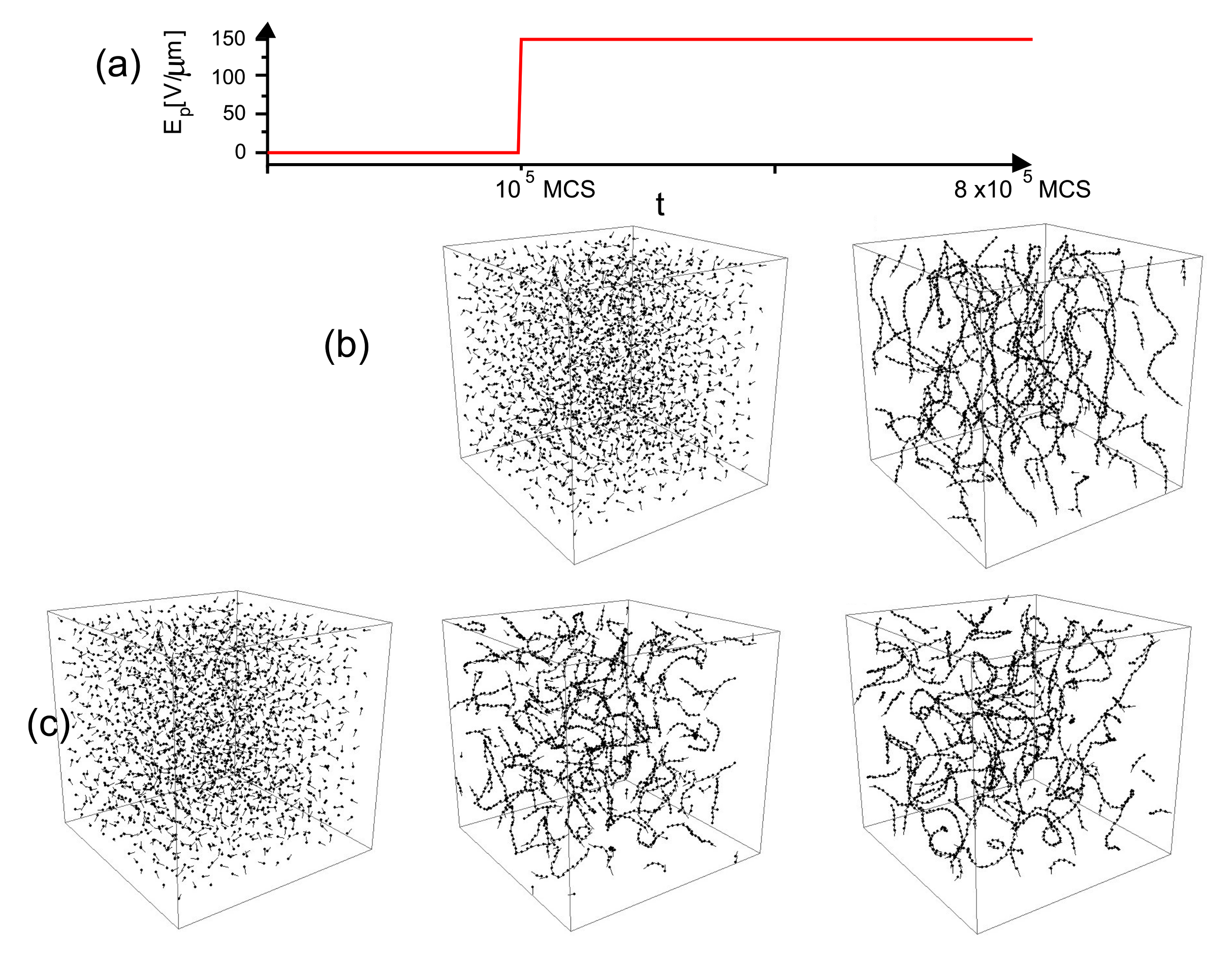
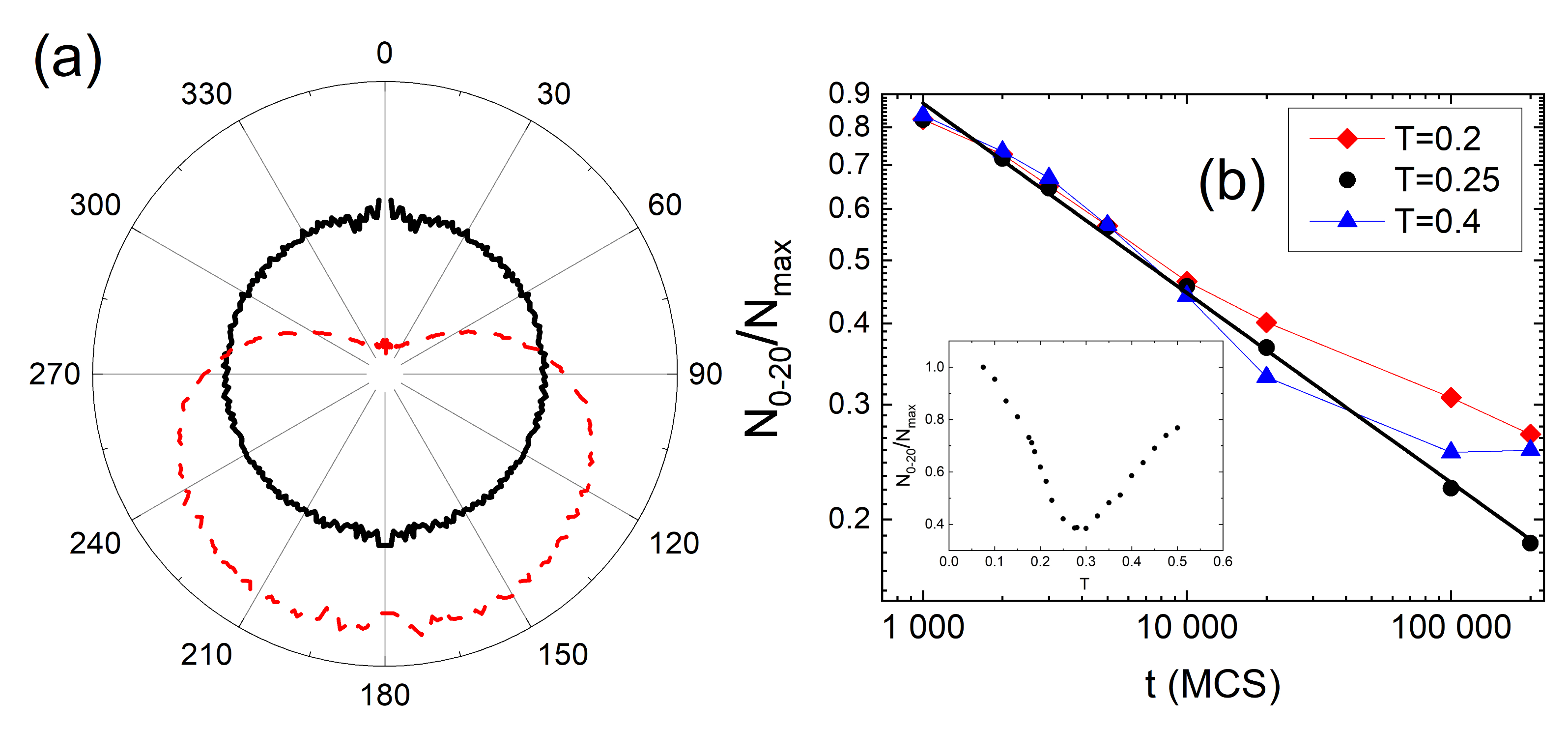
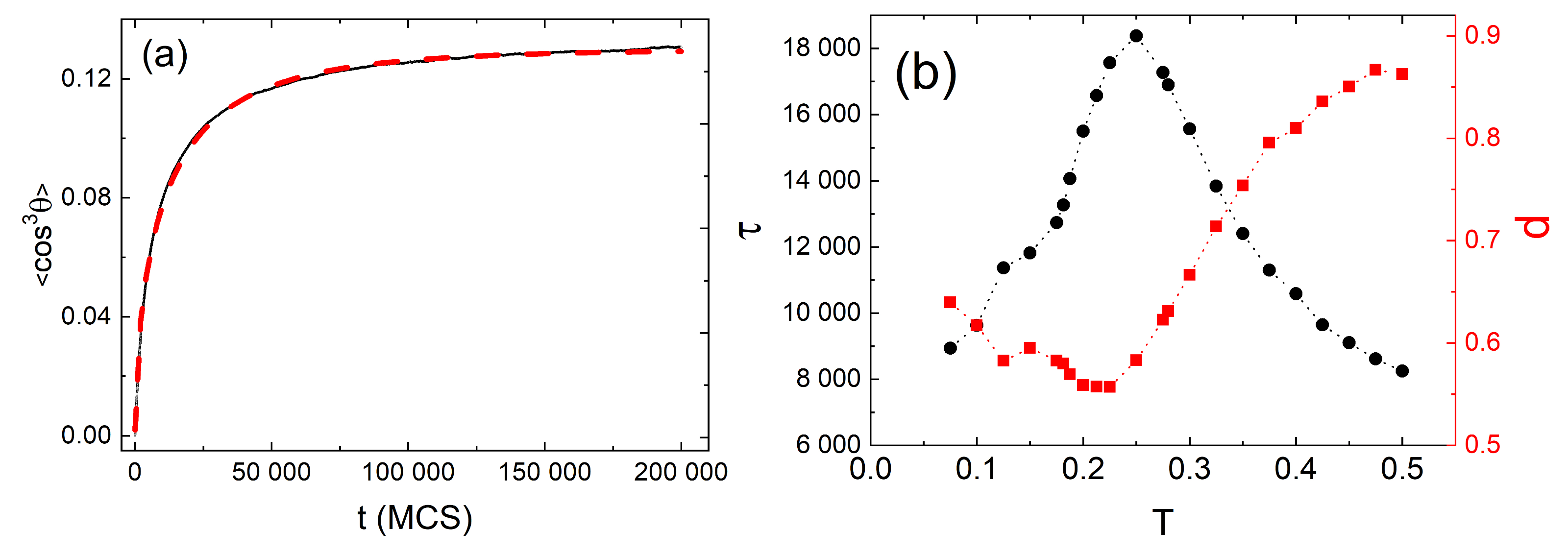
3.4. Complex Dynamics of Photoinduced Mass Transport in 3D and 2D
3.4.1. 3D
3.4.2. 2D
3.5. Inscription of Diffraction Gratings in Polymers and Bio-Polymers
3.5.1. Polymers with Embedded Azo-Dye Molecules
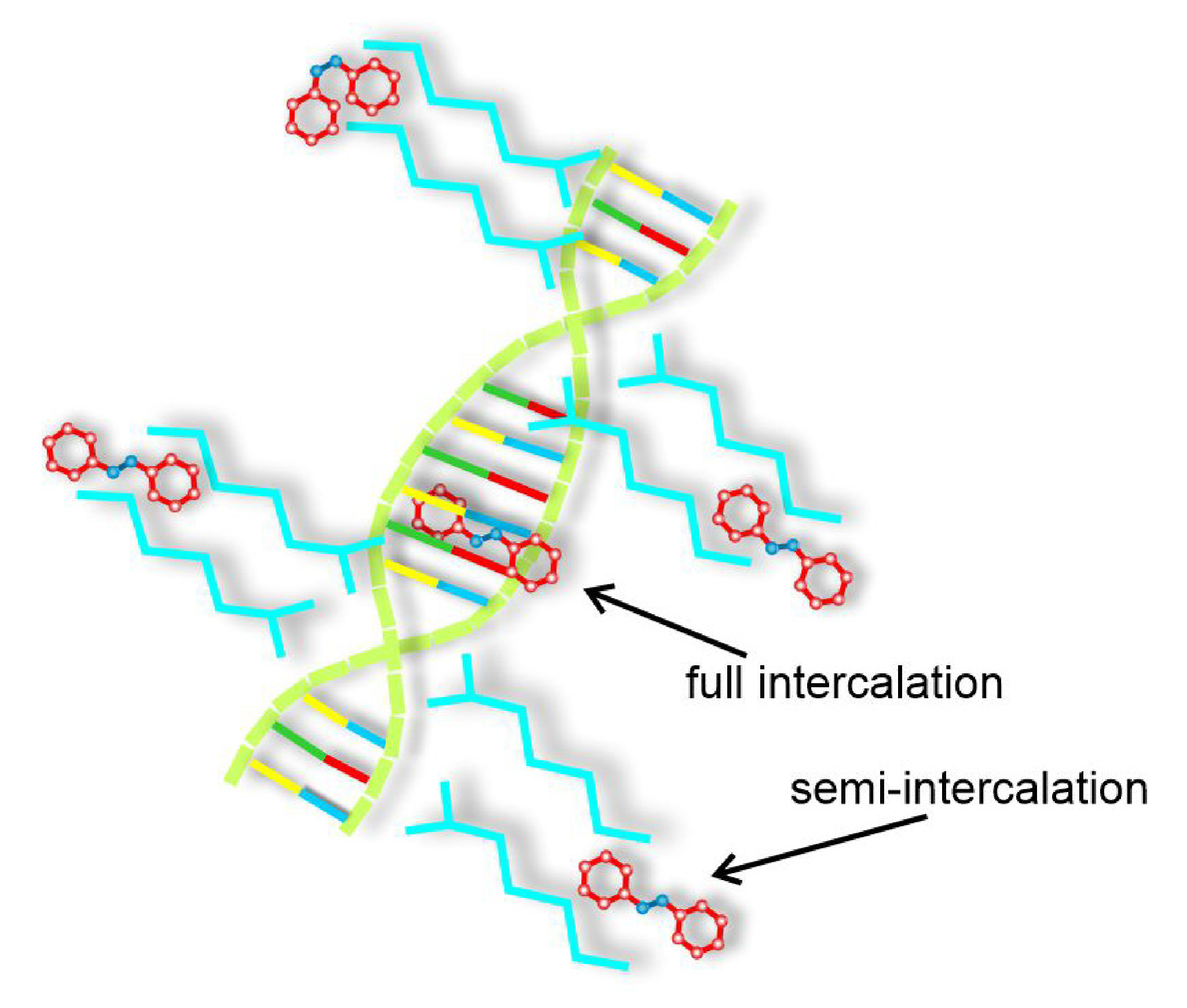
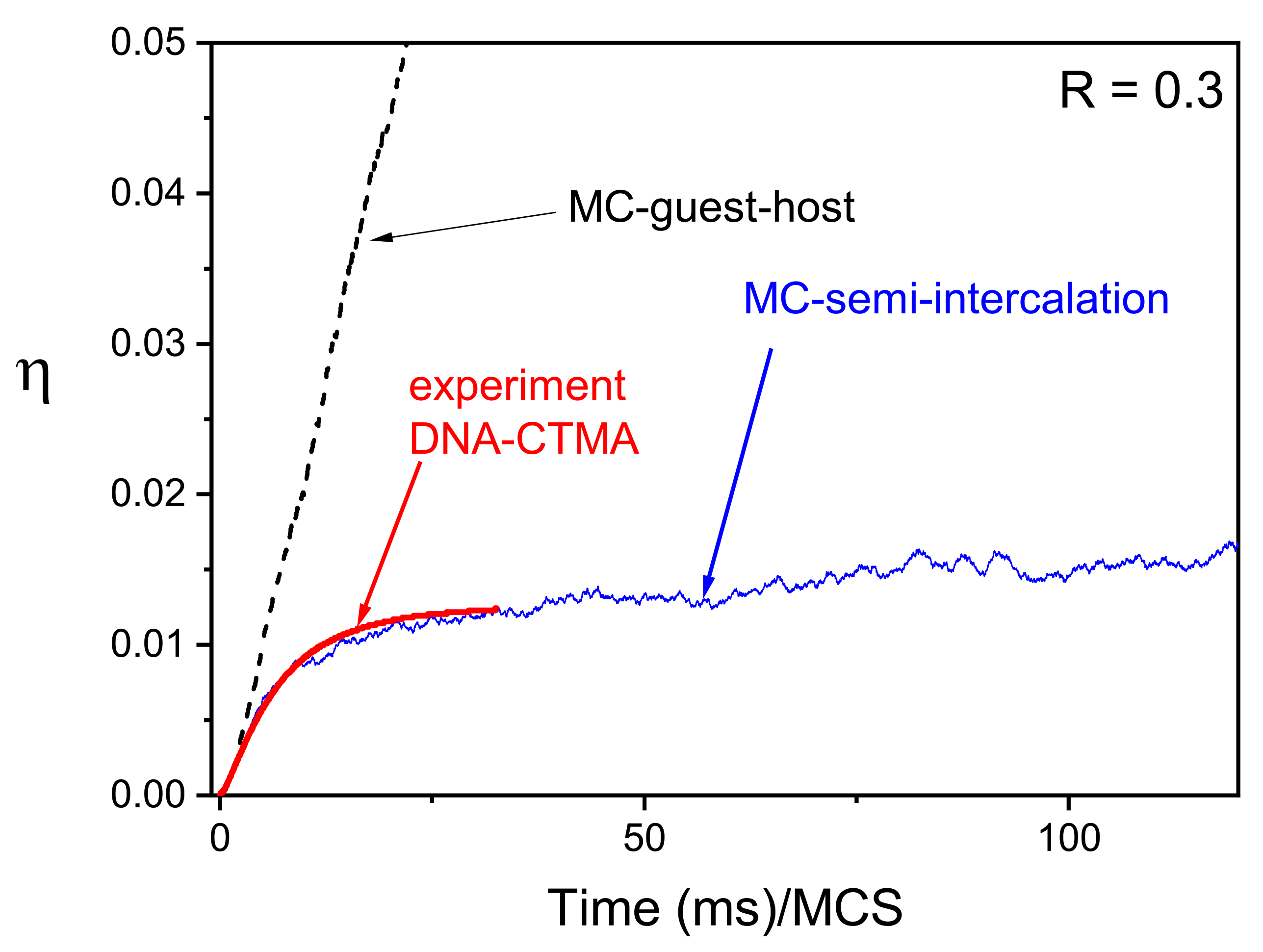
3.5.2. Bio-Polymers: Semi-Intercalation Scenario
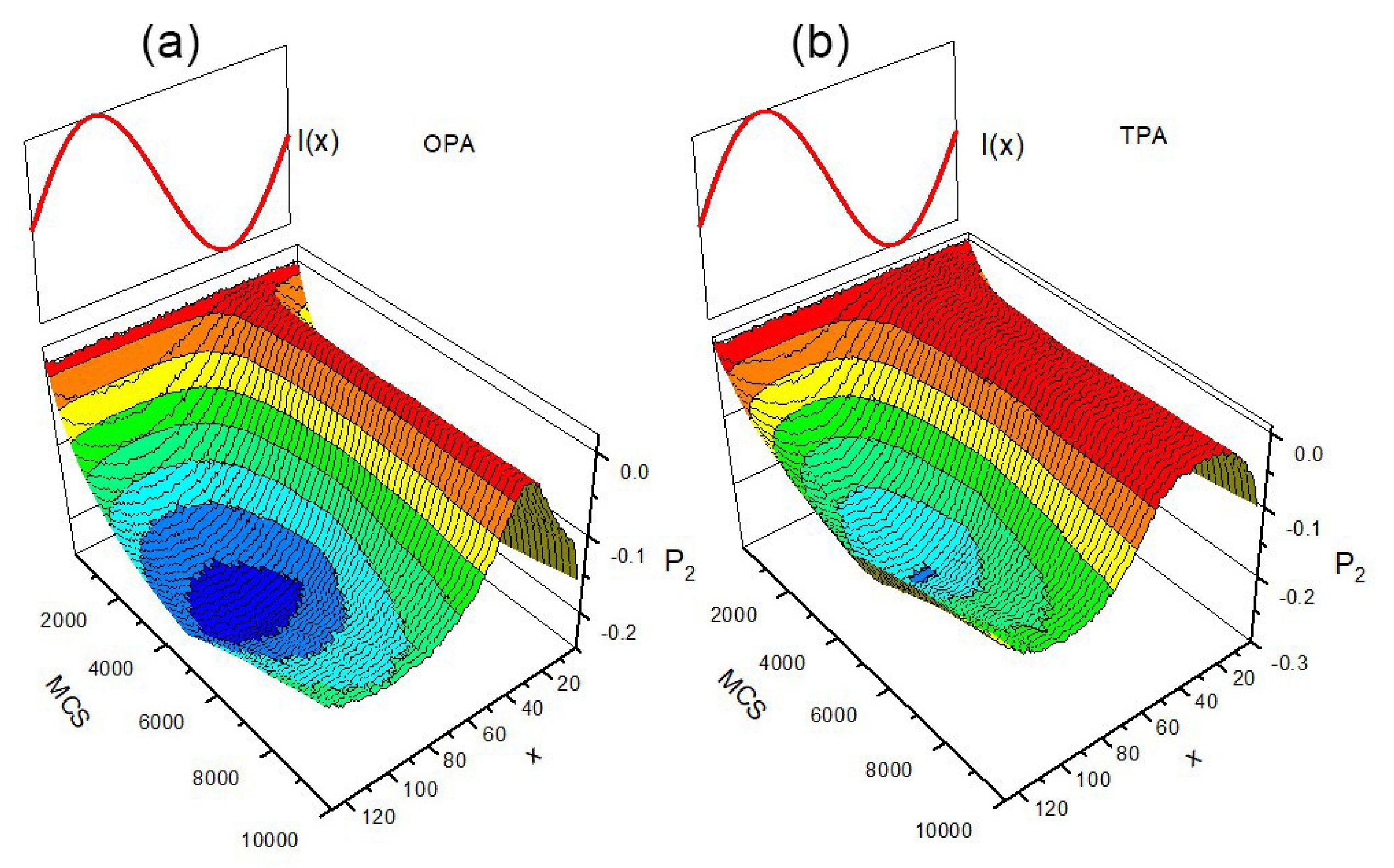
3.6. SHG in Poled Polymers: Pre-Poling History Paradigm
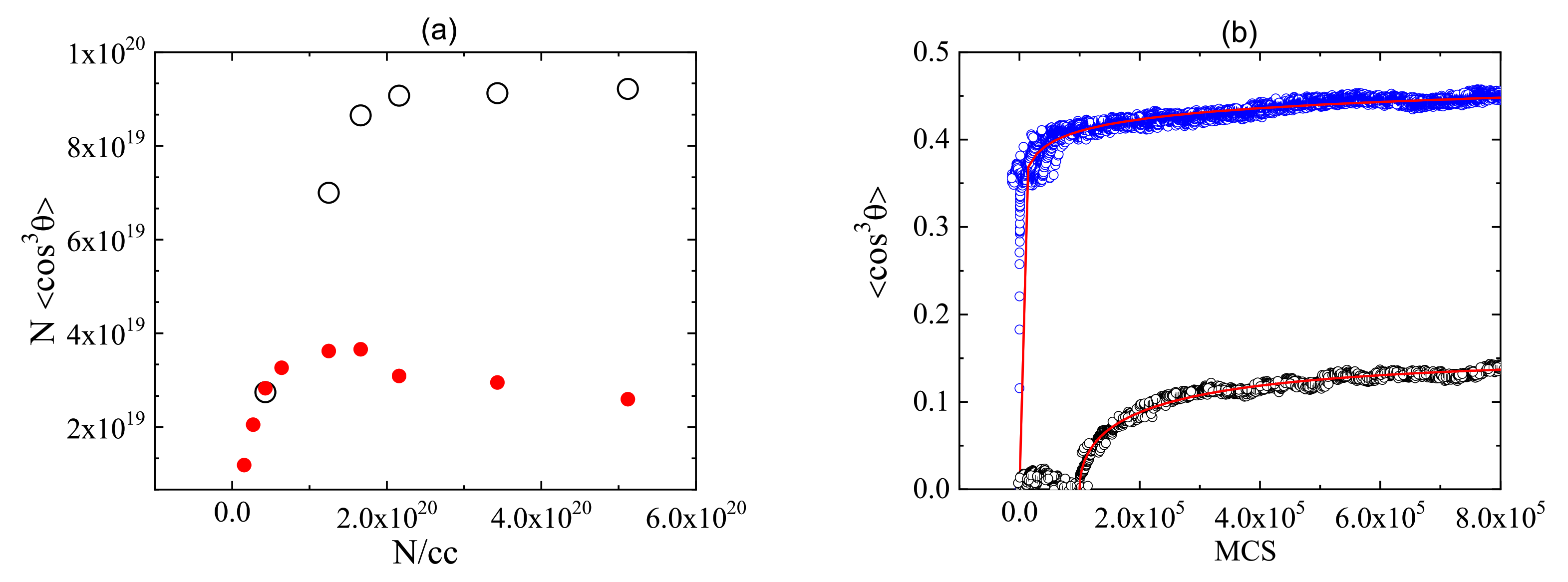
3.7. All Optical Poling
3.8. Photomechanical Effect in Polymeric Fibers
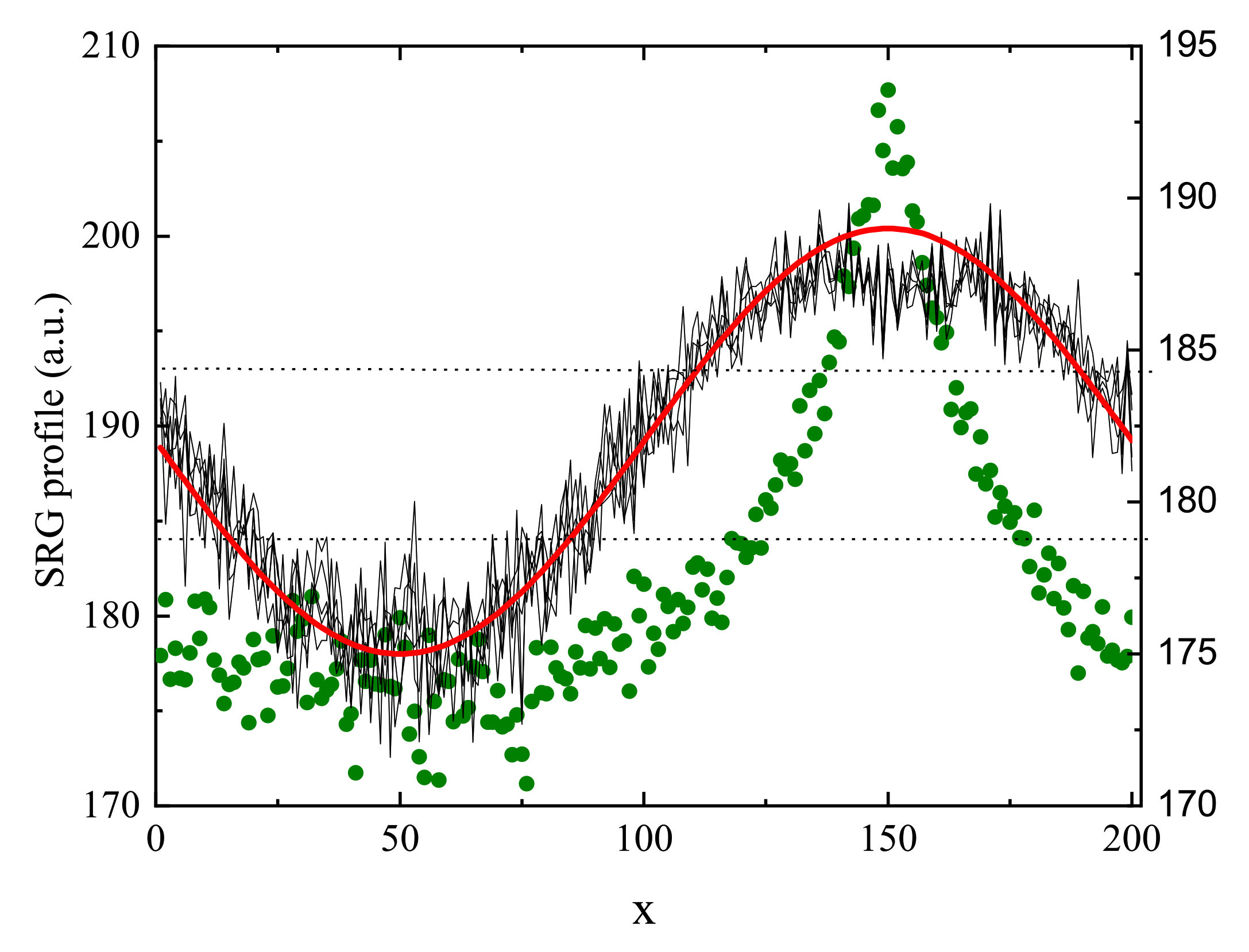
3.9. Continuous Time Random Walk and Toy Model of SRG Inscription
4. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhao, Y.; Ikeda, T. Smart Lightresponsive Materials: Azobenzene-Containing Polymers and Liquid Crystals; Wiley: Hoboken, NJ, USA, 2009. [Google Scholar]
- Boyd, R. Nonlinear Optics; Academic Press: Cambridge, MA, USA, 2000. [Google Scholar]
- Stegeman, G.I.; Stegeman, R.A. Nonlinear Optics: Phenomena, Materials and Devices; Wiley: Hoboken, NJ, USA, 2012. [Google Scholar]
- Doi, M.; Edwards, S.F. The Theory of Polymer Dynamics; Clarendon Press: Oxford, UK; Oxford University Press: New York, NY, USA, 1986. [Google Scholar]
- Kawakatsu, K. Statistical Physics of Polymers; Springer: Berlin/Heidelberg, Germany, 2004. [Google Scholar]
- Grosberg, A.Y.; Khoklov, A.R. Statistical Physics of Macromolecules; AIP Press: Woodbury, NY, USA, 1994. [Google Scholar]
- Pokrovskii, V.N. The Mesoscopic Theory of Polymer Dynamics; Kluwer Academic Publishers: New York, NY, USA, 2002. [Google Scholar]
- De Gennes, P.-G. Scaling Concepts in Polymer Physics; Cornell University Press: New York, NY, USA, 1979. [Google Scholar]
- Patashinski, A.Z.; Mitus, A.C.; Ratner, M.A. Towards understanding the local structure of liquids. Phys. Rep. 1997, 288, 409–434. [Google Scholar] [CrossRef]
- Binder, K. Monte Carlo and Molecular Dynamics Simulations in Polymer Science; Oxford University Press: Oxford, UK, 1995. [Google Scholar]
- Allen, M.P.; Tildesley, D.J. Computer Simulation of Liquids; Oxford Science Publishing: Oxford, UK, 1987. [Google Scholar]
- Frenkel, D.; Smit, B. Understanding Molecular Simulation; Academic Press: Cambridge, MA, USA, 2002. [Google Scholar]
- Kremer, K. Computer Simulations for Macromolecular Science. Macromol. Chem. Phys. 2003, 204, 257–264. [Google Scholar] [CrossRef]
- Binder, K.; Ciccotti, G. Monte Carlo and Molecular Dynamics of Condensed Matter Systems; Italian Physical Society: Bologna, Italy, 1996. [Google Scholar]
- Baumgartner, A.; Binder, K. Dynamics of entangled polymer melts: A computer simulation. J. Chem. Phys. 1981, 75, 29943. [Google Scholar] [CrossRef]
- Carmesin, I.; Kremer, K. The bond fluctuation method: A new effective algorithm for the dynamics of polymers in all spatial dimensions. Macromolecules 1988, 21, 2819–2823. [Google Scholar] [CrossRef]
- Wittmann, H.-P.; Kremer, K. Vectorized version of the bond fluctuation method for lattice polymers. Comp. Phys. Commun. 1990, 61, 309–330. [Google Scholar] [CrossRef]
- Paul, W.; Binder, K.; Heermann, D.W.; Kremer, K. Dynamics of polymer solutions and melts. Reptation predictions and scaling of relaxation times. J. Chem. Phys. 1991, 95, 7726. [Google Scholar] [CrossRef]
- Deutsch, H.; Dickman, R. Equation of state for athermal lattice chains in a 3d fluctuating bond model. J. Chem. Phys. 1990, 93, 8983. [Google Scholar] [CrossRef]
- Deutsch, H.-P.; Binder, K. Interdiffusion and self-diffusion in polymer mixtures: A Monte Carlo study. J. Chem. Phys. 1991, 94, 2294–2304. [Google Scholar] [CrossRef]
- Muller, M.; Paul, W. Measuring the chemical potential of polymer solutions and melts in computer simulations. J. Chem. Phys. 1994, 100, 719. [Google Scholar] [CrossRef]
- Wilding, N.; Muller, M. Accurate measurements of the chemical potential of polymeric systems by Monte Carlo simulation. J. Chem. Phys. 1994, 101, 4324. [Google Scholar] [CrossRef] [Green Version]
- Stukan, M.; Ivanov, V.; Muller, M.; Paul, W.; Binder, K. Finite size effects in pressure measurements for Monte Carlo simulations of lattice polymer models. J. Chem. Phys. 2002, 117, 9934. [Google Scholar] [CrossRef]
- Wittmer, J.P.; Meyer, H.; Baschnagel, J.; Johner, A.; Obukhov, S.P.; Mattioni, L.; Muller, M.; Semenov, A.N. Long Range Bond-Bond Correlations in Dense Polymer Solutions. Phys. Rev. Lett. 2004, 93, 147801. [Google Scholar] [CrossRef] [Green Version]
- Wittmer, J.P.; Beckrich, P.; Meyer, H.; Cavallo, A.; Johner, A.; Baschnagel, J.; Johner, A.; Semenov, A.N.; Obukhov, S.P.; Benoit, H. Intramolecular long-range correlations in polymer melts: The segmental size distribution and its moments. Phys. Rev. E 2007, 76, 011803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer, H.; Wittmer, J.P.; Kreer, T.; Beckrich, P.; Johner, A.; Farago, J.; Baschnagel, J. Static Rouse modes and related quantities: Corrections to chain ideality in polymer melts. Eur. Phys. J. E 2008, 26, 25. [Google Scholar] [CrossRef] [PubMed]
- Muller, M.; Binder, K.; Schafer, L. Intra- and Interchain Correlations in Semidilute Polymer Solutions: Monte Carlo Simulations and Renormalization Group Results. Macromolecules 2000, 33, 4568. [Google Scholar] [CrossRef]
- Wittmer, J.P.; Beckrich, P.; Johner, A.; Semenov, A.N.; Obukhov, S.P.; Meyer, H.; Baschnagel, J. Why polymer chains in a melt are not random walks. Europhys. Lett. 2007, 77, 56003. [Google Scholar] [CrossRef] [Green Version]
- Beckrich, P.; Johner, A.; Semenov, A.N.; Obukhov, S.P.; Benoit, H.; Wittmer, J.P. Intramolecular form factor in dense polymer systems: Systematic deviations from the Debye formula. Macromolecules 2007, 40, 3805. [Google Scholar] [CrossRef] [Green Version]
- Wittmer, J.P.; Paul, W.; Binder, K. Rouse and reptation dynamics at finite temperatures: A Monte Carlo simulation. Macromolecules 1992, 25, 7211. [Google Scholar] [CrossRef]
- Kreer, T.; Baschnagel, J.; Muller, M.; Binder, K. Monte Carlo Simulation of Long Chain Polymer Melts: Crossover from Rouse to Reptation Dynamics. Macromolecules 2001, 34, 1105. [Google Scholar] [CrossRef] [Green Version]
- Mattioni, L.; Wittmer, J.P.; Baschnagel, J.; Barrat, J.-L.; Luijten, E. Dynamical properties of the slithering-snake algorithm: A numerical test of the activated-reptation hypothesis. Eur. Phys. J. E 2003, 10, 369. [Google Scholar] [CrossRef]
- Azuma, R.; Takayama, H. Diffusion of single long polymers in fixed and low density matrix of obstacles confined to two dimensions. J. Chem. Phys. 1999, 111, 8666. [Google Scholar] [CrossRef] [Green Version]
- Muller, M.; Wittmer, J.P.; Cates, M.E. Topological effects in ring polymers: A computer simulation study. Phys. Rev. E 1996, 53, 5063. [Google Scholar] [CrossRef] [Green Version]
- Muller, M. Miscibility behavior and single chain properties in polymer blends: A bond fluctuation model study. Macromol. Theory Simul. 1999, 8, 343. [Google Scholar] [CrossRef]
- Cavallo, A.; Muller, M.; Binder, K. Anomalous scaling of the critical temperature of unmixing with chain length for two-dimensional polymer blends. Europhys. Lett. 2003, 61, 214. [Google Scholar] [CrossRef]
- Sommer, J.-U.; Saalwachter, K. Segmental order in end-linked polymer networks: A Monte Carlo study. Eur. Phys. J. E 2005, 18, 167. [Google Scholar] [CrossRef] [PubMed]
- Binder, K.; Baschnagel, J.; Paul, W. Glass transition of polymer melts: Test of theoretical concepts by computer simulation. Prog. Polym. Sci. 2003, 28, 115. [Google Scholar] [CrossRef]
- Baschnagel, J.; Binder, K.; Wittmann, H.-P. The influence of the cooling rate on the glass transition and the glassy state in three-dimensional dense polymer melts: A Monte Carlo study. J. Phys. Condens. Matter 1993, 5, 1597. [Google Scholar] [CrossRef]
- Wittmann, H.-P.; Kremer, K.; Binder, K. Glass transition of polymer melts: A two-dimensional Monte Carlo study in the framework of the bond fluctuation method. J. Chem. Phys. 1996, 96, 6291. [Google Scholar] [CrossRef]
- Deutsch, H.-P.; Binder, K. Critical Behavior and Crossover Scaling in Symmetric Polymer Mixtures: A Monte Carlo Investigation. Macromolecules 1992, 25, 6214. [Google Scholar] [CrossRef]
- Werner, A.; Schmid, F.; Muller, M. Monte Carlo simulations of copolymers at homopolymer interfaces: Interfacial structure as a function of the copolymer density. J. Chem. Phys. 1999, 110, 5370. [Google Scholar] [CrossRef] [Green Version]
- Lang, M.; Werner, M.; Dockhorn, R.; Kreer, T. Arm Retraction Dynamics in Dense Polymer Brushes. Macromolecules 2016, 49, 5190–5201. [Google Scholar] [CrossRef]
- Lang, M.; Hoffmann, M.; Dockhorn, R.; Werner, M.; Sommer, J.-U. Fluctuation driven height reduction of crosslinked polymer brushes: A Monte Carlo study. J. Chem. Phys. 2013, 139, 164903. [Google Scholar] [CrossRef]
- Lai, P.-Y.; Binder, K. Structure and dynamics of grafted polymer layers: A Monte Carlo simulation. J. Chem. Phys. 1991, 95, 9288. [Google Scholar] [CrossRef]
- Lai, P.-Y. Grafted polymer layers with chain exchange: A Monte Carlo simulation. J. Chem. Phys. 1993, 98, 669. [Google Scholar] [CrossRef]
- Wittmer, J.; Johner, A.; Joanny, J.F.; Binder, K. Chain desorption from a semidilute polymer brush: A Monte Carlo simulation. J. Chem. Phys. 1994, 101, 4379. [Google Scholar] [CrossRef] [Green Version]
- Kopf, A.; Baschnagel, J.; Wittmer, J.; Binder, K. On the Adsorption Process in Polymer Brushes: A Monte Carlo Study. Macromolecules 1996, 29, 1433. [Google Scholar] [CrossRef]
- Wittmer, J.P.; Cates, M.E.; Johner, A.; Turner, M.S. Diffusive growth of a polymer layer by in situ polymerization. Europhys. Lett. 1996, 33, 397. [Google Scholar] [CrossRef] [Green Version]
- Khalatur, P.; Khokhlov, A.; Prokhorova, S.; Sheiko, S.; Moller, M.; Reineker, P.; Shirvanyanz, D. Unusual conformation of molecular cylindrical brushes strongly adsorbed on a flat solid Surface. Eur. Phys. J. E 2000, 1, 99. [Google Scholar] [CrossRef]
- Mischler, C.; Baschnagel, J.; Binder, K. Polymer films in the normal-liquid and supercooled state: A review of recent Monte Carlo simulation results. Adv. Colloid Interface Sci. 2001, 94, 197. [Google Scholar] [CrossRef] [Green Version]
- Cavallo, A.; Müller, M.; Wittmer, J.P.; Johner, A. Single chain structure in thin polymer films: Corrections to Flory’s and Silberberg’s hypotheses. J. Phys. Condens. Matter. 2005, 17, S1697. [Google Scholar] [CrossRef]
- Wittmer, J.P.; Milchev, A.; Cates, M.E. Dynamical Monte Carlo study of equilibrium polymers: Static properties. J. Chem. Phys. 1998, 109, 834. [Google Scholar] [CrossRef] [Green Version]
- Wittmer, J.P.; Beckrich, P.; Crevel, F.; Huang, C.C.; Cavallo, A.; Kreer, T.; Meyer, H. Are polymer melts ideal? Comput. Phys. Commun. 2007, 177, 146. [Google Scholar] [CrossRef] [Green Version]
- Cavallo, A.; Muller, M.; Binder, K. Formation of Micelles in Homopolymer-Copolymer Mixtures: Quantitative Comparison between Simulations of Long Chains and Self-Consistent Field Calculations. Macromolecules 2006, 39, 9539. [Google Scholar] [CrossRef]
- Cavallo, A.; Muller, M.; Binder, K. Monte Carlo Simulation of a Homopolymer-Copolymer Mixture Interacting with a Surface: Bulk versus Surface Micelles and Brush Formation. Macromolecules 2008, 41, 4937. [Google Scholar] [CrossRef]
- Wengenmayr, M.; Dockhorn, R.; Sommer, J.-U. Multicore Unimolecular Structure Formation in Single Dendritic-Linear Copolymers under Selective Solvent Conditions. Macromolecules 2016, 49, 9215. [Google Scholar] [CrossRef]
- Lang, M.; Müller, T. Analysis of the Gel Point of Polymer Model Networks by Computer Simulations. Macromolecules 2020, 53, 498–512. [Google Scholar] [CrossRef]
- Muller, T.; Sommer, J.-U.; Lang, M. Tendomers—force sensitive bis-rotaxanes with jump-like deformation behavior. Soft Matter 2019, 15, 3671–3679. [Google Scholar] [CrossRef]
- Rabbel, H.; Breier, P.; Sommer, J.-U. Swelling Behavior of Single-Chain Polymer Nanoparticles: Theory and Simulation. Macromolecules 2017, 50, 7410–7418. [Google Scholar] [CrossRef]
- Lang, M.; Schwenke, K.; Sommer, J.-U. Short Cyclic Structures in Polymer Model Networks: A Test of Mean Field Approximation by Monte Carlo Simulations. Macromolecules 2012, 45, 4886–4895. [Google Scholar] [CrossRef]
- Lang, M.; Fischer, J.; Werner, M.; Sommer, J.-U. Olympic Gels: Concatenation and Swelling. Macromol. Symp. 2015, 358, 140–147. [Google Scholar] [CrossRef]
- Fischer, J.; Lang, M.; Sommer, J.-U. The formation and structure of Olympic gels. J. Chem. Phys. 2015, 143, 243114. [Google Scholar] [CrossRef]
- Lang, M.; Fischer, J.; Werner, M.; Sommer, J.-U. Swelling of Olympic Gels. Phys. Rev. Lett. 2014, 112, 238001. [Google Scholar] [CrossRef]
- Dockhorn, R.; Pluuschke, L.; Geisler, M.; Zessin, J.; Lindner, P.; Mundil, R.; Merna, J.; Sommer, J.-U.; Lederer, A. Polyolefins Formed by Chain Walking Catalysis—A Matter of Branching Density Only? J. Am. Chem. Soc. 2019, 141, 1558615596. [Google Scholar] [CrossRef]
- Jurjiu, A.; Dockhorn, R.; Mironova, O.; Sommer, J.-U. Two universality classes for random hyperbranched polymers. Soft Matter 2014, 10, 4935–4946. [Google Scholar] [CrossRef] [Green Version]
- Wengenmayr, M.; Dockhorn, R.; Sommer, J.-U. Dendrimers in Solution of Linear Polymers: Crowding Effects. Macromolecules 2019, 52, 2616–2626. [Google Scholar] [CrossRef]
- Klos, J.S.; Sommer, J.-U. Dendrimer solutions: A Monte Carlo study. Soft Matter 2016, 12, 9007–9013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sommer, J.-U.; Klos, J.S.; Mironova, O. Adsorption of branched and dendritic polymers onto flat surfaces: A Monte Carlo study. J. Chem. Phys. 2013, 139, 244903. [Google Scholar] [CrossRef] [PubMed]
- Klos, J.S.; Sommer, J.-U. Simulations of Terminally Charged Dendrimers with Flexible Spacer Chains and Explicit Counterions. Macromolecules 2010, 43, 4418. [Google Scholar] [CrossRef]
- Checkervarty, A.; Werner, M.; Sommer, J.-U. Formation and stabilization of pores in bilayermembranes by peptide-like amphiphilic polymers. Soft Matter 2018, 14, 2526–2534. [Google Scholar] [CrossRef] [PubMed]
- Rabbel, H.; Werner, M.; Sommer, J.-U. Interactions of Amphiphilic Triblock Copolymers with Lipid Membranes: Modes of Interaction and Effect on Permeability Examined by Generic Monte Carlo Simulations. Macromolecules 2015, 48, 4724. [Google Scholar] [CrossRef]
- Werner, M.; Sommer, J.-U. Translocation and Induced Permeability of Random AmphiphilicCopolymers Interacting with Lipid Bilayer Membranes. Biomacromolecules 2015, 16, 125–135. [Google Scholar] [CrossRef]
- Sommer, J.-U.; Werner, M.; Baulin, V.A. Critical adsorption controls translocation of polymer chains through lipid bilayers and permeation of solvent. Europhys. Lett. 2012, 98, 18003. [Google Scholar] [CrossRef]
- Baschnagel, J.; Wittmer, J.P.; Meyer, H. From Synthetic Polymers to Proteins. Comput. Soft Matter 2004, 23, 83. [Google Scholar]
- Muller, M. Handbook of Materials Modeling; Springer: New York, NY, USA, 2005. [Google Scholar]
- Wittmer, J.P.; Cavallo, A.; Kreer, T.; Baschnagel, J.; Johner, A. A finite excluded volume bond-fluctuation model: Static properties of dense polimer melts revisited. J. Chem. Phys. 2009, 131, 064901. [Google Scholar] [CrossRef] [Green Version]
- Wani, O.M.; Zeng, H.; Priimagi, A. A light-driven artificial flytrap. Nat. Commun. 2017, 8, 15546. [Google Scholar] [CrossRef]
- Gelebart, A.H.; Mulder, D.J.; Varga, M.; Konya, A.; Vantomme, G.; Meijer, E.W.; Selinger, R.L.B.; Broer, D.J. Making waves in a photoactive polymer film. Nature 2017, 546, 632. [Google Scholar] [CrossRef] [Green Version]
- Kuzyk, M.G. Polymer Fiber Optics; CRC Press: Boca Raton, FL, USA, 2007. [Google Scholar]
- Kuzyk, M.G.; Dawson, N.J. Photomechanical materials and applications: A tutorial. Adv. Opt. Phot. 2020, 12, 847. [Google Scholar] [CrossRef]
- Priimagi, A.; Shevchenko, A. Azopolymer-based micro- and nanopatterning for photonic applications. J. Polym. Sci. Pol. Phys. 2014, 52, 163–182. [Google Scholar] [CrossRef]
- Oscurato, S.L.; Salvatore, M.; Maddalena, P.; Ambrosio, A. From nanoscopic to macroscopic photo-driven motion in azobenzene-containing materials. Nanophotonics 2018, 7, 1387–1422. [Google Scholar] [CrossRef]
- Barrett, C.J.; Rochon, P.L.; Natansohn, A.L. Model of laser-driven mass transport in thin films of dye-functionalized polymers. J. Chem. Phys. 1998, 109, 1505–1516. [Google Scholar] [CrossRef]
- Kumar, J.; Li, L.; Jiang, X.L.; Kim, D.-Y.; Lee, T.S.; Tripathy, S. Gradient force: The mechanism for surface relief grating formation in azobenzene functionalized polymers. Appl. Phys. Lett. 1998, 72, 2096–2098. [Google Scholar] [CrossRef]
- Lefin, P.; Fiorini, C.; Nunzi, J.M. Anisotropy of the photoinduced translation diffusion of azo-dyes. Opt. Mater. 1998, 9, 323–328. [Google Scholar] [CrossRef]
- Pedersen, T.G.; Johansen, P.M.; Holme, N.C.R.; Ramanujam, P.S.; Hvilsted, S. Mean-Field Theory of Photoinduced Formation of Surface Relief in Side-Chain Azobenzene Polymers. Phys. Rev. Lett. 1998, 80, 89–92. [Google Scholar] [CrossRef]
- Baldus, O.; Zilker, S.J. Surface Relief Gratings in Photoaddressable Polymers Generated by CW Holography. Appl. Phys. B 2001, 72, 425–427. [Google Scholar] [CrossRef]
- Gaididei, Y.B.; Christiansen, P.L.; Ramanujam, P.S. Theory of Photoinduced Deformation of Molecular Films. Appl. Phys. B Lasers Opt. 2002, 74, 139–146. [Google Scholar] [CrossRef]
- Saphiannikova, M.; Neher, D. Thermodynamic Theory of Light-Induced Material Transport in Amorphous Azobenzene Polymer Films. J. Phys. Chem. B 2005, 109, 19428–19436. [Google Scholar] [CrossRef] [PubMed]
- Bellini, B.; Ackermann, J.; Klein, H.; Grave, C.; Dumas, P.; Safarov, V. Light-induced molecular motion of azobenzene-containing molecules: A random-walk model. J. Phys. Condens. Matter 2006, 18, 1817–1835. [Google Scholar] [CrossRef]
- Juan, M.L.; Plain, J.; Bachelot, R.; Royer, P.; Gray, S.K.; Wiederrecht, G.P. Multiscale model for photoinduced molecular motion in azo polymers. ACS Nano 2009, 3, 1573–1579. [Google Scholar] [CrossRef] [PubMed]
- Bublitz, D.; Fleck, B.; Wenke, L. A Model for Surface-Relief Formation in Azobenzene Polymers. Appl. Phys. B 2001, 72, 931–936. [Google Scholar] [CrossRef]
- Karageorgiev, P.; Neher, D.; Schulz, B.; Stiller, B.; Pietsch, U.; Giersig, M.; Brehmer, L. From anisotropic photo-fluidity towards nanomanipulation in the optical near-field. Nat. Mater. 2005, 4, 699–703. [Google Scholar] [CrossRef]
- Lee, S.; Kang, H.S.; Park, J.-K. Directional photofluidization lithography: Micro/ nanostructural evolution by photofluidic motions of azobenzene materials. Adv. Mater. 2012, 24, 2069–2103. [Google Scholar] [CrossRef]
- Fang, G.J.; Maclennan, J.E.; Yi, Y.; Glaser, M.A.; Farrow, M.; Korblova, E.; Walba, D.M.; Furtak, T.E.; Clark, N.A. Athermal photofluidization of glasses. Nat. Commun. 2013, 4, 1521. [Google Scholar] [CrossRef] [Green Version]
- Hurduc, N.; Donose, B.C.; Macovei, A.; Paius, C.; Ibanescu, C.; Scutaru, D.; Hamel, M.; Branza-Nichita, N.; Rocha, L. Direct observation of athermal photofluidisation in azopolymer films. Soft Matter 2014, 10, 4640–4647. [Google Scholar] [CrossRef]
- Veer, P.U.; Pietsch, U.; Rochon, P.L.; Saphiannikova, M. Temperature dependent analysis of grating formation on azobenzene polymer films. Mol. Cryst. Liquid Cryst. 2008, 486, 1108–1120. [Google Scholar] [CrossRef]
- Mechau, N.; Saphiannikova, M.; Neher, D. Dielectric and mechanical properties of azobenzene polymer layers under visible and ultraviolet irradiation. Macromolecules 2005, 38, 3894–3902. [Google Scholar] [CrossRef]
- Mechau, N.; Saphiannikova, M.; Neher, D. Molecular tracer diffusion in thin azobenzene polymer layers. Appl. Phys. Lett. 2006, 89, 251902. [Google Scholar] [CrossRef]
- Srikhirin, T.; Laschitsch, A.; Neher, D.; Johannsmann, D. Light-induced softening of azobenzene dye-doped polymer films probed with quartz crystal resonators. Appl. Phys. Lett. 2000, 77, 963–965. [Google Scholar] [CrossRef]
- Saphiannikova, M.; Toshchevikov, V. Optical deformations of azobenzene polymers: Orientation approach vs. photofluidization concept. J. Soc. Inf. Disp. 2015, 23, 146–153. [Google Scholar] [CrossRef]
- Lee, S.; Kang, H.S.; Ambrosio, A.; Park, J.-K.; Marrucci, L. Directional Superficial Photofluidization for Deterministic Shaping of Complex 3D Architectures. ACD Appl. Mater. Inter. 2015, 7, 8209–8217. [Google Scholar] [CrossRef]
- Pirani, F.; Angelini, A.; Frascella, F.; Rizzo, R.; Ricciardi, S.; Descrovi, E. Light-Driven Reversible Shaping of Individual Azopolymeric Micro-Pillars. Sci. Rep. 2016, 6, 31702. [Google Scholar] [CrossRef] [Green Version]
- Ambrosio, A.; Borbone, F.; Carella, A.; Centore, R.; Fusco, S.; Kuball, H.-G.; Maddalena, P.; Romano, C.; Roviello, A.; Stolte, M. Cis–trans isomerization and optical laser writing in new heterocycle based azo-polyurethanes. Opt. Mater. 2012, 34, 724–728. [Google Scholar] [CrossRef]
- Uchida, E.; Azumi, R.; Norikane, Y. Light-induced crawling of crystals on a glass surface. Nat. Commun. 2015, 6, 7310. [Google Scholar] [CrossRef] [PubMed]
- Nath, N.K.; Panda, M.K.; Sahoo, S.C.; Naumov, P. Thermally induced and photoinduced mechanical effects in molecular single crystals—A revival. Cryst. Eng. Commun. 2014, 16, 1850–1858. [Google Scholar] [CrossRef]
- Zhou, H.; Xue, C.; Weis, P.; Suzuki, Y.; Huang, S.; Koynov, K.; Auernhammer, G.K.; Rüdiger, B.; Butt, H.-J.; Wu, S. Photoswitching of glass transition temperatures of azobenzene-containing polymers induces reversible solid-to-liquid transitions. Nat. Chem. 2017, 9, 145–151. [Google Scholar] [CrossRef]
- Wen-Cong, X.; Shaodong, S.; Wu, S. Photoinduced Reversible Solid-to-Liquid Transitions for Photoswitchable Materials. Angew. Chem. Int. Edit. 2019, 58, 9712–9740. [Google Scholar]
- Yang, B.; Cai, F.; Huang, S.; Yu, H. Athermal and Soft Multi-Nanopatterning of Azopolymers: Phototunable Mechanical Properties. Angew. Chem. Int. Ed. 2020, 59, 4188. [Google Scholar] [CrossRef] [Green Version]
- Kopyshev, A.; Galvin, C.J.; Genzer, J.; Lomadze, N.; Santer, S. Opto-Mechanical Scission of Polymer Chains in Photosensitive Diblock-Copolymer Brushes. Langmuir 2013, 29, 13967–13974. [Google Scholar] [CrossRef]
- Yadavalli, N.S.; Linde, F.; Kopyshev, A.; Santer, S. Soft matter beats hard matter: Rupturing of thin metallic films induced by mass transport in photosensitive polymer films. ACS Appl. Mater. Interfaces 2013, 5, 7743–7747. [Google Scholar] [CrossRef]
- Di Florio, G.; Brundermann, E.; Yadavalli, N.S.; Santer, S.; Havenith, M. Graphene multilayer as nanosized optical strain gauge for polymer surface relief gratings. Nano Lett. 2014, 14, 5754–5760. [Google Scholar] [CrossRef]
- Toshchevikov, V.; Saphiannikova, M.; Heinrich, G. Microscopic theory of light-induced deformation in amorphous side-chain azobenzene polymers. J. Phys. Chem. B 2009, 113, 5032–5045. [Google Scholar] [CrossRef]
- Ilnytskyi, J.M.; Saphiannikova, M. Reorientation dynamics of chromophores in photosensitive polymers by means of coarse-grained modeling. ChemPhysChem 2015, 16, 3180. [Google Scholar] [CrossRef]
- Ilnytskyi, J.M.; Toshchevikov, V.; Saphiannikova, M. Modeling of the photo-induced stress in azobenzene polymers by combining theory and computer simulations. Soft Matter 2019, 15, 9894. [Google Scholar] [CrossRef] [PubMed]
- Toshchevikov, V.; Ilnytskyi, J.; Saphiannikova, M. Photoisomerization kinetics and mechanical stress in azobenzene-containing materials. J. Phys. Chem. Lett. 2017, 8, 1094–1098. [Google Scholar] [CrossRef]
- Toshchevikov, V.; Petrova, T.; Saphiannikova, M. Kinetics of light-induced ordering and deformation in lc azobenzene-containing materials. Soft Matter 2017, 13, 2823–2835. [Google Scholar] [CrossRef] [PubMed]
- Yadav, B.; Domurath, J.; Kim, K.; Lee, S.; Saphiannikova, M. Orientation approach to directional photodeformations in glassy side-chain azopolymers. J. Phys. Chem. B 2019, 123, 3337–3347. [Google Scholar] [CrossRef] [PubMed]
- Yadav, B.; Domurath, J.; Saphiannikova, M. Modeling of stripe patterns in photosensitive azopolymers. Polymers 2020, 12, 735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loebner, S.; Lomadze, N.; Kopyshev, A.; Koch, M.; Guskova, O.; Saphiannikova, M.; Santer, S. Light-induced deformation of azobenzene-containing colloidal spheres: Calculation and measurement of opto-mechanical stresses. J. Phys. Chem. B 2018, 122, 2001–2009. [Google Scholar] [CrossRef]
- Yadavalli, N.S.; Saphiannikova, M.; Lomadze, N.; Goldenberg, L.M.; Santer, S. Structuring of photosensitive material below diffraction limit using far field irradiation. Appl. Phys. A 2013, 113, 263–272. [Google Scholar] [CrossRef]
- Yadavalli, N.S.; Saphiannikova, M.; Santer, S. Photosensitive response of azobenzene containing films towards pure intensity or polarization interference patterns. Appl. Phys. Lett. 2014, 105, 051601. [Google Scholar] [CrossRef]
- Juan, M.L.; Plain, J.; Bachelot, R.; Royer, P.; Gray, S.K.; Wiederrecht, G.P. Stochastic model for photoinduced surface relief grating formation through molecular transport in polymer films. Appl. Phys. Lett. 2008, 93, 153304. [Google Scholar] [CrossRef]
- Plain, J.; Wiederrecht, G.P.; Gray, S.K.; Royer, P.; Bachelot, R. Multiscale optical imaging of complex fields based on the use of azobenzene nanomotors. J. Phys. Chem. Lett. 2013, 4, 2124–2132. [Google Scholar] [CrossRef]
- Dumont, M.; El Osman, A. On spontaneous and photoinduced orientational mobility of dye molecules in polymers. Chem. Phys. 1999, 245, 437–462. [Google Scholar] [CrossRef]
- Tiberio, G.; Muccioli, L.; Berardi, R.; Zannoni, C. How Does the Trans-Cis Photoisomerization of Azobenzene Take Place in Organic Solvents? ChemPhysChem 2010, 11, 1018–1028. [Google Scholar] [CrossRef] [PubMed]
- Chigrinov, V.; Pikin, S.; Verevochnikov, A.; Kozenkov, V.; Khazimullin, M.; Ho, J.; Huang, D.D.; Kwok, H.S. Diffusion Model of Photoaligning Azo-Dye Layers. Phys. Rev. E Stat. Nonlinear Soft Matter Phys. 2004, 69, 061713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ilnytskyi, J.; Saphiannikova, M.; Neher, D. Photo-Induced Deformations in Azobenzene-Containing Side-Chain Polymers: Molecular Dynamics Study. Condens. Matter Phys. 2006, 9, 87–94. [Google Scholar] [CrossRef] [Green Version]
- Toshchevikov, V.; Saphiannikova, M.; Heinrich, G. Theory of light-induced deformations in azobenzene polymers: Structure-property relationship. Proc. SPIE 2009, 7487, 74870B. [Google Scholar]
- Saphiannikova, M.; Toshchevikov, V.; Ilnytskyi, J. Photoinduced Deformations in Azobenzene Polymer Films. Nonlinear Opt. Quant. Opt. 2010, 41, 27–57. [Google Scholar]
- Toshchevikov, V.; Saphiannikova, M.; Heinrich, G. Light-induced deformation of azobenzene elastomers: A regular cubic network model. J. Phys. Chem. B 2012, 116, 913–924. [Google Scholar] [CrossRef]
- Toshchevikov, V.P.; Saphiannikova, M.; Heinrich, G. Theory of light-induced deformation of azobenzene elastomers: Influence of network structure. J. Chem. Phys. 2012, 137, 024903. [Google Scholar] [CrossRef]
- Toshchevikov, V.; Saphiannikova, M.; Heinrich, G. Effects of the liquid-crystalline order on the light-induced deformation of azobenzene elastomers. Proc. SPIE 2012, 8545, 854507. [Google Scholar]
- Toshchevikov, V.; Saphiannikova, M. Theory of light-induced deformation of azobenzene elastomers: Effects of the liquid-crystalline interactions and biaxiality. J. Phys. Chem. B 2014, 118, 12297–12309. [Google Scholar] [CrossRef] [PubMed]
- Petrova, T.; Toshchevikov, V.; Saphiannikova, M. Light-induced deformation of polymer networks containing azobenzene chromophores and liquid crystalline mesogens. Soft Matter 2015, 11, 3412–3423. [Google Scholar] [CrossRef] [PubMed]
- Toshchevikov, V.; Petrova, T.; Saphiannikova, M. Light-induced deformation of liquid crystalline polymer networks containing azobenzene chromophores. Proc. SPIE 2015, 9565, 956504. [Google Scholar]
- Toshchevikov, V.; Petrova, T.; Saphiannikova, M. Kinetics of Ordering and Deformation in Photosensitive Azobenzene LC Networks. Polymers 2018, 10, 531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jelken, J.; Henkel, C.; Santer, S. Formation of half-period surface relief gratings in azobenzene containing polymer films. Appl. Phys. B 2020, 126, 149. [Google Scholar] [CrossRef]
- Saad, B. Linearly and circularly polarized laser photoinduced molecular order in azo dye doped polymer films. In Proceedings of the Nanophotonics and Micro/Nano Optics International Conference—Nanop 2016, Paris, France, 7–9 December 2016; Volume 139, p. 00012. [Google Scholar]
- Larson, L.G. The Structure and Rheology of Complex Fluids; Oxford University Press: New York, NY, USA, 1999. [Google Scholar]
- Kang, H.S.; Kim, H.T.; Park, J.K.; Lee, S. Light-Powered Healing of a Wearable Electrical Conductor. Adv. Funct. Mater. 2014, 24, 7273–7283. [Google Scholar] [CrossRef]
- Ambrosio, A.; Camposeo, A.; Carella, A.; Borbone, F.; Pisignano, D.; Roviello, A.; Maddalena, P. Realization of submicrometer structuresby a confocal system on azopolymer filmscontaining photoluminescent chromophores. J. Appl. Phys. 2010, 107, 083110. [Google Scholar] [CrossRef] [Green Version]
- Pawlik, G.; Mitus, A.C.; Miniewicz, A.; Kajzar, F. Kinetics of diffraction gratings formation in a polymer matrix containing azobenzene chromophores: Experiments and Monte Carlo simulations. J. Chem. Phys. 2003, 119, 6789. [Google Scholar] [CrossRef]
- Pawlik, G.; Mitus, A.C.; Miniewicz, A.; Kajzar, F. Monte Carlo simulations of temperature dependence of the kinetics of diffraction gratings formation in a polymer matrix containing azobenzene chromophores. J. Non. Opt. Phys. Mater. 2004, 13, 481–489. [Google Scholar] [CrossRef]
- Metropolis, N.; Rosenbluth, A.W.; Rosenbluth, M.N.; Teller, A.N.; Teller, E. Equation of state calculations by fast computing machines. J. Chem. Phys. 1953, 21, 1087–1092. [Google Scholar] [CrossRef] [Green Version]
- Landau, D.P.; Binder, K. A Guide to Monte Carlo Simulations in Statistical Physics; Cambridge University Press: Cambridge, UK, 2000. [Google Scholar]
- Soto, M.; Esteva, M.; Martinez-Romero, O.; Baez, J.; Elias-Zñiga, A. Modeling Percolation in Polymer Nanocomposites by Stochastic Microstructuring. Materials 2015, 8, 6697–6718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruan, C. “Skin-Core-Skin” Structure of Polymer Crystallization Investigated by Multiscale Simulation. Materials 2018, 11, 610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radosz, W.; Pawlik, G.; Mitus, A.C. Complex Dynamics of Photo-Switchable Guest Molecules in All-Optical Poling Close to the Glass Transition: Kinetic Monte Carlo Modeling. J. Phys. Chem. B 2018, 122, 1756–1765. [Google Scholar] [CrossRef] [PubMed]
- Pawlik, G.; Radosz, W.; Mitus, A.C.; Mysliwiec, J.; Miniewicz, A.; Kajzar, F.; Rau, I. Holographic grating inscription in DR1: DNA-CTMA thin films: The puzzle of time scales. Cent. Eur. J. Chem. 2014, 12, 886–892. [Google Scholar] [CrossRef]
- Pawlik, G.; Mitus, A.C.; Miniewicz, A.; Sobolewska, A.; Kajzar, F. Temperature dependence of the kinetics of diffraction gratings formation in a polymer matrix containing azobenzene chromophores: Monte Carlo simulations and experiment. Mol. Cryst. Liq. Cryst. 2005, 426, 243–252. [Google Scholar] [CrossRef]
- Pawlik, G.; Wrobel, P.; Mitus, A.C.; Kuzyk, M.G. Towards Monte Carlo simulation of the photomechanical effect in polymeric fibers. Proc. SPIE 2011, 8113. [Google Scholar] [CrossRef]
- Pawlik, G.; Miniewicz, A.; Sobolewska, A.; Mitus, A.C. Generic stochastic Monte Carlo model of the photoinduced mass transport in azo-polymers and fine structure of Surface Relief Gratings. Europhys. Lett. 2014, 105, 26002. [Google Scholar] [CrossRef]
- Pawlik, G.; Mitus, A.C.; Mysliwiec, J.; Miniewicz, A.; Grote, J.G. Photochromic dye semi-intercalation into DNA-based polymeric matrix: Computer modeling and experiment. Chem. Phys. Lett. 2010, 484, 321–323. [Google Scholar] [CrossRef]
- Pawlik, G.; Rau, I.; Kajzar, F.; Mitus, A.C. Second-harmonic generation in poled polymers: Pre-poling history paradigm. Opt. Express 2010, 18, 18793. [Google Scholar] [CrossRef] [Green Version]
- Radosz, W.; Orlik, R.; Pawlik, G.; Mitus, A.C. On complex structure of local free volume in bond fluctuation model of polymer matrix. Polymer 2019, 177, 1–9. [Google Scholar] [CrossRef]
- Guo, J.; Andre, P.; Adam, M.; Panyukov, S.; Rubinstein, M.; DeSimone, J.M. Solution Properties of a Fluorinated Alkyl Methacrylate Polymer in Carbon Dioxide. Macromolecules 2006, 39, 3427–3434. [Google Scholar] [CrossRef]
- Kiselev, A.D.; Chigrinov, V.G.; Kwok, H.-S. Kinetics of photoinduced ordering in azo-dye films: Two-state and diffusion models. Phys. Rev. E 2009, 80, 011706. [Google Scholar] [CrossRef] [Green Version]
- Tavarone, R.; Charbonneau, P.; Stark, H. Kinetic Monte Carlo simulations for birefringence relaxation of photo-switchable molecules on a surface. J. Chem. Phys 2016, 144, 104703. [Google Scholar] [CrossRef] [Green Version]
- Pawlik, G.; Mitus, A.C.; Kajzar, F. Monte Carlo simulation of two-photon induced molecular orientation in solid polymer films. Proc. SPIE 2006, 6330, 633003. [Google Scholar]
- Sekkat, Z.; Knoll, W. Photoreactive Organic Thin Films; Academic Press: Cambridge, MA, USA, 2002. [Google Scholar]
- Pawlik, G.; Wysoczanski, T.; Mitus, A.C. Complex Dynamics of Photoinduced Mass Transport and Surface Relief Gratings Formation. Nanomaterials 2019, 9, 352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pawlik, G.; Mitus, A.C. Photoinduced Mass Transport in Azo-Polymers in 2D: Monte Carlo Study of Polarization Effects. Materials 2020, 13, 4724. [Google Scholar] [CrossRef]
- Rau, I.; Kajzar, F. Second harmonic generation and its applications. Nonl. Opt. Quant. Opt. 2008, 38, 99–140. [Google Scholar]
- Bublitz, D.; Helgert, M.; Fleck, B.; Wenke, L.; Hvilstedt, S.; Ramanujam, P.S. Photoinduced Deformation of Azobenzene Polyester Films. Appl. Phys. B 2000, 70, 863–865. [Google Scholar] [CrossRef]
- Priimagi, A.; Shimamura, A.; Kondo, M.; Hiraoka, T.; Kubo, S.; Mamiya, J.I.; Kinoshita, M.; Ikeda, T.; Shishido, A. Location of the azobenzene moieties within the cross-linked liquid-crystalline polymers can dictate the direction of photoinduced bending. ACS Macro. Lett. 2012, 1, 96–99. [Google Scholar] [CrossRef]
- Wang, D.H.; Lee, K.M.; Yu, Z.N.; Koerner, H.; Vaia, R.A.; White, T.J.; Tan, L.S. Photomechanical response of glassy azobenzene polyimide networks. Macromolecules 2011, 44, 3840–3846. [Google Scholar] [CrossRef]
- Han, M.; Morino, S.; Ichimura, K. Factors Affecting In-Plane and Out-of-Plane Photoorientation of Azobenzene Side Chains Attached to Liquid Crystalline Polymers Induced by Irradiation with Linearly Polarized Light. Macromolecules 2000, 33, 6360–6371. [Google Scholar] [CrossRef]
- Buffeteau, T.; Labarthet, F.L.; Sourisseau, C.; Kostromine, S.; Bieringer, T. Biaxial Orientation Induced in a Photoaddressable Azopolymer Thin Film As Evidenced by Polarized UV-Visible, Infrared, and Raman Spectra. Macromolecules 2004, 37, 2880–2889. [Google Scholar] [CrossRef]
- Jung, C.C.; Rosenhauer, R.; Rutloh, M.; Kempe, C.; Stumpe, J. The Generation of Three-Dimensional Anisotropies in Thin Polymer Films by Angular Selective Photoproduct Formation and Annealing. Macromolecules 2005, 38, 4324–4330. [Google Scholar] [CrossRef]
- Ilnytskyi, J.; Neher, D.; Saphiannikova, M. Opposite photo-induced deformations in azobenzene-containing polymers with different molecular architecture: Molecular dynamics study. J. Chem. Phys. 2011, 135, 044901. [Google Scholar] [CrossRef] [PubMed]
- Feller, W. An Introduction to Probability Theory and Its Applications; Wiley & Sons: Hoboken, NJ, USA, 1950. [Google Scholar]
- Hansen, J.-P.; McDonald, I.R. Theory of Simple Liquids; Academic Press: Cambridge, MA, USA, 2006. [Google Scholar]
- Stauffer, D.; Aharony, A. Introduction to Percolation Theory; Taylor & Francis: Abingdon, UK, 2003. [Google Scholar]
- Kim, D.Y.; Tripathy, S.K.; Li, L.; Kumar, J. Laser-induced holographic surface relief gratings on nonlinear optical polymer films. Appl. Phys. Lett. 1995, 66, 1166. [Google Scholar] [CrossRef] [Green Version]
- Rochon, P.; Batalla, E.; Natansohn, A. Optically induced surface gratings on azoaromatic polymer films. Appl. Phys. Lett. 1995, 66, 136. [Google Scholar] [CrossRef]
- Barrett, C.J.; Natansohn, A.L.; Rochon, P.L. Mechanism of optically inscribed high-efficiency diffraction gratings in azo polymer films. J. Phys. Chem. 1996, 100, 8836–8842. [Google Scholar] [CrossRef]
- Yager, K.G.; Barrett, C.J. Photomechanical surface patterning in azo-polymer materials. Macromolecules 2006, 39, 9320–9326. [Google Scholar] [CrossRef]
- Henneberg, O.; Geue, T.; Saphiannikova, M.; Pietsch, U.; Rochon, P.; Natansohn, A. Formation and dynamics of polymer surface relief gratings. Appl. Surf. Sci. 2001, 182, 272–279. [Google Scholar] [CrossRef]
- Ashkin, A.; Dziedzic, J.; Bjorkholm, J.E.; Chu, J.E. Observation of a single-beam gradient force optical trap for dielectric particles. Opt. Lett. 1986, 11, 288–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sumaru, K.; Yamanaka, T.; Fukuda, T.; Matsuda, H. Photoinduced surface relief gratings on azopolymer films: Analysis by a fluid mechanics model. Appl. Phys. Lett. 1999, 75, 1878. [Google Scholar] [CrossRef]
- Sumaru, K.; Fukuda, T.; Kimura, T.; Matsuda, H.; Yamanaka, T. Photoinduced surface relief formation on azopolymer films: A driving force and formed relief profile. J. Appl. Phys. 2002, 91, 3421–3424. [Google Scholar] [CrossRef]
- Boeckmann, M.; Doltsinis, N.L. Towards understanding photomigration: Insights from atomistic simulations of azopolymer films explicitely including light-induced isomerization dynamics. J. Chem. Phys. 2016, 145, 154701. [Google Scholar] [CrossRef] [PubMed]
- Mahimwalla, Z.; Yager, K.G.; Mamiya, J.; Shishido, A.; Priimagi, A.; Barrett, C.J. Azobenzene photomechanics: Prospects and potential applications. Polym. Bull. 2012, 69, 967–1006. [Google Scholar] [CrossRef]
- Sobolewska, A.; Bartkiewicz, S. Surface relief grating in azo-polymer obtained for s-s polarization configuration of the writing beams. Appl. Phys. Lett. 2012, 101, 193301. [Google Scholar] [CrossRef]
- Saphiannikova, M.; Toshchevikov, V.; Ilnytskyi, J. Nanoscopic actuators in light-induced deformation of glassy azo-polymers. Proc. SPIE 2013, 8901, 89010X. [Google Scholar]
- Schab-Balcerzak, E.; Siwy, M.; Kawalec, M.; Sobolewska, A.; Chamera, A.; Miniewicz, A. Synthesis, characterization, and study of photoinduced optical anisotropy in polyimides containing side azobenzene units. J. Phys. Chem. A 2009, 113, 8765–8780. [Google Scholar] [CrossRef] [PubMed]
- Fabbri, F.; Garrot, D.; Lahlil, K.; Boilot, J.P.; Lassailly, Y.; Peretti, J. Evidence of two distinct mechanisms driving photoinduced matter motion in thin films containing azobenzene derivatives. J. Phys. Chem. B 2011, 115, 1363–1367. [Google Scholar] [CrossRef]
- Miniewicz, A.; Kochalska, A.; Mysliwiec, J.; Samoc, A.; Samoc, M.; Grote, J.G. Deoxyribonucleic acid-based photochromic material for fast dynamic holography. Appl. Phys. Lett. 2007, 91, 041118. [Google Scholar] [CrossRef] [Green Version]
- Mitus, A.C.; Pawlik, G.; Kochalska, A.; Mysliwiec, J.; Miniewicz, A.; Kajzar, F. Experimental and Monte Carlo studies of diffraction grating inscription in DNA-based materials. Proc. SPIE 2007, 6646, 664601. [Google Scholar]
- Mitus, A.C.; Pawlik, G.; Kajzar, F.; Grote, J.G. Kinetic Monte Carlo study of diffraction grating recording/erasure in DNA-based azo-dye systems. Proc. SPIE 2008, 7040, 70400A. [Google Scholar]
- Sou, H.; Spaeth, H.; Linhard, V.N.L.; Steckl, A.J. Role of Surfactants in the Interaction of Dye Molecules in Natural DNA Polymers. Langmuir 2009, 25, 11698. [Google Scholar]
- Kawabe, Y.; Wang, L.; Horinouchi, S.; Ogata, N. Amplified Spontaneous Emission from Fluorescent-Dye-Doped DNA-Surfactant Complex Films. Adv. Mater. 2000, 12, 1281–1283. [Google Scholar] [CrossRef]
- Samoc, M.; Samoc, A.; Miniewicz, A.; Markowicz, P.P.; Prasad, P.N.; Grote, J.G. Cubic nonlinear optical effects in deoxyribonucleic acid (DNA) based materials containing chromophores. Proc. SPIE 2007, 6646, 66460A. [Google Scholar]
- Lee, Y.-C. Role of Carbohydrates in Oxidative Modification of Fibrinogen and Other Plasma Proteins. In Photoactive Organic Materials: Science and Application; Kajzar, F., Agranovich, V.M., Lee, C.Y.-C., Eds.; NATO ASI Series High Technolog; Kluwer: Dordrecht, The Netherlands, 1995; Volume 9, pp. 175–181. [Google Scholar]
- Dalton, L.R. Nonlinear Optical Polymeric Materials: From Chromophore Design to Commercial Applications. In Polymers for Photonics Applications I, Advances in Polymer Science; Lee, K.S., Ed.; Springer: Berlin/Heidelberg, Germany, 2002; Volume 158, pp. 1–86. [Google Scholar]
- Dalton, L.R.; Robinson, B.H.; Jen, A.K.-Y.; Steier, W.H.; Nielsen, R. Systematic Development of High Bandwidth, Low Drive Voltage Organic Electrooptic Devices and Their Applications. Opt. Mater. 2003, 21, 19–28. [Google Scholar] [CrossRef]
- Rutkis, M.; Jurgis, A.; Kampars, V.; Vembris, A.; Tokmakovs, A.; Kokars, V. New Figure of Merit for Tailoring Optimal Structure of the Second Order NLO Chromophore for Guest-Host Polymers. Mol. Cryst. Liq. Cryst. 2008, 485, 903–914. [Google Scholar] [CrossRef]
- Harper, A.W.; Sun, S.S.; Dalton, L.R.; Garner, S.M.; Chen, A.; Kalluri, S.; Steier, W.H.; Robinson, B.H. Translating Microscopic Optical Nonlinearity to Macroscopic Optical Nonlinearity: The Role of Chromophore-Chromophore Electrostatic Interactions. J. Opt. Soc. Am. B 1998, 15, 329–337. [Google Scholar] [CrossRef]
- Robinson, B.H.; Dalton, L.R.; Harper, A.W.; Ren, A.S.; Wang, F.; Zhang, C.; Todorova, G.; Lee, M.S.; Aniszfeld, R.; Garner, S.M.; et al. The Molecular and Supramolecular Engineering of Polymeric Electro-optic Materials. Chem. Phys. 1999, 245, 35–50. [Google Scholar] [CrossRef]
- Rau, I.; Armatys, P.; Chollet, P.-A.; Kajzar, F.; Bretonniere, Y.; Andraud, C. Aggregation: A new mechanism of relaxation of polar order in electro-optic polymers. Chem. Phys. Lett. 2007, 442, 32–333. [Google Scholar] [CrossRef]
- Pawlik, G.; Mitus, A.C.; Rau, I.; Kajzar, F. Poling of Electro-Optic Materials: Paradigms and Concepts. Nonlinear Opt. Quant. Opt. 2010, 40, 57–63. [Google Scholar]
- Mitus, A.C.; Pawlik, G.; Rau, I.; Kajzar, F. Computer Simulations of Poled Guest-Host Systems. Nonlinear Opt. Quant. Opt. 2008, 38, 141–162. [Google Scholar]
- Dalton, L.R.; Steier, W.H.; Robinson, B.H.; Zhang, C.; Ren, A.S.; Garner, S.M.; Chen, A.; Londergan, T.M.; Irwin, L.; Carlson, B.; et al. From Molecules to Opto-Chips: Organic Electrooptic Materials. J. Chem. Mater. 1999, 9, 1905–1920. [Google Scholar] [CrossRef]
- Robinson, B.H.; Dalton, L.R. Monte Carlo Statistical Mechanical Simulations of the Competition of Intermolecular Electrostatic and Poling Field Interactions in Defining Macroscopic Electrooptic Activity for Organic Chromophore/Polymer Materials. J. Phys. Chem. A 2000, 104, 4785–4795. [Google Scholar] [CrossRef]
- Dalton, L.R. Rational Design of Organic Electrooptic Materials. J. Phys. Condens. Matter 2003, 15, R897–R934. [Google Scholar] [CrossRef]
- Rommel, H.L.; Robinson, B.H. Orientation of Electro-optic Chromophores under Poling Conditions: A Spheroidal Model. J. Phys. Chem. C 2007, 111, 18765–18777. [Google Scholar] [CrossRef]
- Pawlik, G.; Mitus, A.C.; Rau, I.; Kajzar, F. Monte Carlo Modeling of Chosen Non-Linear Optical Effects for Systems of Guest Molecules in Polymeric and Liquid-Crystal Matrices. Nonlinear Opt. Quant. Opt. 2009, 38, 227–244. [Google Scholar]
- Makowska-Janusik, M.; Reis, H.; Papadopoulos, M.G.; Economou, I.; Zacharopoulos, N.J. Molecular Dynamics Simulations of Electric Field Poled Nonlinear Optical Chromophores Incorporated in a Polymer Matrix. J. Phys. Chem. B 2004, 108, 588–596. [Google Scholar] [CrossRef]
- Leahy-Hoppa, M.R.; Cunningham, P.D.; French, J.A.; Hayden, L.M. Atomistic Molecular Modeling of the Effect of Chromophore Concentration on the Electro-optic Coefficient in Nonlinear Optical Polymers. J. Phys. Chem. A 2006, 110, 5792–5797. [Google Scholar] [CrossRef]
- Reis, H.; Makowska-Janusik, M.; Papadopoulos, M.G. Nonlinear optical susceptibilities of poled guest-host systems: A computational study. J. Phys. Chem. B 2004, 108, 8931–8940. [Google Scholar] [CrossRef]
- Pereverzev, Y.V.; Prezhdo, O.V. Mean-field theory of acentric order of dipolar chromophores in polymeric electro-optic materials. Phys. Rev. E 2000, 62, 8324–8334. [Google Scholar] [CrossRef]
- Pereverzev, Y.V.; Prezhdo, O.V.; Dalton, L.R. Mean-field theory of acentric order of chromophores with displaced dipoles. Chem. Phys. Lett. 2001, 340, 328–335. [Google Scholar] [CrossRef]
- Won-Kook, K.; Hayden, L.M. Fully atomistic modeling of an electric field poled guest-host nonlinear optical polymer. J. Chem. Phys. 1999, 111, 5212–5222. [Google Scholar]
- Tu, Y.; Luo, Y.; Agren, H. Molecular Dynamics Simulations Applied to Electric Field Induced Second Harmonic Generation in Dipolar Chromophore Solutions. J. Phys. Chem. B 2006, 110, 8971–8977. [Google Scholar] [CrossRef] [PubMed]
- Tu, Y.; Zhang, Q.; Agren, H. Electric field poled polymeric nonlinear optical systems: Molecular dynamics simulations of poly(methyl methacrylate) doped with disperse red chromophores. J. Phys. Chem. B 2007, 111, 3591–3598. [Google Scholar] [CrossRef] [PubMed]
- Pawlik, G.; Mitus, A.C.; Rau, I.; Kajzar, F. Monte Carlo kinetic study of chromophore distribution in poled guest-host systems. Proc. SPIE 2008, 6891, 68910A-1. [Google Scholar]
- Pawlik, G.; Wronski, D.; Mitus, A.C.; Rau, I.; Andraud, C.; Kajzar, F. A new mechanism of relaxation in poled guest-host systems: Monte Carlo analysis of aggregation scenario. Proc. SPIE 2007, 6653, 66530J-1. [Google Scholar]
- Bian, S.; Robinson, D.; Kuzyk, M.G. Optically activated cantilever using photomechanical effects in dye-doped polymer fibers. J. Opt. Soc. Am. B 2006, 23, 697–708. [Google Scholar] [CrossRef] [Green Version]
- Barrett, C.J.; Mamiya, J.; Yager, K.G.; Ikeda, T. Photo-mechanical effects in azobenzene-containing soft materials. Soft Matter 2007, 3, 1249–1261. [Google Scholar] [CrossRef]
- Jiang, C.H.; Kuzyk, M.G.; Ding, J.L.; Johns, W.E.; Welker, D.J. Fabrication and mechanical behavior of dye-doped polymer optical fiber. J. Appl. Phys. 2002, 92, 4–12. [Google Scholar] [CrossRef]
- Metzler, R.; Klafter, J. The random walk’s guide to anomalous diffusion: A fractional dynamics approach. Phys. Rep. 2000, 339, 1–77. [Google Scholar] [CrossRef]
- Fulger, D.; Scalas, E.; Germano, G. Monte Carlo simulation of uncoupled continuous-time random walks yielding a stochastic solution of the space-time fractional diffusion equation. Phys. Rev. E 2008, 77, 021122. [Google Scholar] [CrossRef] [Green Version]
- Falco, M.; Simari, C.; Ferrara, C.; Nair, J.R.; Meligrana, G.; Bella, F.; Nicotera, I.; Mustarelli, P.; Winter, M.; Gerbaldi, C. Understanding the effect of UV-Induced cross-linking on the physicochemical properties of highly performing PEO/LiTFSI-based polymer electrolytes. Langmuir 2019, 35, 8210–8219. [Google Scholar] [CrossRef] [PubMed]
- Falco, M.; Castro, L.; Nair, J.R.; Bella, F.; Barde, F.; Meligrana, G.; Gerbaldi, C. UV-cross-linked composite polymer electrolyte for high-rate, ambient temperature lithium batteries. ACS Appl. Energy Mater. 2019, 2, 1600–1607. [Google Scholar] [CrossRef]
- Scalia, A.; Bella, F.; Lamberti, A.; Gerbaldi, C.; Tresso, E. Innovative multipolymer electrolyte membrane designed by oxygen inhibited UV-crosslinking enables solid-state in plane integration of energy conversion and storage devices. Energy 2019, 166, 789–795. [Google Scholar] [CrossRef]
- Zada, M.H.; Kumar, A.; Elmalak, O.; Mechrez, G.; Domb, A.J. Effect of ethylene oxide and gamma (γ-) sterilization on the properties of a PLCL polymer material in balloon implants. ACS Omega 2019, 4, 21319–21326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perez-Calixto, M.; Diaz-Rodriguez, P.; Concheiro, A.; Alvarez-Lorenzo, C.; Burillo, G. Amino-functionalized polymers by gamma radiation and their influence on macrophage polarization. React. Funct. Polym. 2020, 151, 104568. [Google Scholar] [CrossRef]
- Sacco, A.; Bella, F.; De La Pierre, S.; Castellino, M.; Bianco, S.; Bongiovanni, R.; Pirri, C.F. Electrodes/electrolyte interfaces in the presence of a surface-modified photopolymer electrolyte: Application in dye-sensitized solar cells. ChemPhysChem 2015, 16, 960–969. [Google Scholar] [CrossRef] [PubMed]
- Tishkevich, D.I.; Grabchikov, S.S.; Lastovskii, S.B.; Trukhanov, S.V.; Zubar, T.I.; Vasin, D.S.; Trukhanov, A.V.; Kozlovskiy, A.L.; Zdorovets, M.M. Effect of the Synthesis Conditions and Microstructure for Highly Effective Electron Shields Production Based on Bi Coatings. ACS Appl. Energy Mater. 2018, 1, 1695–1702. [Google Scholar] [CrossRef]
- Tishkevich, D.I.; Grabchikov, S.S.; Lastovskii, S.B.; Trukhanov, S.V.; Vasin, D.S.; Zubar, T.I.; Kozlovskiy, A.L.; Zdorovets, M.V.; Sivakov, V.A.; Muradyan, T.R.; et al. Function composites materials for shielding applications: Correlation between phase separation and attenuation properties. J. Alloy. Comp. 2019, 771, 238–245. [Google Scholar] [CrossRef]
- Tishkevich, D.I.; Grabchikov, S.S.; Grabchikova, E.A.; Vasin, D.S.; Lastovskiy, S.B.; Yakushevich, A.S.; Vinnik, D.A.; Zubar, T.I.; Kalagin, I.V.; Mitrofanov, S.V.; et al. Modeling of paths and energy losses of high-energy ions in single-layered and multilayered materials. IOP Conf. Ser. Mater. Sci. Eng. 2020, 848, 012089. [Google Scholar] [CrossRef]






























Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mitus, A.C.; Saphiannikova, M.; Radosz, W.; Toshchevikov, V.; Pawlik, G. Modeling of Nonlinear Optical Phenomena in Host-Guest Systems Using Bond Fluctuation Monte Carlo Model: A Review. Materials 2021, 14, 1454. https://doi.org/10.3390/ma14061454
Mitus AC, Saphiannikova M, Radosz W, Toshchevikov V, Pawlik G. Modeling of Nonlinear Optical Phenomena in Host-Guest Systems Using Bond Fluctuation Monte Carlo Model: A Review. Materials. 2021; 14(6):1454. https://doi.org/10.3390/ma14061454
Chicago/Turabian StyleMitus, Antoni C., Marina Saphiannikova, Wojciech Radosz, Vladimir Toshchevikov, and Grzegorz Pawlik. 2021. "Modeling of Nonlinear Optical Phenomena in Host-Guest Systems Using Bond Fluctuation Monte Carlo Model: A Review" Materials 14, no. 6: 1454. https://doi.org/10.3390/ma14061454
APA StyleMitus, A. C., Saphiannikova, M., Radosz, W., Toshchevikov, V., & Pawlik, G. (2021). Modeling of Nonlinear Optical Phenomena in Host-Guest Systems Using Bond Fluctuation Monte Carlo Model: A Review. Materials, 14(6), 1454. https://doi.org/10.3390/ma14061454