
Characterization of Carbon Nanostructures by Photoelectron Spectroscopies


Abstract
:1. Introduction

1.1. X-ray Photoelectron Spectroscopy
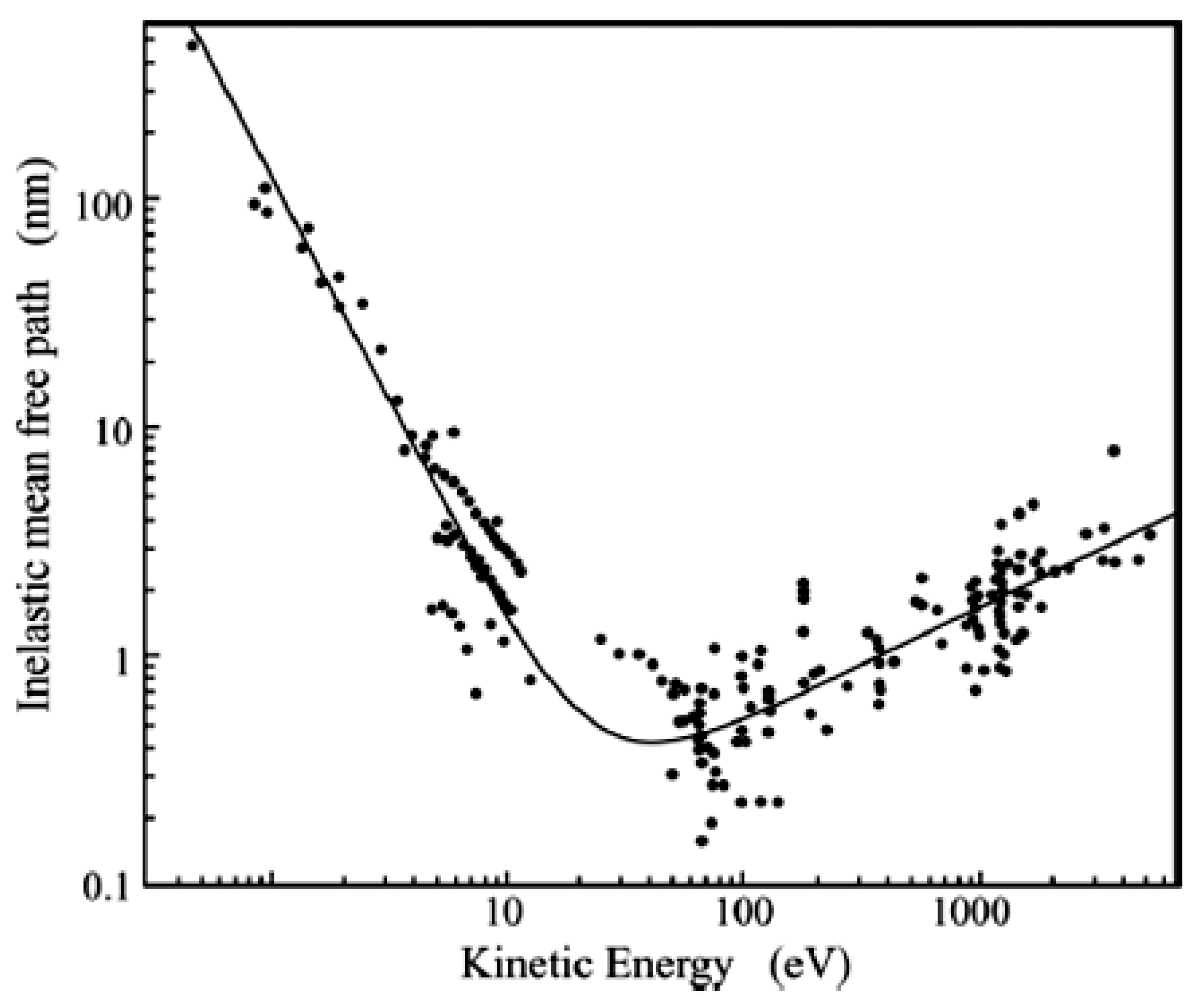
1.2. Surface Sensitivity of the Photoelectron Spectroscopies
1.3. Angle-Resolved XPS
2. Quantum Confinement
3. Limits of the Photoelectron Spectroscopies
4. Characterization of Carbon Nanostructures
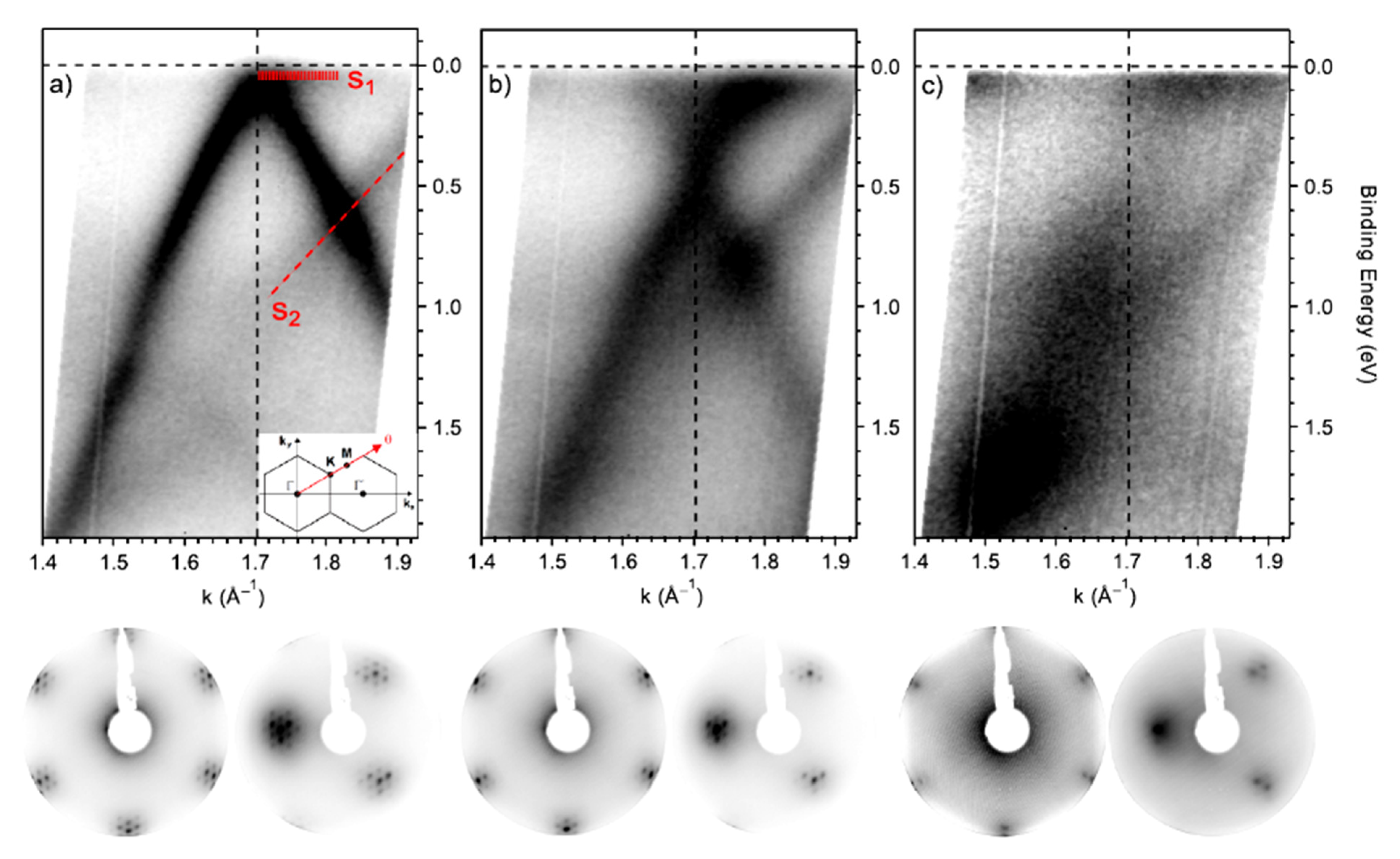
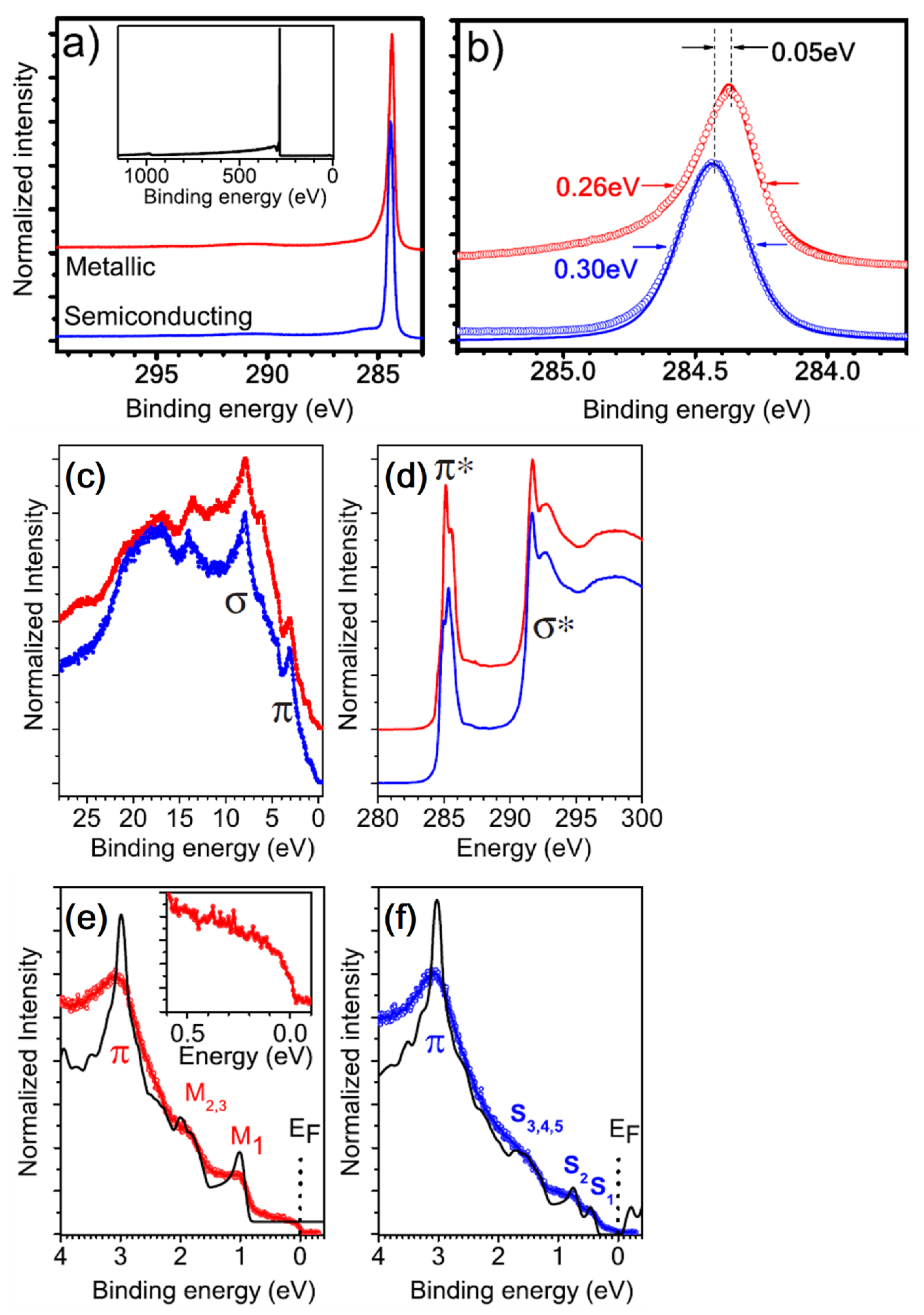
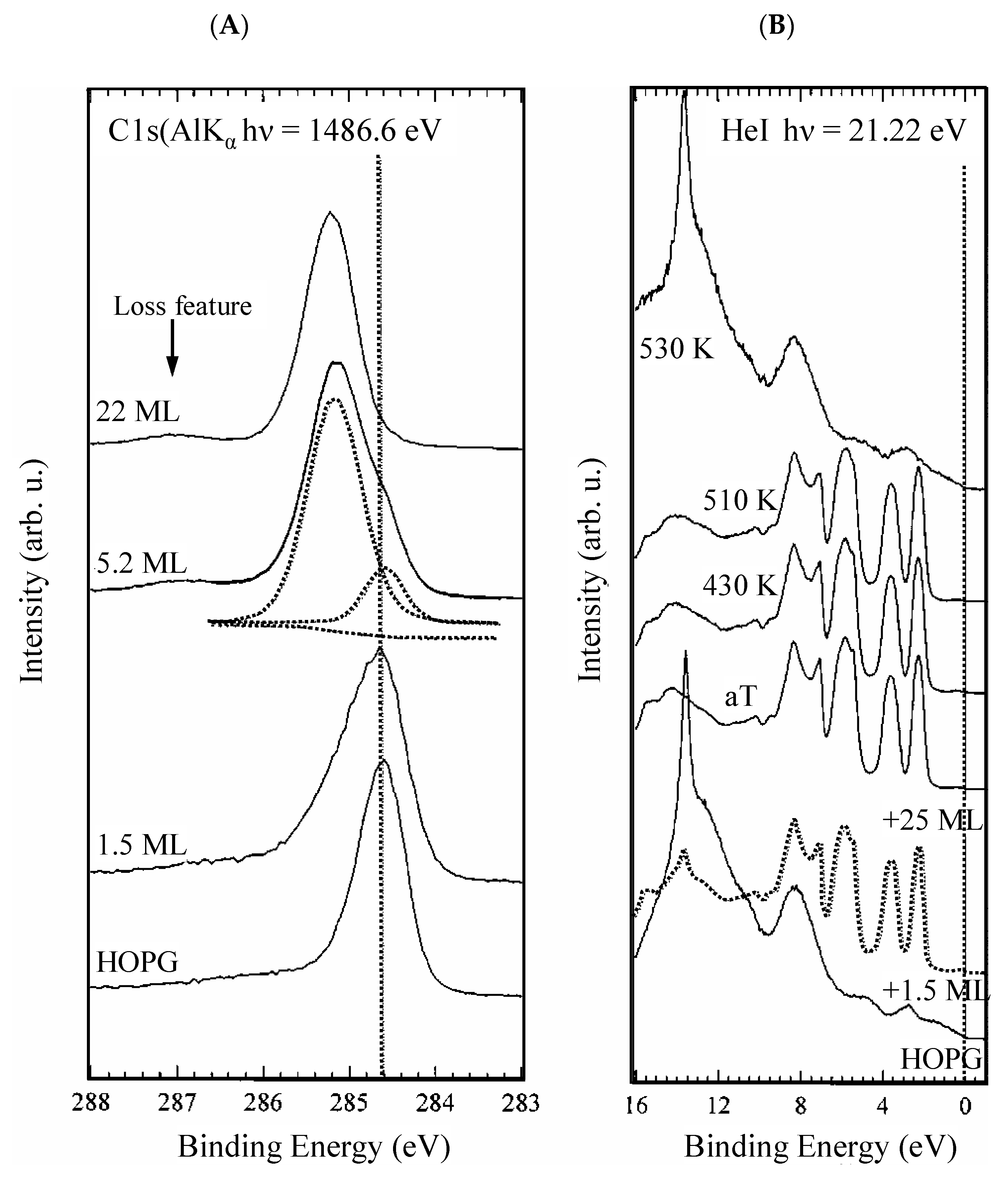
4.1. The Analysis of the Electronic Structure of Graphene
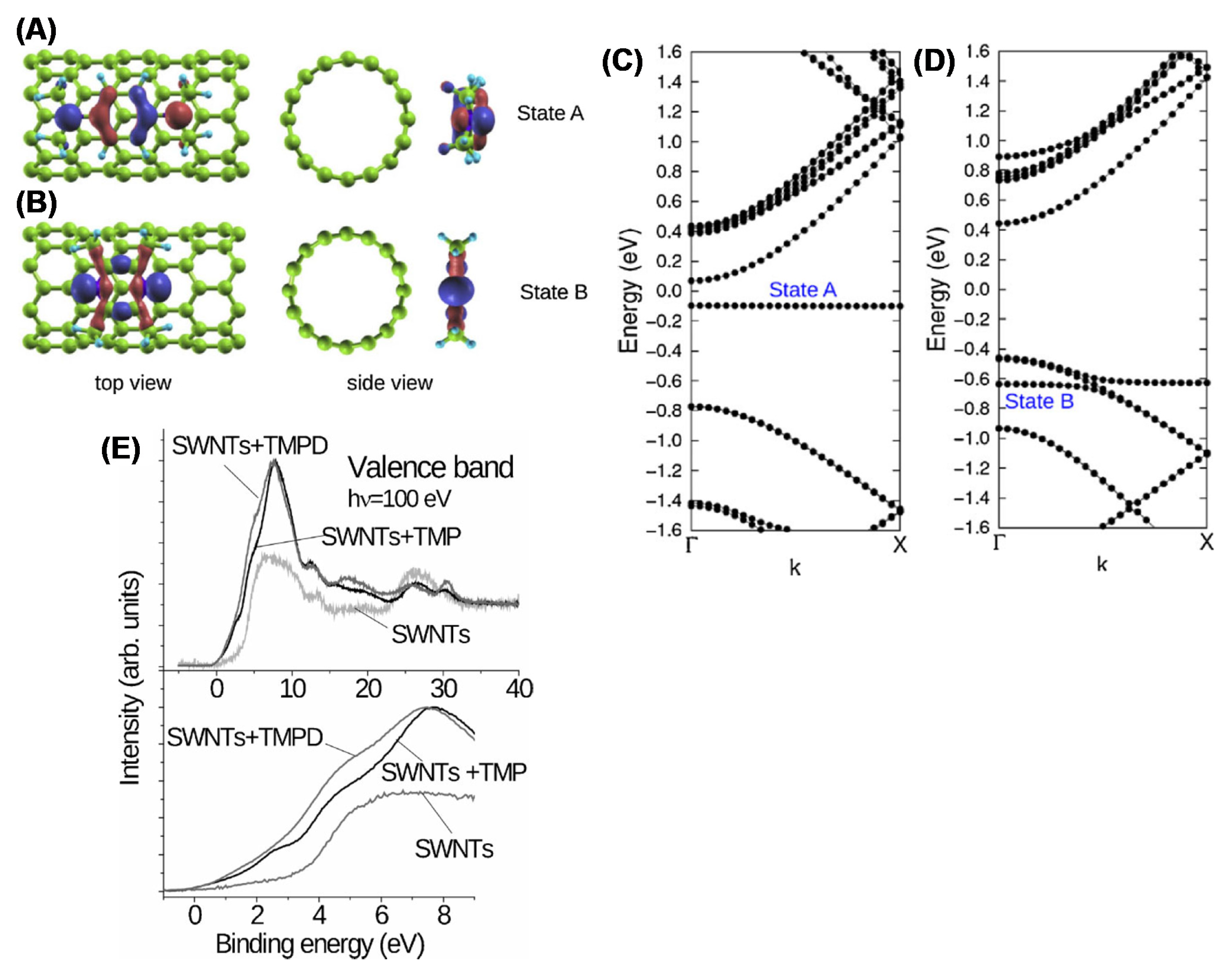
4.2. Characterization of Carbon Nanotubes
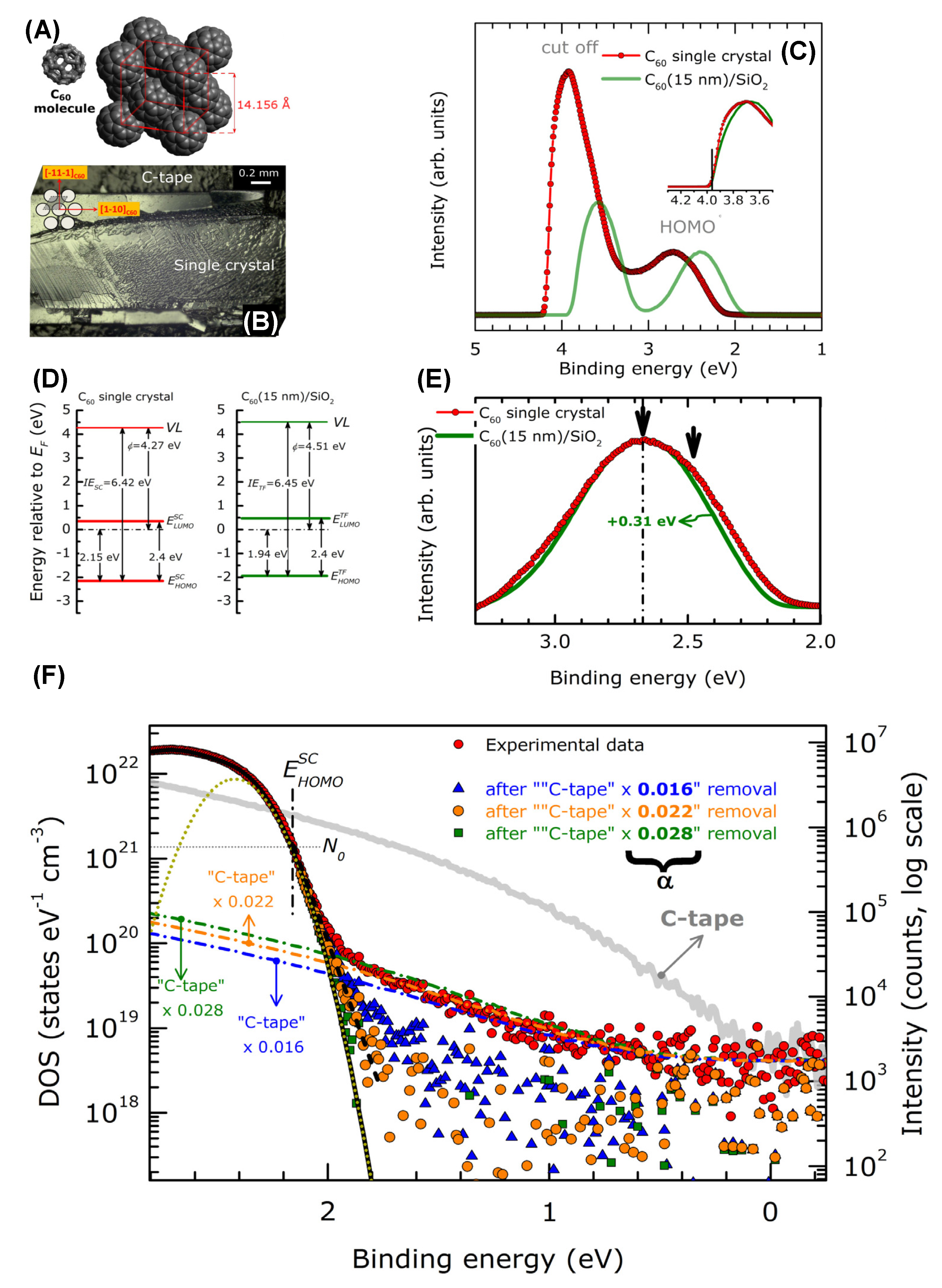
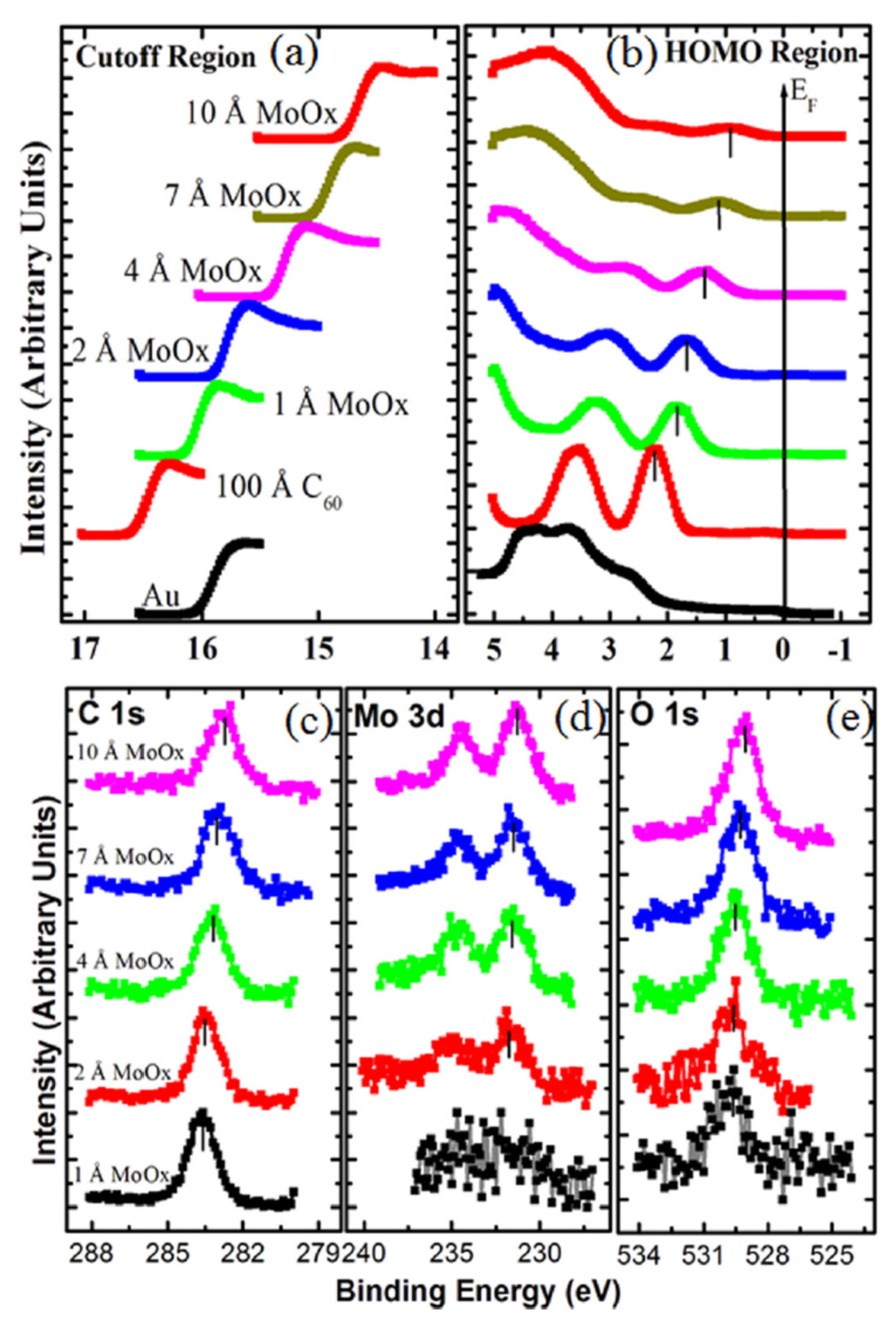
4.3. Quantum Confinement Effects in Fullerenes
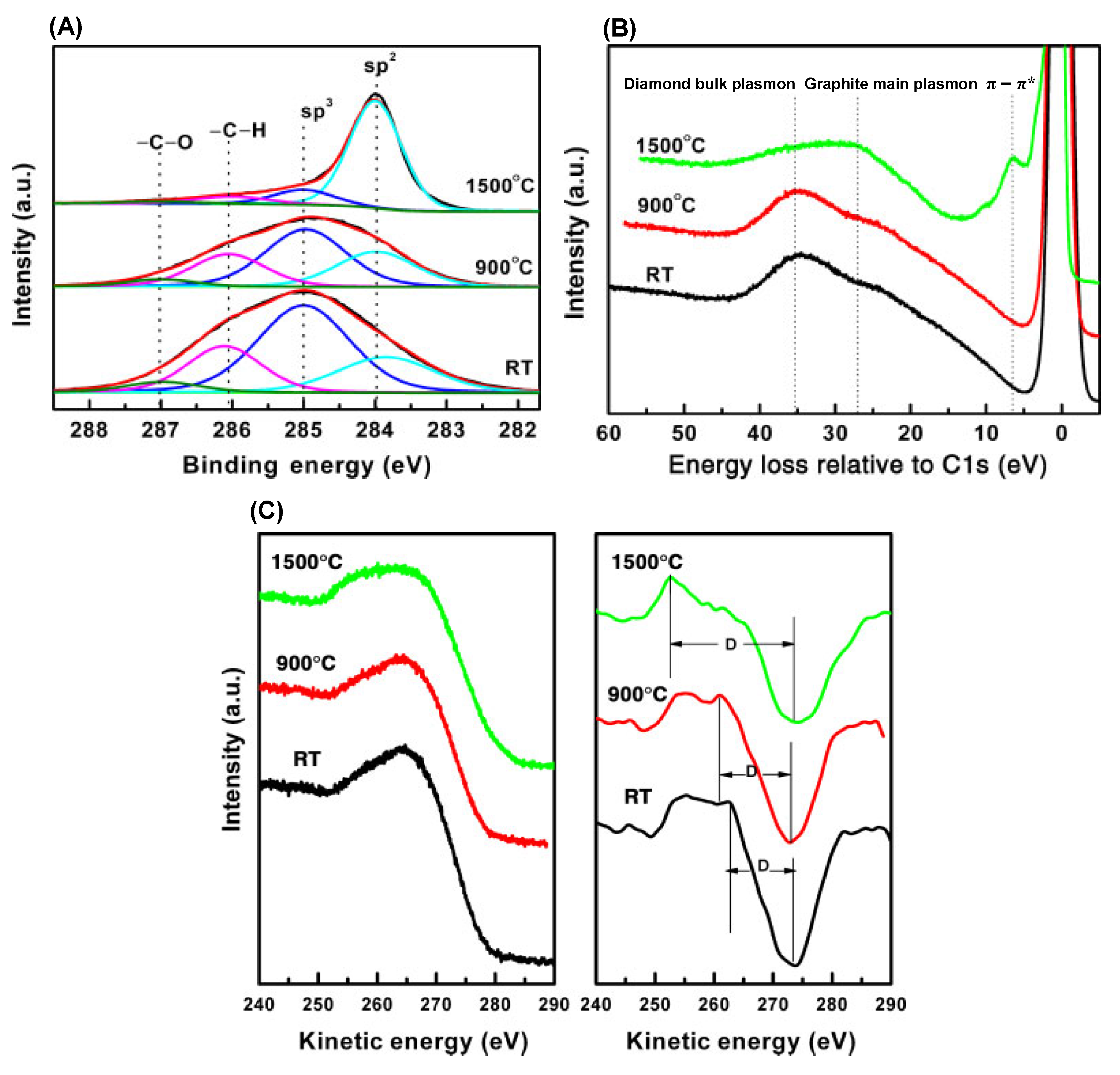
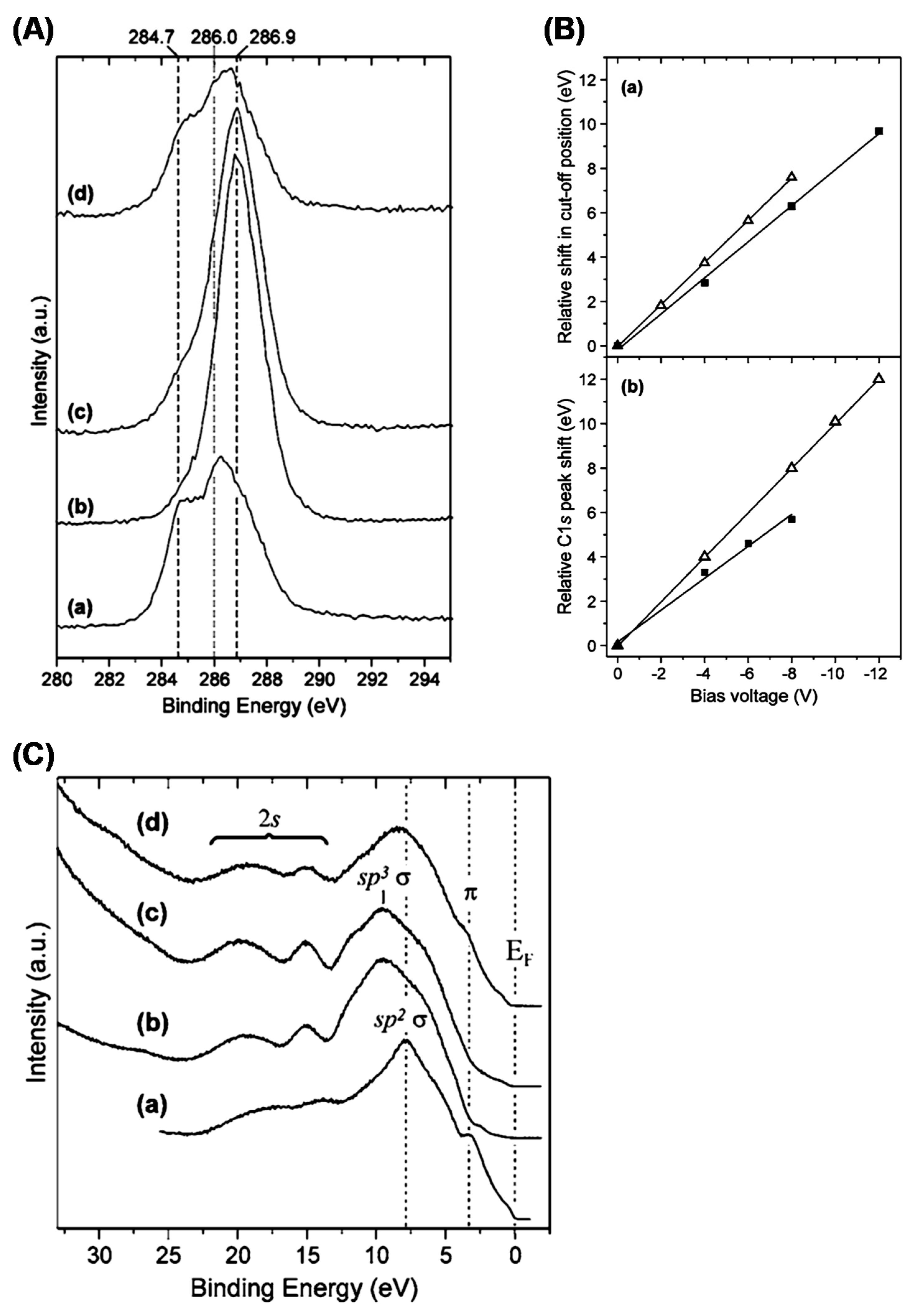
4.4. Surface Chemistry and Properties of Nanodiamonds
4.5. Carbon Dots
4.6. Carbon Nanofibers
4.7. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mohan, V.B.; Lau, K.-T.; Hui, D.; Bhattacharyya, B. Graphene-based materials and their composites: A review on production, applications and product limitations. Compos. Part B Eng. 2018, 142, 200–220. [Google Scholar] [CrossRef]
- Backes, C.; Abdelkader, A.M.; Alonso, C.; Andrieux-Ledier, A.; Arenal, R.; Azpeitia, J.; Balakrishnan, N.; Banszerus, L.; Barjon, J.; Bartali, R.; et al. Production and processing of graphene and related materials. 2D Mater. 2020, 7, 022001. [Google Scholar] [CrossRef]
- Mochalin, V.N.; Shenderova, O.; Ho, D.; Gogotsi, Y. The properties and applications of nanodiamonds. Nat. Nanotechnol. 2011, 7, 11–23. [Google Scholar] [CrossRef] [PubMed]
- Aharonovich, I.; Neu, E. Diamond Nanophotonics. Adv. Opt. Mater. 2014, 1408, 5451. [Google Scholar] [CrossRef]
- Georgakilas, V.; Perman, J.A.; Tucek, J.; Zboril, R. Broad Family of Carbon Nanoallotropes: Classification, Chemistry, and Applications of Fullerenes, Carbon Dots, Nanotubes, Graphene, Nanodiamonds, and Combined Superstructures. Chem. Rev. 2015, 115, 4744–4822. [Google Scholar] [CrossRef]
- Jariwala, D.; Sangwan, V.K.; Lauhon, L.J.; Marks, T.J.; Hersam, M.C. Carbon nanomaterials for electronics, optoelectronics, photovoltaics, and sensing. Chem. Soc. Rev. 2013, 42, 2824–2860. [Google Scholar] [CrossRef] [Green Version]
- Ruppender, H.J.; Grunze, M.; Kong, C.W.; Wilmers, M. In situ X-ray photoelectron spectroscopy of surfaces at pressures up to 1 mbar. Surf. Interface Anal. 1990, 15, 245–253. [Google Scholar] [CrossRef]
- Trotochaud, L.; Head, A.R.; Karslglu, O.; Kyhl, L.; Bluhm, H. Ambient pressure XPS Practical considerations and experimental frontiers. J. Phys. Condens. Matter 2017, 29, 053002. [Google Scholar] [CrossRef]
- Moulde, J.F. The Impact of the Scanning XPS Microprobe on Industrial Applications of X-ray Photoelectron Spectroscopy. J. Electron Spectrosc. Relat. Phenom. 2019, 231, 43–49. [Google Scholar] [CrossRef]
- Roberts, A.J.; Moffitt, C.E. Trends in XPS instrumentation for industrial surface analysis and materials characterization. J. Electron Spectrosc. Relat. Phenom. 2019, 231, 68–74. [Google Scholar] [CrossRef]
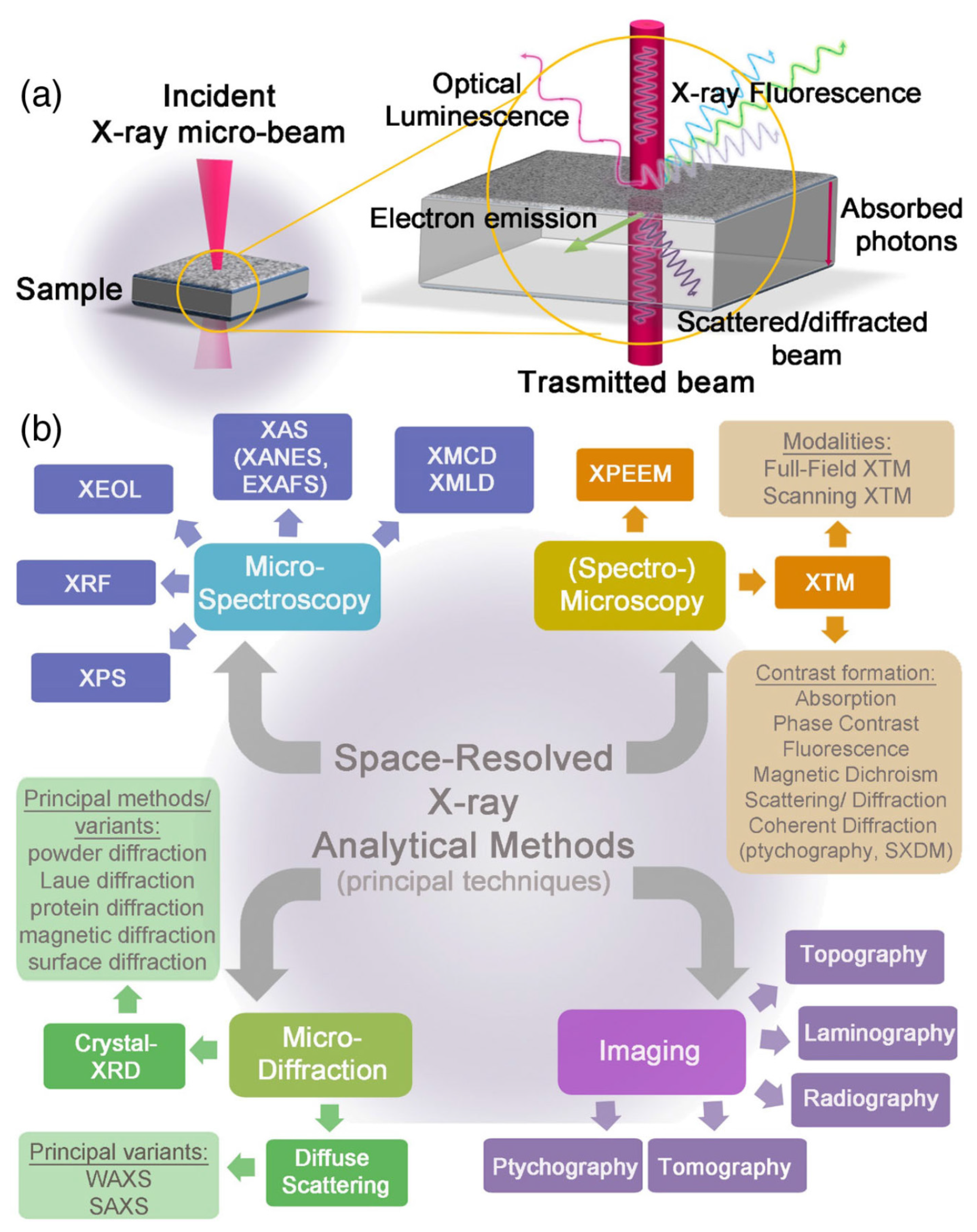
- Mino, L.; Borfecchia, E.; Segura-Ruiz, J.; Giannini, C.; Martinez-Criado, G.; Lamberti, C. Materials characterization by synchrotron x-ray microprobes and nanoprobes. Rev. Mod. Phys. 2018, 90, 025007. [Google Scholar] [CrossRef]
- Egelhoff, W.F. Core-level binding-energy shifts at surfaces and in solids. Surf. Sci. Rep. 1987, 6, 253–451. [Google Scholar] [CrossRef]
- Schattke, W.; van Hove, M.A. Solid-State Photoemission and Related Methods: Theory and Experiment; Wiley: Weinheim, Germany, 2003. [Google Scholar]
- Miron, C.; Morin, P. Handbook of High-Resolution Spectroscopy; John Wiley & Sons, Ltd.: New York, NY, USA, 2011. [Google Scholar]
- Susi, T.; Pichler, T.; Ayala, P. X-ray photoelectron spectroscopy of graphitic carbon nanomaterials doped with heteroatoms. Beilstein J. Nanotechnol. 2015, 6, 177–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weiland, C.; Rumaniz, A.K.; Pianetta, P.; Woicik, J.C. Recent applications of hard X-ray photoelectron spectroscopy. J. Vac. Sci. Technol. A 2016, 34, 030801. [Google Scholar] [CrossRef]
- Nordling, C.; Sokolowski, E.; Siegbahn, K. Precision Method for Obtaining Absolute Values of Atomic Binding Energies. Phys. Rev. 1957, 105, 1676. [Google Scholar] [CrossRef]
- Fadley, C.S. X-ray photoelectron spectroscopy: From origins to future directions. Nucl. Instrum. Methods Phys. Res. A 2009, 601, 8–39. [Google Scholar] [CrossRef]
- Hill, J.P. Ultraviolet Photoelectron Spectroscopy. In Characterization of Materials; Kaufmann, E.N., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2003. [Google Scholar]
- Rabalais, J.W. Principles of Ultraviolet Photoelectron Spectroscopy; Wiley-Interscience: New York, NY, USA, 1977. [Google Scholar]
- Powell, C.J.; Seah, M.P. Precision, accuracy, and uncertainty in quantitative surface analyses by Auger-electron spectroscopy and X-ray photoelectron spectroscopy. J. Vac. Sci. Technol. A 1990, 8, 735. [Google Scholar] [CrossRef]
- Tilinin, I.S.; Jablonski, A.; Werner, W.S.M. Quantitative surface analysis by Auger and X-ray photoelectron spectroscopy. Prog. Surf. Sci. 1996, 52, 193–335. [Google Scholar] [CrossRef]
- Seah, M.P. Quantification of AES and XPS. In Practical Surface Analysis Vol. 1 Auger and X-ray Photoelectron Spectroscopy; Briggs, D., Seah, M.P., Eds.; J. Wiley & Sons: Chichester, UK, 1990; pp. 206–251. [Google Scholar]
- Fadley, C.S. X-ray photoelectron spectroscopy: Progress and perspectives. J. Electron Spectrosc. Relat. Phenom. 2010, 178–179, 2–32. [Google Scholar] [CrossRef]
- Reilman, R.F.; Msezane, A.; Manson, S.T. Relative intensities in photoelectron spectroscopy of atoms and molecules. J. Electron Spectrosc. Relat. Phenom. 1976, 8, 389–394. [Google Scholar] [CrossRef]
- Scofield, J.H. Hartree-Slater subshell photoionization cross-sections at 1254 and 1487 eV. J. Electron Spectrosc. Relat. Phenom. 1976, 8, 129–137. [Google Scholar] [CrossRef]
- Wagner, C.D.; Davis, L.E.; Zeller, M.V.; Taylor, J.A.; Raymond, R.H.; Gale, L.H. Empirical atomic sensitivity factors for quantitative analysis by electron spectroscopy for chemical analysis. Surf. Interface Anal. 1981, 3, 211–225. [Google Scholar] [CrossRef]
- Brundle, C.R.; Crist, B.V. X-ray photoelectron spectroscopy: A perspective on quantitation accuracy for composition analysis of homogeneous materials. J. Vac. Sci. Technol. A 2020, 38, 041001. [Google Scholar] [CrossRef]
- Briggs, D. Surface Analysis of Polymers by XPS and Static SIMS; Cambridge University Press: Cambridge, UK, 1998. [Google Scholar]
- Seah, M.P.; Dench, W.A. Quantitative electron spectroscopy of surfaces: A standard data base for electron inelastic mean free paths in solids. Surf. Interface Anal. 1979, 1, 2–11. [Google Scholar] [CrossRef]
- Mason, M.G. Electronic structure of supported small metal clusters. Phys. Rev. B 1983, 27, 748. [Google Scholar] [CrossRef]
- Watson, R.E.; Perlman, L. X-ray photoelectron spectroscopy application to metals and alloys. In Structure and Bonding; Springer: Berlin/Heidelberg, Germany, 1975; pp. 83–112. [Google Scholar]
- Wertheim, G.K.; DiCenzo, S.B.; Youngquist, S.E. Unit Charge on Supported Gold Clusters in Photoemission Final State. Phys. Rev. Lett. 1983, 51, 2310. [Google Scholar] [CrossRef]
- Wertheim, G.K.; DiCenzo, S.B. Cluster growth and core-electron binding energies in supported metal clusters. Phys. Rev. B 1988, 37, 844. [Google Scholar] [CrossRef]
- Calliari, L.; Speranza, G.; Minati, L.; Micheli, V.; Baranov, A.; Fanchenko, S. Composition and structure of a-C: Au nanocomposites obtained by physical vapour deposition. Appl. Surf. Sci. 2008, 255, 2214–2218. [Google Scholar] [CrossRef]
- Videnovic, I.; Oelhafen, P. Photoelectron spectroscopy study of metallic nanocluster arrangement at the surface of reactively sputtered amorphous hydrogenated carbon. J. Appl. Phys. 2005, 97, 074308. [Google Scholar] [CrossRef]
- Takahiro, K.; Oizumi, S.; Terai, A.; Kawatsura, K.; Tsuchiya, B.; Nagata, S.; Yamamoto, S.; Naramoto, H.; Narumi, K.; Sasase, M. Core level and valence band photoemission spectra of Au clusters embedded in carbon. J. Appl. Phys. 2006, 100, 084325. [Google Scholar] [CrossRef]
- Minati, L.; Speranza, G.; Calliari, L.; Micheli, V.; Baranov, A.; Fanchenko, S. The Influence of Metal Nanoparticle Size Distribution in Photoelectron Spectroscopy. J. Phys. Chem. A 2008, 112, 7856–7861. [Google Scholar] [CrossRef] [PubMed]
- Zorn, G.; Dave, S.R.; Gao, X.; Castner, D.G. Method for Determining the Elemental Composition and Distribution in Semiconductor Core-Shell Quantum Dots. Anal. Chem. 2011, 83, 866–873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kono, S.; Kageura, T.; Hayashi, Y.; Ri, S.-G.; Teraji, T.; Takeuchi, D.; Ogura, M.; Kodama, H.; Sawabe, A.; Inaba, M.; et al. Carbon 1s X-ray photoelectron spectra of realistic samples of hydrogen-terminated and oxygen-terminated CVD diamond (111) and (001). Diam. Relat. Mater. 2019, 93, 105–130. [Google Scholar] [CrossRef]
- Powell, C.J.; Jackson, A. NIST Electron Inelastic-Mean-Free-Path Database; Version 11; NIST: Gaithersburg, MD, USA, 2000. [Google Scholar]
- Brzhezinskaya, M.M.; Muradyan, V.E.; Vinogradov, N.A.; Preobrajenski, A.B.; Gudat, W.; Vinogradov, A.S. Electronic structure of fluorinated multiwalled carbon nanotubes studied using X-ray absorption and photoelectron spectroscopy. Phys. Rev. B 2009, 79, 155439. [Google Scholar] [CrossRef]
- Baer, D.R. Guide to making XPS measurements on nanoparticles. J. Vac. Sci. Technol. A 2020, 38, 031201. [Google Scholar] [CrossRef] [Green Version]
- Briggs, D.; Seah, M.P. Practical Surface Analysis; J. Wiley & Sons: Chichester, UK, 1990. [Google Scholar]
- Briggs, D.; Grant, J. (Eds.) Surface Analysis by Auger and X-ray Photoelectron Spectroscopy; IMP Publications: London, UK, 2003. [Google Scholar]
- Briggs, D. Handbook of X-ray and Ultraviolet Photoelectron Spectroscopy; Heyden: London, UK; Philadelphia, PA, USA, 1977. [Google Scholar]
- Hufner, S. Photoelectron Spectroscopy: Principles and Applications, 3rd ed.; Springer: Berlin/Heidelberg, Germany, 2003. [Google Scholar]
- Kalha, C.; Fernando, N.K.; Bhatt, P.; Johansson, F.O.L.; Lindblad, A.; Rensmo, H.; Zendejas-Medina, L.; Lindblad, R.; Sio, S.; Jeurgens, L.P.H.; et al. Hard X-ray photoelectron spectroscopy: A snapshot of the state-of-the-art in 2020. J. Phys. Condens. Matter 2021, 33, 233001. [Google Scholar] [CrossRef]
- Weinhardt, L.; Steininger, R.; Kreikemeyer-Lorenzo, D.; Mangold, S.; Hauschild, D.; Batchelor, D.; Spangenberg, T.; Heske, C. X-SPEC: A 70 eV to 15 keV undulator beamline for X-ray and electron spectroscopies. J. Synchrotron Radiat. 2021, 28, 609–617. [Google Scholar] [CrossRef]
- Baer, D.R.; Engelhard, M.H. XPS analysis of nanostructured materials and biological surfaces. J. Electron Spectrosc. Relat. Phenom. 2010, 178–179, 415–432. [Google Scholar] [CrossRef]
- Doniach, S.; Sunjic, P. Many-electron singularity in X-ray photoemission and X-ray line spectra from metals. J. Phys. C Solid State Phys. 1970, 3, 285–291. [Google Scholar] [CrossRef]
- Speranza, G.; Minati, L. The surface and bulk core lines in crystalline and disordered polycrystalline graphite. Surf. Sci. 2006, 600, 4438–4444. [Google Scholar] [CrossRef]
- Speranza, G.; Miinati, L.; Anderle, M. The C1s core line in irradiated graphite. J. Appl. Phys. 2007, 102, 043504. [Google Scholar] [CrossRef]
- Prince, K.C.; Ulrych, I.; Peloi, M.; Ressel, B.; Chab, V.; Crotti, C.; Comicioli, C. Core-level photoemission from graphite. Phys. Rev. B 2000, 62, 6866–6868. [Google Scholar] [CrossRef]
- Lesiak, B.; Zemek, J.; Houdkova, J.; Kromka, A.; Jozwik, A. Electron spectra Line shape analysis of Highly oriented Pyrolytic Graphite and Nanocrystalline diamond. Anal. Sci. 2010, 26, 217–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balasubramanian, T.; Andersen, J.N.; Walladen, L. Surface-bulk core-level splitting in graphite. Phys. Rev. B 2001, 64, 205420. [Google Scholar] [CrossRef]
- Smith, R.A.P.; Armstrong, C.W.; Smith, G.C.; Weightman, P. Observation of a surface chemical shift in carbon 1s core-level photoemission from highly oriented pyrolytic graphite. Phys. Rev. B 2002, 66, 245409. [Google Scholar] [CrossRef]
- Yang, D.-Q.; Sacher, E. Carbon 1s X-ray Photoemission Line Shape Analysis of Highly Oriented Pyrolytic Graphite: The Influence of Structural Damage on Peak Asymmetry. Langmuir 2006, 22, 860–862. [Google Scholar] [CrossRef]
- Geim, A.K.; Novoselov, K.S. The rise of graphene. Nat. Mater. 2007, 6, 183–191. [Google Scholar] [CrossRef]
- Lee, C.; Wei, X.; Kysar, J.W.; Hone, J. Measurement of the Elastic Properties and Intrinsic Strength of Monolayer Graphene. Science 2008, 321, 385–388. [Google Scholar] [CrossRef]
- Bolotin, K.I.; Sikes, K.J.; Jiang, Z.; Klima, M.; Fudenberg, G.; Hone, J.; Kim, P.; Stormer, H.L. Ultrahigh electron mobility in suspended graphene. Solid State Commun. 2008, 146, 351–355. [Google Scholar] [CrossRef] [Green Version]
- Züttel, A.; Sudan, P.; Mauron, P.; Wenger, P. Model for the hydrogen adsorption on carbon nanostructures. Appl. Phys. A 2004, 78, 941–946. [Google Scholar] [CrossRef]
- Nair, R.R.; Blake, P.; Grigorenko, A.N.; Novoselov, K.S.; Booth, T.J.; Stauber, T.; Peres, N.M.R.; Geim, A.K. Fine Structure Constant Defines Visual Transparency of Graphene. Science 2008, 320, 1308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, H.; Chen, S.; Wee, A.T.S.; Chen, W. Chap 1-Epitaxial growth of graphene on silicon carbide (SiC). In Graphene-Properties, Preparation, Characterization and Devices; Woodhead Publishing Ltd.: Sawston, UK, 2014; pp. 3–26. [Google Scholar]
- Wang, J.-B.; Ren, Z.; Hou, Y.; Yan, X.-L.; Liu, P.-Z.; Hua, Z.; Zhang, H.X.; Guo, J.J. A review of graphene synthesis at low temperatures by CVD methods. New Carbon Mater. 2020, 35, 193–208. [Google Scholar] [CrossRef]
- Novoselov, K.S.; Geim, A.K.; Morozov, S.V.; Jiang, D.; Zhang, Y.; Dubonos, S.V.; Grigorieva, I.V.; Firsov, A.A. Electric Field Effect in Atomically Thin Carbon Films. Science 2004, 306, 666–669. [Google Scholar] [CrossRef] [Green Version]
- Hernandez, Y.; Nicolosi, V.; Lotya, M.; Blighe, F.M.; Sun, Z.; De, S.; McGovern, I.T.; Holland, B.; Byrne, M.; Gun’Ko, Y.K.; et al. High-yield production of graphene by liquid-phase exfoliation of graphite. Nat. Nanotechnol. 2008, 3, 563–568. [Google Scholar] [CrossRef] [Green Version]
- Knieke, C.; Berger, A.; Voigt, M.; Taylor, R.N.K.; Röhrl, J.; Peukert, W. Scalable production of graphene sheets by mechanical delamination. Carbon 2010, 48, 3196–3204. [Google Scholar] [CrossRef]
- Lv, Y.; Yu, L.; Jiang, C.; Chen, S.; Nie, Z. Synthesis of graphene nanosheet powder with layer number control via a soluble salt-assisted route. RSC Adv. 2014, 4, 13350–13354. [Google Scholar] [CrossRef]
- Hummers, W.B.; Offeman, R.E. Preparation of graphitic oxide. J. Am. Chem. Soc. 1958, 80, 1339. [Google Scholar] [CrossRef]
- Guex, L.G.; Sacchi, B.; Peuvot, K.F.; Andersson, R.L.; Pourrahimi, A.M.; Strom, V.; Farris, S.; Olsson, R.T. Experimental review: Chemical reduction of graphene oxide (GO) to reduced graphene oxide (rGO) by aqueous chemistry. Nanoscale 2017, 9, 9562–9571. [Google Scholar] [CrossRef] [Green Version]
- Saleem, H.; Haneef, M.; Abbasi, H.Y. Synthesis route of reduced graphene oxide via thermal reduction of chemically exfoliated graphene oxide. Mater. Chem. Phys. 2018, 204, 1–7. [Google Scholar] [CrossRef]
- Uz, M.; Jackson, K.; Donta, M.S.; Jung, J.; Lentner, M.T.; Hondred, J.A.; Claussen, J.C.; Mallapragada, S.K. Fabrication of High-resolution Graphene-based Flexible Electronics via Polymer Casting. Sci. Rep. 2019, 9, 10595. [Google Scholar] [CrossRef]
- Deng, D.; Novoselov, K.S.; Fu, Q.; Zheng, N.; Tian, Z.; Bao, X. Catalysis with two-dimensional materials and their heterostructures. Nat. Nanotechnol. 2016, 11, 218. [Google Scholar] [CrossRef] [PubMed]
- Nag, A.; Mitra, A.; Chandra, M. Graphene and its sensor-based applications: A review. Sens. Actuators A 2018, 270, 177–194. [Google Scholar] [CrossRef]
- Peña-Bahamond, J.; Nguyen, H.N.; Fanourakis, S.K.; Rodrigues, D.F. Recent advances in graphene-based biosensor technology with applications in life sciences. J. Nanobiotechnol. 2018, 16, 75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer, J.C. Chapter 5: Transmission electron microscopy (TEM) of graphene. In Graphene Properties, Preparation, Characterisation and Devices; Woodhead Publishing Ltd.: Sawston, UK, 2014; pp. 101–123. [Google Scholar]
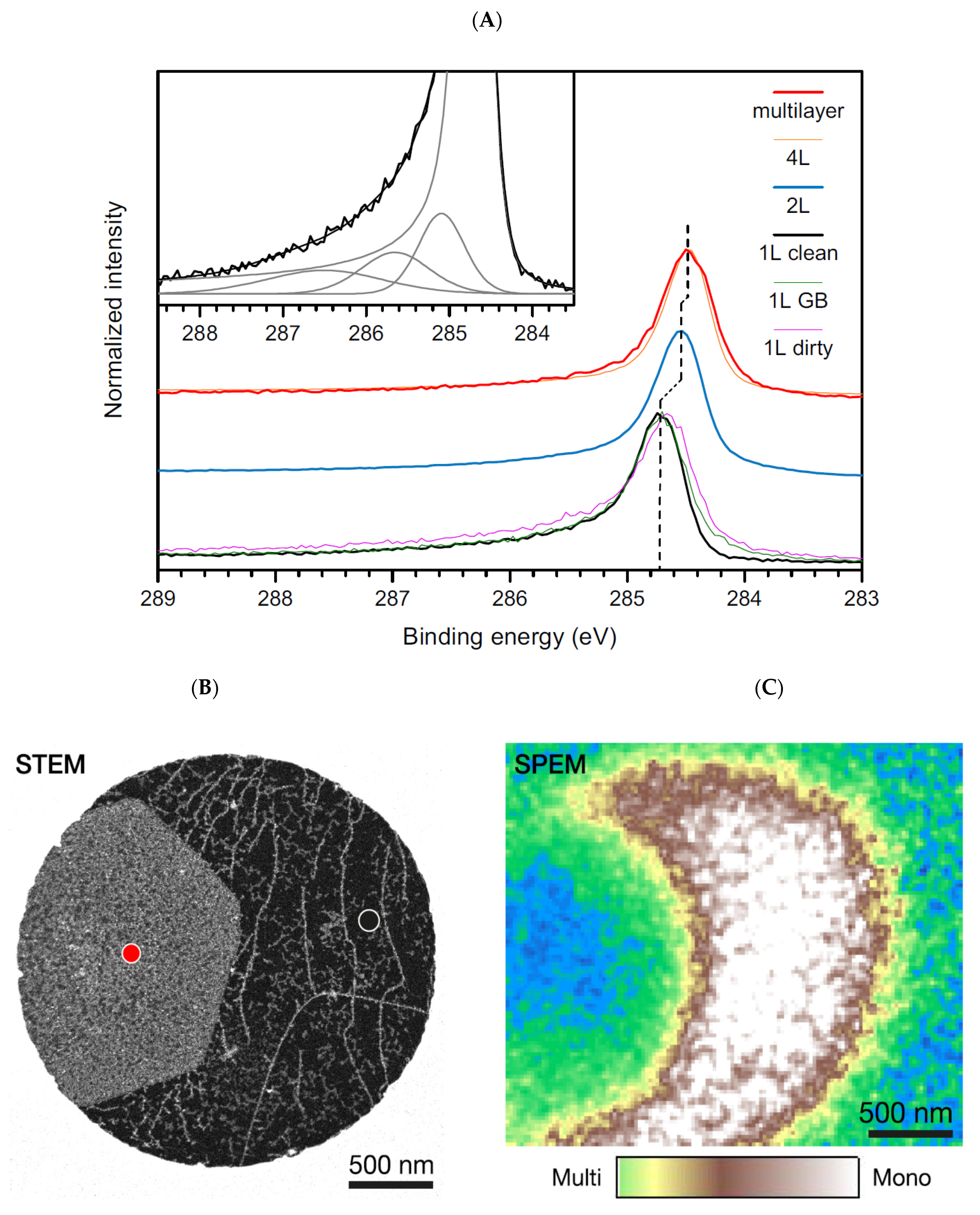
- Toma, S.; Scardamaglia, M.; Mustonen, K.; Tripathi, M.; Mittelberger, M.; Al-Hada, M.; Amati, M.; Sezen, H.; Zeller, P.; Larsen, A.H.; et al. Intrinsic core level photoemission of suspended monolayer graphene. Phys. Rev. Mater. 2018, 2, 074005. [Google Scholar]
- Hibino, H.; Kageshima, H.; Kotsugi, M.; Meda, F.; Guo, F.-Z.; Watanabe, Y. Dependence of electronic properties of epitaxial few-layer graphene on the number of layers investigated by photoelectron emission microscopy. Phys. Rev. B 2009, 79, 125437. [Google Scholar] [CrossRef]
- Gupta, B.; Notarianni, M.; Mishra, N.; Shafiei, M.; Iacopi, F.; Motta, N. Evolution of epitaxial graphene layers on 3C SiC/Si(111) as a function of annealing temperature in UHV. Carbon 2014, 68, 563–572. [Google Scholar] [CrossRef] [Green Version]
- Niesner, D.; Fauster, T. Image-potential states and work function of graphene. J. Phys. Condens. Matter 2014, 26, 393001. [Google Scholar] [CrossRef] [Green Version]
- Kralj, M.; Pletikosic, I.; Petrovic, M.; Pervan, P.; Milun, M.; N’Diaye, A.T.; Busse, C.; Michely, T.; Fujii, J.; Vobornik, I. Graphene on Ir(111) characterized by angle-resolved photoemission. Phys. Rev. B 2011, 84, 075427. [Google Scholar] [CrossRef] [Green Version]
- Tatti, R.; Aversa, L.; Verrucchi, R.; Cavaliere, E.; Garberoglio, G.; Pugno, N.M.; Speranza, G.; Taioli, S. Synthesis of single layer graphene on Cu(111) by C60 supersonic molecular beam epitaxy. RSC Adv. 2016, 6, 37982. [Google Scholar] [CrossRef]
- Kraus, J.; Reichelt, R.; Gunther, S.; Gregoratti, L.; Amati, M.; Kiskinova, M.; Yulaev, A.; Vlassioukd, I.; Kolmakov, A. Photoelectron spectroscopy of wet and gaseous samples through graphene membranes. Nanoscale 2014, 6, 14394. [Google Scholar] [CrossRef]
- Xu, M.; Fujita, D.; Gao, J.; Hanagata, N. Auger Electron Spectroscopy: A Rational Method for Determining Thickness of Graphene Films. ACS Nano 2010, 4, 2937–2945. [Google Scholar] [CrossRef] [PubMed]
- Georgakilas, V.; Otyepka, M.; Bourlinos, A.B.; Chandra, V.; Kim, N.; Kemp, K.C.; Hobza, P.; Zboril, R.; Kim, K.S. Functionalization of Graphene: Covalent and Non-Covalent Approaches, Derivatives and Applications. Chem. Rev. 2012, 112, 6156–6214. [Google Scholar] [CrossRef] [PubMed]
- Speranza, G. The Role of Functionalization in the Applications of Carbon Materials: An Overview. C-J. Carbon Res. 2019, 5, 84. [Google Scholar] [CrossRef] [Green Version]
- Yu, W.; Sisi, L.; Haiyan, Y.; Jie, L. Progress in the functional modification of graphene/graphene oxide: A review. RSC Adv. 2020, 10, 15328–15345. [Google Scholar] [CrossRef]
- Guo, Z.; Chakraborty, S.; Monikh, F.A.; Varsou, M.D.; Chetwynd, A.J.; Afantitis, A.; Lynch, I.; Zhang, P. Surface Functionalization of Graphene-Based Materials: Biological Behavior, Toxicology, and Safe-by-Design Aspects. Adv. Biol. 2021, 5, 2100637. [Google Scholar] [CrossRef]
- Peng, C.; Zhang, X. Chemical Functionalization of Graphene Nanoplatelets with Hydroxyl, Amino, and Carboxylic Terminal Groups. Chemistry 2021, 3, 873–888. [Google Scholar] [CrossRef]
- Santos, E.J.G.; Ayuela, A.; Sanchez-Portal, D. Universal magnetic properties of sp3-type defects in covalently functionalized graphene. New J. Phys. 2012, 14, 043022. [Google Scholar] [CrossRef] [Green Version]
- Elias, D.C.; Nair, R.R.; Mohiuddin, T.M.G.; Morozov, S.V.; Blake, P.; Halsall, M.P.; Ferrari, A.C.; Boukhvalov, D.W.; Katsnelson, M.I.; Geim, A.K.; et al. Control of Graphene’s Properties by Reversible Hydrogenation: Evidence for Graphane. Science 2009, 323, 610–613. [Google Scholar] [CrossRef] [Green Version]
- Ngyen, P.; Berry, V. Graphene Interfaced with Biological Cells: Opportunities and Challenges. J. Phys. Chem. Lett. 2012, 3, 1024–1029. [Google Scholar] [CrossRef]
- Xia, J.; Chen, F.; Li, J.; Tao, N. Measurement of the quantum capacitance of graphene. Nat. Nanotechnol. 2009, 4, 505–509. [Google Scholar] [CrossRef]
- Dreyer, D.R.; Park, S.; Bielawski, C.W.; Ruoff, R.S. The chemistry of graphene oxide. Chem. Soc. Rev. 2010, 39, 228–240. [Google Scholar] [CrossRef] [PubMed]
- Sreeprasad, T.S.; Berry, V. How Do the Electrical Properties of Graphene Change with its Functionalization? Small 2013, 9, 341–350. [Google Scholar] [CrossRef] [PubMed]
- Susi, T.; Kaukonen, M.; Havu, P.; Ljungberg, M.P.; Ayala, P.; Kauippen, M. Core level binding energies of functionalized and defective graphene. Beilstein J. Nanotechnol. 2014, 5, 121–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bulusheva, L.G.; Arkhipov, V.E.; Popov, K.M.; Sysoev, V.I.; Makarova, A.A.; Okotrub, A.V. Electronic Structure of Nitrogen- and Phosphorus-Doped Graphenes Grown by Chemical Vapor Deposition Method. Materials 2020, 13, 1173. [Google Scholar] [CrossRef] [Green Version]
- Fates, R.; Bouridah, H.; Raskin, J.-P. Probing carrier concentration in gated single, bi- and tri-layer CVD graphene using Raman spectroscopy. Carbon 2019, 149, 390–399. [Google Scholar] [CrossRef]
- Das, A.; Pisana, S.; Chakraborty, B.; Piscanec, S.; Saha, S.K.; Waghmare, U.V.; Novoselov, K.S.; Krishnamurthy, H.R.; Geim, A.K.; Ferrari, A.C.; et al. Monitoring dopants by Raman scattering in an electrochemically top-gated graphene transistor. Nat. Nanotechnol. 2008, 3, 210–215. [Google Scholar] [CrossRef] [Green Version]
- Scardamaglia, M.; Toma, S.; Struzzi, C.; Snyders, R.; di Santo, G.; Petaccia, L.; Bittencourt, C. Spectroscopic observation of oxygen dissociation on nitrogendoped graphene. Sci. Rep. 2017, 7, 7960. [Google Scholar] [CrossRef] [Green Version]
- Charoenpakdee, J.; Suntijitrungruang, O.; Boonchui, S. Chirality effects on an electron transport in single-walled carbon nanotube. Sci. Rep. 2020, 10, 18949. [Google Scholar] [CrossRef]
- Rathinavel, S.; Priyadharshini, K.; Panda, D. A review on carbon nanotube: An overview of synthesis, properties, functionalization, characterization, and the application. Mater. Sci. Eng. B 2021, 268, 115095. [Google Scholar] [CrossRef]
- Eatemadi, A.; Daraee, H.; Karimkhanloo, H.; Kouhi, M.; Zarghami, N.; Akbarzadeh, A.; Abasi, M.; Henifehpour, Y.; Joo, S.W. Carbon nanotubes: Properties, synthesis, purification, and medical applications. Nanoscale Res. Lett. 2014, 9, 393. [Google Scholar] [CrossRef] [Green Version]
- Arora, N.; Sharma, N.N. Arc discharge synthesis of carbon nanotubes: Comprehensive review. Diam. Relat. Mater. 2014, 50, 135–150. [Google Scholar] [CrossRef]
- Prasek, J.; Drbohlavova, J.; Chomoucka, J.; Hubalek, J.; Jasek, O.; Adamc, V.; Kizek, R. Methods for carbon nanotubes synthesis—Review. J. Mater. Chem. 2011, 21, 15872–15884. [Google Scholar] [CrossRef]
- Rahman, G.; Najaf, Z.; Mehmood, A.; Bilal, S.; ul Haq Ali Shah, A.; Mian, S.A.; Ali, G. An Overview of the Recent Progress in the Synthesis and Applications of Carbon Nanotubes. C-J. Carbon Res. 2019, 5, 3. [Google Scholar] [CrossRef] [Green Version]
- Kumar, M.; Ando, Y. Chemical vapor deposition of carbon nanotubes: A review on growth mechanism and mass production. J. Nanosci. Nanotechnol. 2010, 10, 3739–3758. [Google Scholar] [CrossRef] [Green Version]
- Hamzah, N.; Yasin, M.F.M.; Yusop, M.Z.M.; Saatac, A.; Subhae, N.A.M. Rapid production of carbon nanotubes: A review on advancement in growth control and morphology manipulations of flame synthesis. J. Mater. Chem. A 2017, 5, 25144–25170. [Google Scholar] [CrossRef]
- Yoshida, H.; Takeda, S.; Uchiyama, T.; Kohno, H.; Homma, Y. Atomic-Scale In-situ Observation of Carbon Nanotube Growth from Solid State Iron Carbide Nanoparticles. Nano Lett. 2008, 8, 2082–2086. [Google Scholar] [CrossRef]
- Adamska, M.; Narkiewicz, U. Purification of Carbon Nanotubes A Review of Methodology. Nanosci. Nanotechnol. Lett. 2018, 10, 1329–1342. [Google Scholar] [CrossRef]
- Hou, P.-X.; Liu, C.; Cheng, H.-M. Purification of carbon nanotubes. Carbon 2008, 46, 2003–2025. [Google Scholar] [CrossRef]
- Chen, P.; Wu, X.; Sun, X.; Lin, J.; Tan, K.L. Electronic Structure and Optical Limiting Behavior of Carbon Nanotubes. Phys. Rev. Lett. 1999, 82, 2548. [Google Scholar] [CrossRef] [Green Version]
- Schafer, J.; Ristein, J.; Graupner, R.; Ley, L.; Stephan, U.; Frauenheim, T.; Veersamy, V.S.; Amaratunga, G.A.J.; Weiler, M.; Ehrhardt, H. Photoemission study of amorphous carbon modifications and comparison with calculated densities of states. Phys. Rev. B 1996, 53, 7762. [Google Scholar] [CrossRef]
- Umari, P.; Petrenko, O.; Taioli, S.; de Souza, M.M. Communication: Electronic band gaps of semiconducting zig-zag carbon nanotubes from many-body perturbation theory calculations. J. Chem. Phys. 2012, 136, 181101. [Google Scholar] [CrossRef] [PubMed]
- Soncini, C.; Bondino, F.; Magnano, E.; Bhardwaj, S.; Kumar, M.; Cepek, C.; Pedio, M. Electronic properties of carbon nanotubes as detected by photoemission and inverse photoemission. Nanotechnology 2021, 32, 105703. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, S.; Bower, C.; Kiyokura, T.; Nath, K.G. Photoemission spectroscopy of single-walled carbon nanotube bundles. J. Electron Spectrosc. Relat. Phenom. 2001, 114–116, 225–228. [Google Scholar] [CrossRef]
- Schiessling, J.; Kjeldgaard, L.; Rohmund, F.; Falk, L.K.L.; Campbell, E.E.B.; Nordgren, J.; Bruhwiler, P.A. Synchrotron radiation study of the electronic structure of multiwalled carbon nanotubes. J. Phys. Condens. Matter 2003, 15, 6563–6579. [Google Scholar] [CrossRef]
- Li, Z.; Zheng, L.; Yan, W.; Pan, Z.; Wei, S. Spectroscopic Characteristics of Differently Produced Single-Walled Carbon Nanotubes. ChemPhysChem 2009, 10, 2296–2304. [Google Scholar] [CrossRef]
- Bennich, P.; Puglia, C.; Bruhwiler, P.A.; Nilsson, A.; Maxwell, A.J.; Sandell, A.; Martensson, N.; Rudolf, P. Photoemission study of K on graphite. Phys. Rev. B 1999, 59, 8292. [Google Scholar] [CrossRef] [Green Version]
- Bohm, D.; Pines, D. A Collective Description of Electron Interactions I Magnetic Interactions. Phys. Rev. 1951, 82, 625–634. [Google Scholar] [CrossRef]
- Ayala, P.; Miyata, Y.; de Blauwe, K.; Shiozawa, H.; Feng, Y.; Yanagi, K.; Kramberger, C.; Silva, S.R.P.; Follath, R.; Kataura, H.; et al. Disentanglement of the electronic properties of metallicity-selected single-walled carbon nanotubes. Phys. Rev. B 2009, 80, 205427. [Google Scholar] [CrossRef]
- Castrucci, P.; Scarselli, M.; de Crescenzi, M.; el Khahanib, A.; Rosei, F. Probing the electronic structure of carbon nanotubes by nanoscale spectroscopy. Nanoscale 2010, 2, 1611–1625. [Google Scholar] [CrossRef]
- Kramberger, C.; Rauf, H.; Shiozawa, H.; Knupfer, M.; Buchner, B.; Pichler, T.; Batchelor, D.; Kataura, H. Unraveling van Hove singularities in X-ray absorption response of single-wall carbon nanotubes. Phys. Rev. B 2007, 75, 235437. [Google Scholar] [CrossRef]
- Shearer, C.J.; Yu, L.P.; Blanch, A.J.; Zheng, M.; Andersson, G.G.; Shapter, J.G. Broadening of van Hove Singularities Measured by Photoemission Spectroscopy of Single- and Mixed-Chirality Single-Walled Carbon Nanotubes. J. Phys. Chem. C 2019, 123, 26683–26694. [Google Scholar] [CrossRef]
- Okpalugo, T.I.T.; Papakonstantinou, P.; Murphy, H.; McLaughlin, J.; Brown, N.M.D. High resolution XPS characterization of chemical functionalised MWCNTs and SWCNTs. Carbon 2005, 43, 153–161. [Google Scholar] [CrossRef]
- Wespasnick, K.A.; Smith, B.A.; Bitter, J.L.; Fairbrother, D.H. Chemical and structural characterization of carbon nanotube surfaces. Anal. Bioanal. Chem. 2010, 396, 1003–1014. [Google Scholar] [CrossRef] [PubMed]
- Xia, W.; Wang, Y.; Bergstraßer, R.; Kundu, S.; Muhler, M. Surface characterization of oxygen-functionalized multi-walled carbon nanotubes by high-resolution X-ray photoelectron spectroscopy and temperature-programmed desorption. Appl. Surf. Sci. 2007, 254, 247–250. [Google Scholar] [CrossRef]
- Balasubramanian, K.; Burghard, M. Chemically functionalized carbon nanotubes. Small 2005, 1, 180–192. [Google Scholar] [CrossRef]
- Zhao, J.; Park, H.; Han, J.; Lu, J.P. Electronic Properties of Carbon Nanotubes with Covalent Sidewall Functionalization. J. Phys. Chem. B 2004, 108, 4227–4230. [Google Scholar] [CrossRef]
- Lim, S.H.; Lin, J.; Liu, L.; Pan, H.; Pan, H.L.; Ji, W.; Feng, Y.P.; Shen, Z. Functionalization effect on the electronic properties of single walled carbon nanotubes. Funct. Mater. Lett. 2008, 1, 1–6. [Google Scholar] [CrossRef]
- Duclaux, L. Review of the doping of carbon nanotubes (multiwalled and single-walled). Carbon 2002, 40, 1751–1764. [Google Scholar] [CrossRef]
- Park, Y.R.; Ko, M.J.; Song, Y.-H.; Lee, C.J. Surface electronic structure of nitrogen-doped semiconducting single-walled carbon nanotube networks. J. Appl. Phys. 2013, 114, 153516. [Google Scholar] [CrossRef]
- Dettlaff-Weglikowsk, U.; Kim, G.; Bulusheva, L.G.; Roth, S. Modification of the electronic structure in single-walled carbon nanotubes with aromatic amines. Phys. Status Solidi B 2011, 248, 2458–2461. [Google Scholar] [CrossRef]
- Dementjev, A.P.; Eletskii, A.V.; Maslakov, K.I.; Rakov, E.G.; Sukhoverhov, V.F.; Naumkin, A.V. Fluorination of Carbon Nanostructures and Their Comparative Investigation by XPS and XAES Spectroscopy. Fuller. Nanotub. Carbon Nonstruct. 2006, 14, 287–296. [Google Scholar] [CrossRef]
- Osawa, E. Superaromaticity. Kagaku 1970, 25, 854–863. [Google Scholar]
- Castro, E.; Garcia, A.H.; Zavala, G.; Echegoyen, L. Fullerenes in biology and medicine. J. Mater. Chem. B 2017, 5, 6523–6535. [Google Scholar] [CrossRef] [PubMed]
- Lieber, C.M.; Chen, C.-C. Preparation of Fullerenes and Fullerene-Based Materials. Solid State Phys. 1994, 48, 109–148. [Google Scholar]
- Hare, J.P.; Kroto, H.W.; Taylor, R. Preparation and UV/visible spectra of fullerenes C60 and C70. Chem. Phys. Lett. 1991, 177, 394–398. [Google Scholar] [CrossRef] [Green Version]
- Taylor, R.; Langley, G.J.; Kroto, H.W.; Walton, D.R.M. Formation of C60 by pyrolysis of naphthalene. Nature 1993, 366, 728–731. [Google Scholar] [CrossRef]
- Howard, J.B.; McKinnon, J.T.; Makarovsky, Y.; Lafleur, A.L.; Johnson, M.E. Fullerenes C60 and C70 in flames. Nature 1991, 352, 139–141. [Google Scholar] [CrossRef]
- Kroto, H.W. Space, Stars, C60, and Soot. Science 1988, 242, 1139–1145. [Google Scholar] [CrossRef]
- Stalling, D.L.; Kuo, K.C.; Guo, C.Y.; Saim, S. Separation of Fullerenes C60, C70, and C76-84 on Polystyrene Divinylbenzene Columns. J. Liq. Chromatogr. 1993, 16, 699–722. [Google Scholar] [CrossRef]
- Guldi, D.M.; Illescas, B.M.; Atienza, C.M.; Wielopolski, M.; Martin, N. Fullerene for organic electronics. Chem. Soc. Rev. 2009, 38, 1587–1597. [Google Scholar] [CrossRef]
- Prato, M. Fullerene chemistry for materials science applications. J. Mater. Chem. 1997, 7, 1097–1109. [Google Scholar] [CrossRef]
- Martin, N. New challenges in fullerene chemistry. Chem. Commun. 2006, 20, 2093–2104. [Google Scholar] [CrossRef] [PubMed]
- Hebard, A.F.; Rosseinski, M.J.; Haddon, R.C.; Murphy, D.W.; Glarum, S.H.; Palstra, T.T.M.; Ramirez, A.P.; Kortan, A.R. Superconductivity at 18 K in potassium-doped C60. Nature 1991, 350, 600–601. [Google Scholar] [CrossRef]
- Holczer, K.; Klein, O.; Huang, S.-M.; Kaner, R.B.; Fu, K.-J.; Whetten, R.L.; Diederich, F. Alkali-Fulleride Superconductors: Synthesis, Composition, and Diamagnetic Shielding. Science 1991, 252, 1154–1157. [Google Scholar] [CrossRef]
- Allemand, P.M.; Khemani, K.C.; Koch, A.; Wudl, F.; Holczer, K.; Donovan, S.; Gruner, G.; Thompson, J.D. Organic Molecular Soft Ferromagnetism in a Fullerene C60. Science 1991, 253, 301–302. [Google Scholar] [CrossRef]
- Tutt, L.W.; Kost, A. Optical limiting performance of C60 and C70 solutions. Nature 1992, 356, 225–226. [Google Scholar] [CrossRef]
- Pana, Y.; Liu, X.; Zhang, W.; Liu, Z.; Zeng, G.; Shao, B.; Liang, Q.; He, Q.; Yuan, X.; Huang, D.; et al. Advances in photocatalysis based on fullerene C60 and its derivatives: Properties, mechanism, synthesis, and applications. Appl. Catal. B 2020, 265, 118579. [Google Scholar] [CrossRef]
- Wen, C.; Li, J.; Kitazawa, K.; Aida, T.; Honma, I.; Komiyama, H.; Yamada, K. Electrical conductivity of a pure C60 single crystal. Appl. Phys. Lett. 1992, 61, 2162. [Google Scholar] [CrossRef]
- Mort, J.; Ziolo, R.; Machonkin, M.; Huffman, D.R.; Ferguson, M.I. Electrical conductivity studies of undoped solid films of C60, C70. Chem. Phys. Lett. 1991, 186, 284–286. [Google Scholar] [CrossRef]
- Gueorguiev, G.; Pacheco, J.M.; Tomanek, D. Quantum Size Effects in the Polarizability of Carbon Fullerenes. Phys. Rev. Lett. 2004, 92, 215501. [Google Scholar] [CrossRef] [Green Version]
- Matveeva, L.A.; Venger, E.F.; Kolyadina, E.Y.; Neluba, P.L. Quantum-size effects in semiconductor heterosystems. Semicond. Phys. Quantum Electron. Optoelectron. 2017, 20, 224–230. [Google Scholar] [CrossRef] [Green Version]
- Haddon, R.C.; Pasquarello, A. Magnetism of carbon clusters. Phys. Rev. B 1994, 50, 16459–16463. [Google Scholar] [CrossRef] [PubMed]
- Reinke, P.; Feldermann, H.; Oelhafen, P. C60 bonding to graphite and boron nitride surfaces. J. Chem. Phys. 2003, 119, 12547–12552. [Google Scholar] [CrossRef]
- Yu, J.; Kalia, R.; Vashishta, P. Phonon dispersion and density of states of solid C60. Appl. Phys. Lett. 1993, 63, 3152–3154. [Google Scholar] [CrossRef]
- Hua, X.; Cagin, T.; Che, J.; Goddard, W.A. QM(DFT) and MD studies on formation mechanisms of C60 fullerenes. Nanotechnology 2000, 11, 85–88. [Google Scholar] [CrossRef] [Green Version]
- Hasegawa, M.; Ohno, K. Density functional theory for the phase diagram of rigid C60 molecules. Phys. Rev. E 1996, 54, 3928–3932. [Google Scholar] [CrossRef]
- Feng, M.; Zhao, J.; Petek, H. Atomlike Hollow-Core-Bound Molecular Orbitals of C60. Science 2008, 320, 359–362. [Google Scholar] [CrossRef]
- Bilan, S.; Zotti, L.A.; Pauly, F.; Cuevas, J.C. Theoretical study of the charge transport through C60-based single-molecule junctions. Phys. Rev. B 2012, 85, 205403. [Google Scholar] [CrossRef] [Green Version]
- Endo, K.; Koizumi, S.; Otsuka, T.; Ida, T.; Morohashi, T.; Onoe, J.; Nakao, A.; Kurmaev, E.Z.; Moewes, A.; Chong, D.P. Analysis of Electron Spectra of Carbon Allotropes (Diamond, Graphite, Fullerene) by Density Functional Theory Calculations Using the Model Molecules. J. Phys. Chem. A 2003, 107, 9403–9408. [Google Scholar] [CrossRef]
- Kalb, W.L.; Haas, S.; Krellner, C.; Mathis, T.; Batlogg, B. Trap density of states in small-molecule organic semiconductors: A quantitative comparison of thin-film transistors with single crystals. Phys. Rev. B 2010, 81, 155315. [Google Scholar] [CrossRef] [Green Version]
- Yogev, S.; Matsubara, R.; Nakamura, M.; Zschieschang, U.; Klauk, H.; Rosenwaks, Y. Fermi Level Pinning by Gap States in Organic Semiconductors. Phys. Rev. Lett. 2013, 110, 036803. [Google Scholar] [CrossRef] [PubMed]
- Bussolotti, F.; Yang, J.; Hiramoto, M.; Kaji, T.; Kera, S.; Ueno, N. Direct detection of density of gap states in C60 single crystals by photoemission spectroscopy. Phys. Rev. B 2015, 92, 115102. [Google Scholar] [CrossRef]
- Hwang, J.; Wan, A.; Khan, A. Energetics of metal-organic interfaces: New experiments and assessment of the field. Mater. Sci. Eng. R 2009, 64, 1–31. [Google Scholar] [CrossRef]
- Wang, C.; Liu, X.; Wang, C.; Kauppi, J.; Gao, Y. Electronic structure evolution in doping of fullerene (C60) by ultra-thin layer molybdenum trioxide. J. Appl. Phys. 2015, 118, 085304. [Google Scholar] [CrossRef]
- Weibo, Y.; Seifermann, S.M.; Pierrat, P.; Brase, S. Synthesis of highly functionalized C60 fullerene derivatives and their applications in material and life sciences. Org. Biomol. Chem. 2015, 13, 25–54. [Google Scholar]
- Rašović, I. Water-soluble fullerenes for medical applications. Mater. Sci. Technol. 2017, 33, 777–794. [Google Scholar] [CrossRef]
- Lin, H.-L.; Yutaka, M. Functionalization of fullerene through fullerene cation intermediates. Chem. Commun. 2018, 54, 11244–11259. [Google Scholar] [CrossRef]
- Alonso, A.M.; Bonifazi, D.; Prato, M. Functionalization and applications of fullerene-Chap 7. In Carbon Nanotechnologies; Dai, L., Ed.; Elsevier: Amsterdam, The Netherlands, 2006; pp. 155–189. [Google Scholar]
- Fukuzumi, S. Nanocarbons as Electron Donors and Acceptors in Photoinduced Electron-Transfer Reactions. ECS J. Solid State Sci. Technol. 2017, 6, M3055–M3061. [Google Scholar] [CrossRef] [Green Version]
- Tkachenko, N.V.; Rantala, L.; Tauber, A.Y.; Helaja, J.; Hynninen, P.H.; Lemmetyinen, H. Photoinduced Electron Transfer in Phytochlorin−Fullerene Dyads. J. Am. Chem. Soc. 1999, 121, 9378–9387. [Google Scholar] [CrossRef]
- Koeppe, R.; Sariciftci, N.S. Photoinduced charge and energy transfer involving fullerene derivatives. Photochem. Photobiol. Sci. 2005, 5, 1122–1131. [Google Scholar] [CrossRef]
- Mikami, K.; Matsumoto, S.; Tonoi, T.; Okubo, Y.; Suenobu, T.; Fukuzumi, S. Solid state photochemistry for fullerene functionalization: Solid state photoinduced electron transfer in the Diels-Alder reaction with anthracenes. Tetrahedron Lett. 1998, 39, 3733–3736. [Google Scholar] [CrossRef]
- Mroz, P.; Tegos, G.P.; Gali, H.; Wharton, T.; Sarna, T.; Hamblin, M.R. Photodynamic therapy with fullerenes. Photochem. Photobiol. Sci. 2007, 6, 1139–1149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- da Ros, T.; Prato, M. Medicinal chemistry with fullerenes and fullerene derivatives. Chem. Commun. 1999, 8, 663–669. [Google Scholar] [CrossRef]
- Zygouri, P.; Spyrou, K.; Mitsari, E.; Barrio, M.; Macovez, R.; Patila, M.; Stamatis, H.; Verginadis, I.I.; Velalopoulou, A.P.; Evangelou, A.M.; et al. A facile approach to hydrophilic oxidized fullerenes and their derivatives as cytotoxic agents and supports for nanobiocatalytic systems. Sci. Rep. 2020, 10, 8244. [Google Scholar] [CrossRef]
- Li, B.; Zhen, J.; Wan, Y.; Lei, X.; Jia, L.; Wu, X.; Zeng, H.; Chen, M.; Wang, G.-W.; Yang, S. Steering the electron transport properties of pyridine-functionalized fullerene derivatives in inverted perovskite solar cells: The nitrogen site matters. J. Mater. Chem. A 2020, 8, 3872–3881. [Google Scholar] [CrossRef]
- Liu, J.; Wang, Y.; Jiang, P.; Tu, G. Functionalized Amphiphilic Diblock Fullerene Derivatives as a Cathode Buffer Layer for Efficient Inverted Organic Solar Cells. ACS Omega 2020, 5, 1336–1345. [Google Scholar] [CrossRef] [Green Version]
- Nyga, A.; Motya, R.; Bussetti, G.L.; Calloni, A.; Jagadeeshb, M.S.; Pluczyk-Malek, S.; Data, P.; Blacha-Grzechnik, A. Electrochemically deposited poly(selenophene)-fullerene photoactive layer:Tuning of the spectroscopic properties towards visible light-driven generation of singlet oxygen. Appl. Surf. Sci. 2020, 525, 146594. [Google Scholar] [CrossRef]
- Liu, Z.; Koshino, M.; Suenaga, K.; Mrzel, A.; Kataura, H.; Iijima, S. Transmission Electron Microscopy Imaging of Individual Functional Groups of Fullerene Derivatives. Phys. Rev. Lett. 2006, 96, 088304. [Google Scholar] [CrossRef]
- Balaz, P. High-energy milling-Chap 2. In Mechanochemistry in Nanoscience and Minerals Engineering; Springer: Berlin/Heidelberg, Germany, 2008; pp. 103–132. [Google Scholar]
- Boudou, J.P.; Curmi, P.A.; Lelezko, F.; Wrachtrup, J.; Aubert, P.; Sennour, M.; Balasubramanian, G.; Reuter, R.; Thorel, A.; Gaffet, E. High yield fabrication of fluorescent nanodiamonds. Nanotechnology 2009, 20, 235602. [Google Scholar] [CrossRef]
- Guan, S.; Peng, F.; Liang, H.; Fan, C.; Tan, L.; Wang, Z.; Zhang, Y.; Zhang, J.; Yu, H.; He, D. Fragmentation and stress diversification in diamond powder under high pressure. J. Appl. Phys. 2018, 124, 215902. [Google Scholar] [CrossRef]
- Kumar, A.; Lin, P.A.; Xue, A.; Hao, B.; Yap, Y.K.; Sankaran, M. Formation of nanodiamonds at near-ambient conditions via microplasma dissociation of ethanol vapour. Nat. Commun. 2013, 4, 2618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tzeng, Y.-K.; Zhang, J.L.; Lu, H.; Ishiwata, H.; Dahl, J.; Carlson, R.M.K.; Yan, H.; Schreiner, P.R.; Vuckovic, J.; Shen, Z.-X.; et al. Vertical-Substrate MPCVD Epitaxial Nanodiamond Growth. Nano Lett. 2017, 17, 1489–1495. [Google Scholar] [CrossRef] [PubMed]
- Amans, D.; Chenus, A.C.; Ledoux, G.; Dujardin, C.; Reynaud, C.; Sublemontier, O.; Masenelli-Varlot, K.; Guillois, O. Nanodiamond synthesis by pulsed laser ablation in liquids. Diam. Relat. Mater. 2009, 18, 177–180. [Google Scholar] [CrossRef]
- Bazzanella, N.; Cazzanelli, M.; Dorigoni, C.; Bifone, A.; Miotello, A. The modeling and synthesis of nanodiamonds by laser ablation of graphite and diamond-like carbon in liquid-confined ambient. Appl. Phys. A 2018, 124, 72. [Google Scholar]
- Shenderova, O.; Koscheev, A.; Zaripov, N.; Petrov, I.; Skryabin, Y.; Dektov, P.; Turner, S.; van Tendeloo, G. Surface Chemistry and Properties of Ozone-Purified Detonation Nanodiamonds. J. Phys. Chem. C 2011, 115, 9827–9837. [Google Scholar] [CrossRef]
- Popov, V. Several Aspects of Application of Nanodiamonds as Reinforcements for Metal Matrix Composites. Appl. Sci. 2021, 11, 4695. [Google Scholar] [CrossRef]
- Lesiaka, P.; Kover, L.; Toth, J.; Zemek, J.; Jiricek, P.; Kromka, A.; Rangama, N. C sp2/sp3 hybridisations in carbon nanomaterials-XPS and (X)AES study. Appl. Surf. Sci. 2018, 452, 223–231. [Google Scholar] [CrossRef]
- Xie, Y.; Xie, W.G.; Gong, L.; Zhang, W.H.; Chen, S.H.; Zhang, Q.Z.; Chen, J. Surface characterization on graphitization of nanodiamond powder annealed in nitrogen ambient. Surf. Interface Anal. 2010, 42, 1514–1518. [Google Scholar] [CrossRef]
- Speranza, G.; Laidani, N. Measurement of the relative abundance of sp2 and sp3 hybridised atoms in carbon-based materials by XPS: A critical approachPart I. Diam. Relat. Mater. 2004, 13, 445–450. [Google Scholar] [CrossRef]
- Speranza, G.; Laidani, N. Measurement of the relative abundance of sp2 and sp3 hybridised atoms in carbon-based materials by XPS: A critical approachPart II. Diam. Relat. Mater. 2004, 13, 451–458. [Google Scholar] [CrossRef]
- Osswald, S.; Yushin, G.; Mochalin, V.; Kucheyev, S.O. Control of sp2/sp3 Carbon Ratio and Surface Chemistry of Nanodiamond Powders by Selective Oxidation in Air. J. Am. Chem. Soc. 2006, 128, 11635–11642. [Google Scholar] [CrossRef]
- Butenko, Y.V.; Krishnamurthy, S.; Chakraborty, A.K.; Kuznetsov, V.L.; Dhanak, V.R.; Hunt, M.R.C.; Siller, L. Photoemission study of onionlike carbons produced by annealing nanodiamonds. Phys. Rev. B 2005, 71, 075420. [Google Scholar] [CrossRef] [Green Version]
- Yeganeh, M.; Coxon, P.R.; Brieva, A.C.; Dhanak, V.R.; Siller, L.; Butenko, Y.V. Atomic hydrogen treatment of nanodiamond powder studied with photoemission spectroscopy. Phys. Rev. B 2007, 75, 155404. [Google Scholar] [CrossRef]
- Cui, J.B.; Ristein, J.; Ley, L. Electron Affinity of the Bare and Hydrogen Covered Single Crystal Diamond (111) Surface. Phys. Rev. Lett. 1998, 81, 429. [Google Scholar] [CrossRef]
- Wilson, T.B.; Walton, J.S.; Beamson, G. Analysis of chemical vapour deposited diamond films by X-ray photoelectron spectroscopy. J. Electron Spectrosc. Relat. Phenom. 2001, 121, 183–201. [Google Scholar] [CrossRef]
- Maier, F.; Riedel, M.; Mantel, B.; Riestein, J.; Ley, L. Origin of Surface Conductivity in Diamond. Phys. Rev. Lett. 2000, 85, 3472. [Google Scholar] [CrossRef]
- Zhu, Y.; Li, J.; Zhang, Y.; Yang, X.; Chen, N.; Sun, Y.; Zhao, Y.; Fan, C.; Huang, Q. The Biocompatibility of Nanodiamonds and Their Application in Drug Delivery Systems. Theranostics 2012, 2, 302–312. [Google Scholar] [CrossRef]
- Kozakov, A.T.; Kochur, A.G.; Kumar, N.; Panda, K.; Nikolskii, A.V.; Sidashov, A.V. Determination of sp2 and sp3 phase fractions on the surface of diamond films from C1s, valence band X-ray photoelectron spectra and CKVV X-ray-excited Auger spectra. Appl. Surf. Sci. 2021, 536, 147807. [Google Scholar] [CrossRef]
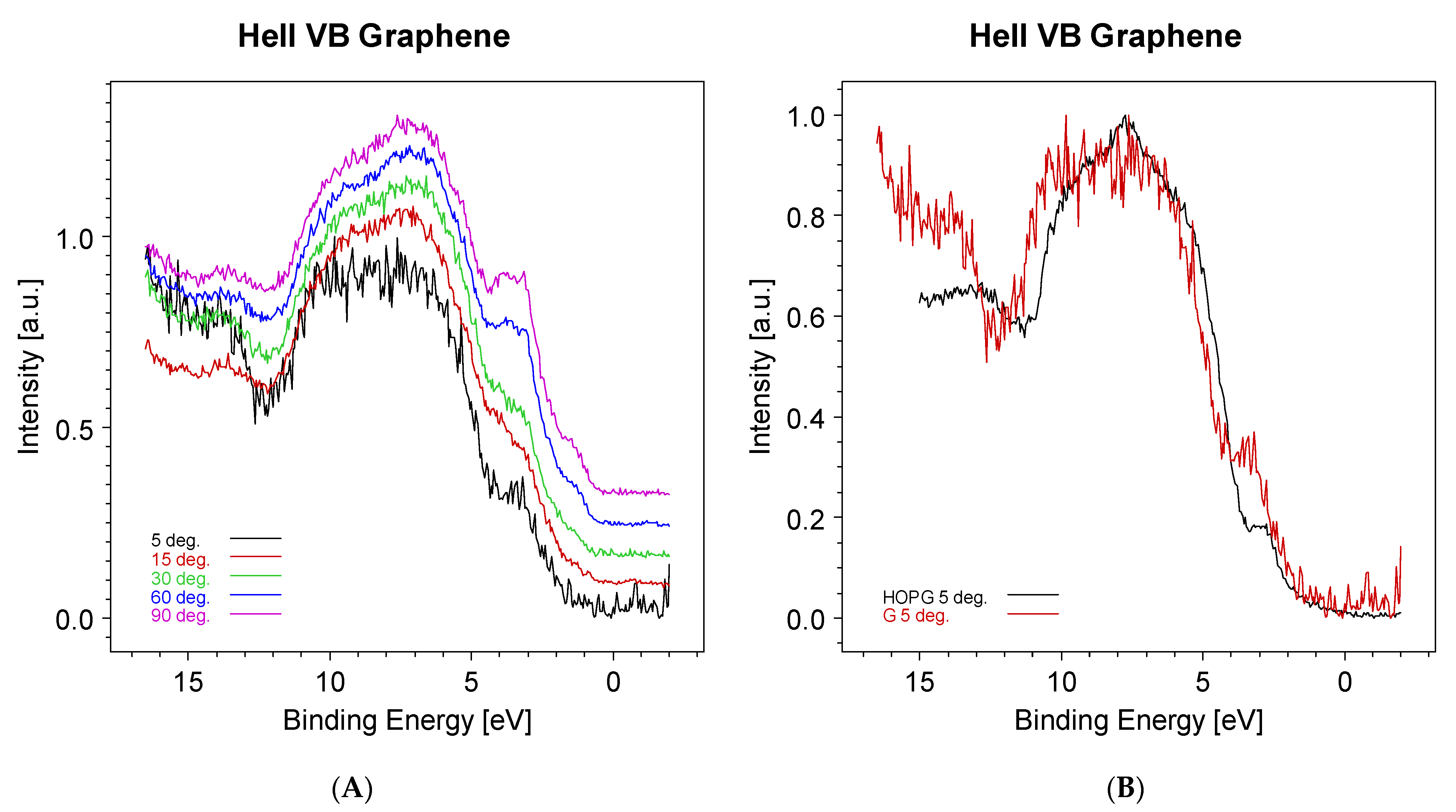
- Bianconi, A.; Hagstrom, S.B.M.; Babrach, R.Z. Photoemission studies of graphite high-energy conduction-band and valence-band states using soft-X-ray synchrotron radiation excitation. Phys. Rev. B 1977, 16, 5543. [Google Scholar] [CrossRef]
- Kundu, R.; Mishra, P.; Sekhar, B.R.; Maniraj, M.; Barman, S.R. Electronic structure of single crystal and highly oriented pyrolytic graphite from ARPES and KRIPES. Phys. B 2012, 407, 827–832. [Google Scholar] [CrossRef]
- Bandis, C.; Pate, B.B. Electron Emission Due to Exciton Breakup from Negative Electron Affinity Diamond. Phys. Rev. Lett. 1995, 74, 777. [Google Scholar] [CrossRef] [PubMed]
- Ristein, J.; Stein, W.; Ley, L. Defect Spectroscopy and Determination of the Electron Diffusion Length in Single Crystal Diamond by Total Photoelectron Yield spectroscopy. Phys. Rev. Lett. 1997, 78, 1803. [Google Scholar] [CrossRef]
- James, M.; Fogarty, F.; Zulkharnay, R.; Fox, N.A.; May, P.W. A review of surface functionalisation of diamond for thermionic emission applications. Carbon 2020, 171, 532–550. [Google Scholar] [CrossRef]
- Nemanich, R.J.; Carlisle, J.A.; Hirata, A. CVD diamond: Research, applications, and challenges. MRS Bull. 2014, 39, 490–494. [Google Scholar] [CrossRef] [Green Version]
- Raty, J.-Y.; Galli, G.; Bosted, C.; van Buuren, T.W.; Terminello, L. Quantum Confinement and Fullerenelike Surface Reconstructions in Nanodiamonds. Phys. Rev. Lett. 2003, 90, 037401. [Google Scholar] [CrossRef] [PubMed]
- Ristein, J. Diamond surfaces: Familiar and amazing. Appl. Phys. A 2005, 82, 377–384. [Google Scholar] [CrossRef]
- Diederich, L.; Kuttel, O.M.; Ruffieux, P.; Pillo, T.; Aebi, P.; Schlapbach, L. Photoelectron emission from nitrogen- and boron-doped diamond(100) surfaces. Surf. Sci. 1998, 417, 41–52. [Google Scholar] [CrossRef] [Green Version]
- Zhirnov, V.V.; Shenderova, O.A.; Jaeger, D.L.; Tyler, T.; Areshkin, D.A.; Brenner, D.W.; Hren, J.J. Electron emission properties of detonation nanodiamonds. Phys. Solid State 2004, 46, 657–661. [Google Scholar] [CrossRef]
- Maillard-Schaller, E.; Kuettel, O.M.; Diederich, L.; Schlapbach, L.; Zhirnov, V.; Belobrov, P.I. Surface properties of nanodiamond films deposited by electrophoresis on Si(100). Diam. Relat. Mater. 1999, 9, 805–808. [Google Scholar] [CrossRef]
- Shenderova, O.; Nunn, N. Chapter 2-Production and purification of nanodiamonds. In Nanodiamonds-Advanced Material Analysis, Properties and Applications-Micro and Nano Technologies; Elsevier: Amsterdam, The Netherlands, 2017; pp. 25–56. [Google Scholar]
- Gismondi, A.; Reina, G.; Orlanducci, S.; Mizzoni, F.; Gay, S.; Terranova, M.L.; Canini, A. Nanodiamonds coupled with plant bioactive metabolites: A nanotech approach for cancer therapy. Biomaterials 2015, 38, 22–35. [Google Scholar] [CrossRef]
- Tinwala, H.; Wairkar, S. Production, surface modification and biomedical applications of nanodiamonds: A sparkling tool for theranostics. Mater. Sci. Eng. C 2019, 97, 913–931. [Google Scholar] [CrossRef] [PubMed]
- Neburkova, J.; Vavra, J.; Cigler, P. Coating nanodiamonds with biocompatible shells for applications in biology and medicine. Curr. Opin. Solid State Mater. Sci. 2017, 21, 21–43. [Google Scholar] [CrossRef]
- Mona, J.; Kuo, C.-J.; Perevedentseva, E.; Priezzhev, A.V.; Cheng, C.-L. Adsorption of human blood plasma on nanodiamond and its influence on activated partial thromboplastin time. Diam. Relat. Mater. 2013, 39, 73–77. [Google Scholar] [CrossRef] [Green Version]
- Li, H.-C.; Hsieh, F.-J.; Chen, C.-P.; Chang, M.-Y.; Hieh, P.C.H.; Chen, C.-C.; Hung, S.-U.; Wu, C.-C.; Chang, H.-C. The hemocompatibility of oxidized diamond nanocrystals for biomedical applications. Sci. Rep. 2013, 3, 3044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, L.-W.; Lin, Y.-C.; Perevedentseva, E.; Lugovtsov, A.; Priezzhev, A.; Cheng, C.-L. Nanodiamonds for medical applications: Interaction with blood in vitro and in vivo. Int. J. Mol. Sci. 2016, 17, 1111. [Google Scholar] [CrossRef] [Green Version]
- Edgington, R.; Spillane, K.M.; Papageorgiou, G.; Wray, W.; Ishiwata, H.; Labarca, M.; Leal-Ortiz, S.; Reid, G.; Webb, M.; Foord, J.; et al. Functionalisation of Detonation Nanodiamond for Monodispersed, Soluble DNA-Nanodiamond Conjugates Using Mixed Silane Bead-Assisted Sonication Disintegration. Sci. Rep. 2018, 8, 728. [Google Scholar] [CrossRef] [Green Version]
- Whitlow, J.; Pacelli, S.; Paul, A. Multifunctional nanodiamonds in regenerative medicine: Recent advances and future directions. J. Control. Release 2017, 261, 62–86. [Google Scholar] [CrossRef]
- Hajiali, F.; Shojaei, A. Silane functionalization of nanodiamond for polymer nanocomposites-effect of degree of silanization. Colloids Surf. A 2016, 506, 254–263. [Google Scholar] [CrossRef]
- Zheng, W.-W.; Hsieh, Y.-H.; Chiu, Y.-C.; Cai, S.-J.; Cheng, C.-L.; Chen, C. Organic functionalization of ultradispersed nanodiamond: Synthesis and applications. J. Mater. Chem. 2009, 19, 8432–8441. [Google Scholar] [CrossRef]
- Krueger, A.; Ozawa, M.; Jarre, G.; Liang, Y.; Stegk, J.; Lu, L. Deagglomeration and functionalisation of detonation diamond. Phys. Status Solid A 2007, 204, 2881–2887. [Google Scholar] [CrossRef]
- Krueger, A.; Lang, D. Functionality is Key: Recent Progress in the Surface Modification of Nanodiamond. Adv. Funct. Mater. 2012, 22, 890–906. [Google Scholar] [CrossRef]
- Barzegar, M.; Olia, A.; Donnelly, P.S.; Hollenberg, L.C.L.; Mulvaney, P.; Simpson, D.A. Advances in the Surface Functionalization of Nanodiamonds for Biological Applications: A Review. ACS Appl. Nano Mater. 2021, 4, 9985–10005. [Google Scholar] [CrossRef]
- Reina, G.; Zhao, L.; Bianco, A.; Komatsu, N. Chemical Functionalization of Nanodiamonds: Opportunities and Challenges Ahead. Angew. Chem. Int. Ed. 2019, 58, 2–14. [Google Scholar] [CrossRef] [PubMed]
- Shenderova, O.; Panich, A.M.; Moseenkov, S.I.; Hens, S.C.; Kuznetsov, V.L.; Vieth, H.-M. Hydroxylated Detonation Nanodiamond: FTIR, XPS, and NMR Studies. J. Phys. Chem. C 2011, 115, 19005–19011. [Google Scholar] [CrossRef]
- Sharin, P.; Sivtseva, A.V.; Popov, V.I. X-ray Photoelectron Spectroscopy of Nanodiamonds Obtained by Grinding and Detonation Synthesis. Technol. Phys. 2021, 66, 275–279. [Google Scholar] [CrossRef]
- Arnault, J.C. X-ray Photoemission Spectroscopy applied to nanodiamonds: From surface chemistry to in situ reactivity. Diam. Relat. Mater. 2018, 84, 157–168. [Google Scholar] [CrossRef]
- Minati, L.; Torrengo, S.; Maniglio, D.; Migliaresi, C.; Speranza, G. Luminescent graphene quantum dots from oxidized multi-walled carbon nanotubes. Mater. Chem. Phys. 2012, 137, 12–16. [Google Scholar] [CrossRef]
- Kahn, Z.; Tsai, T.; Taniguchi, K.; Watanabe, A.; Zettl, F. Imaging electrostatically confined Dirac fermions in graphene quantum dots. Nat. Phys. 2016, 12, 1032. [Google Scholar]
- Gutierrez, C.; Brown, L.; Kim, C.J.; Park, J.; Pasupathy, A.N. Klein tunnelling and electron trapping in nanometre-scale graphene quantum dots. Nat. Phys. 2016, 12, 1069. [Google Scholar] [CrossRef]
- Bai, K.K.; Zhou, J.J.; Wei, Y.C.; Qiao, J.B.; Liu, Y.W.; Liu, H.W.; Jiang, H. Generating atomically sharp p-n junctions in graphene and testing quantum electron optics on the nanoscale. Phys. Rev. B 2018, 97, 045413. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Y.; Mao, J.; Moldovan, D.; Masir, M.R.; Li, G.; Watanabe, K.; Taniguchi, T.; Peeters, F.M.; Andrei, E.Y. Tuning a circular p-n junction in graphene from quantum confinement to optical guiding. Nat. Nanotechnol. 2017, 12, 1045. [Google Scholar] [CrossRef]
- Fu, Z.Q.; Bai, K.K.; Ren, Y.N.; Zhou, J.J.; He, L. Coulomb interaction in quasibound states of graphene quantum dots. Phys. Rev. B 2020, 101, 235310. [Google Scholar] [CrossRef]
- Yuan, F.; Yuan, T.; Sui, L.; Wang, Z.; Xi, Z.; Li, Y.; Li, X.; Fan, L.; Tan, Z.; Chen, A.; et al. Engineering triangular carbon quantum dots with unprecedented narrow bandwidth emission for multicolored LEDs. Nat. Commun. 2018, 9, 2249. [Google Scholar] [CrossRef] [PubMed]
- Yuan, F.; Wang, Z.; Li, X.; Li, Y.; Tan, Z.; Fan, L.; Yang, S. Bright multicolor bandgap fluorescent carbon quantum dots for electroluminescent light-emitting diodes. Adv. Mater. 2017, 29, 1604436. [Google Scholar] [CrossRef] [PubMed]
- Carolan, D.; Rocks, C.; Padmanaban, D.B.; Maguire, P.; Svrcekb, V.; Mariotti, D. Environmentally friendly nitrogen-doped carbon quantum dots for next generation solar cells. Sustain. Energy Fuels 2017, 1, 1611. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.T.; Wang, X.; Wang, H.F.; Lu, F.S.; Luo, P.J.G.; Cao, L.; Meziani, M.J.; Liu, J.H.; Chen, M.; Huang, Y.P.; et al. Carbon Dots as Nontoxic and High-Performance Fluorescence Imaging Agents. J. Phys. Chem. C 2009, 113, 18110–18114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Cao, L.; Lu, F.S.; Meziani, M.J.; Li, H.; Qi, G.; Zhou, B.; Harruff, B.A.; Kerramec, F.; Sun, Y.P. Photoinduced electron transfers with carbon dots. , Chem. Commun. 2009, 2009, 3774–3776. [Google Scholar] [CrossRef]
- Hu, S.L.; Niu, K.Y.; Sun, J.; Yang, J.; Zhao, N.Q.; Du, X.W. One-step synthesis of fluorescent carbon nanoparticles by laser irradiation. J. Mater. Chem. 2009, 19, 484–488. [Google Scholar] [CrossRef]
- Zhao, Q.L.; Zhang, Z.L.; Huang, B.H.; Peng, J.; Zhang, M.; Pang, D.W. Facile preparation of low cytotoxicity fluorescent carbon nanocrystals by electrooxidation of graphite. Chem. Commun. 2008, 2008, 5116–5118. [Google Scholar] [CrossRef]
- Li, H.T.; He, X.D.; Kang, Z.H.; Huang, H.; Liu, Y.; Liu, J.L.; Lian, S.Y.; Tsang, C.H.; Yang, X.B.; Lee, S.T. Water-Soluble Fluorescent Carbon Quantum Dots and Photocatalyst Design, Angew. Angew. Chem. Int. Ed. 2010, 49, 4430–4434. [Google Scholar] [CrossRef]
- Ming, H.; Ma, Z.; Liu, Y.; Pan, K.M.; Yu, H.; Wang, F.; Kang, Z.H. Large scale electrochemical synthesis of high quality carbon nanodots and their photocatalytic property. Dalton Trans. 2012, 41, 9526–9531. [Google Scholar] [CrossRef] [PubMed]
- Gao, R.; Wu, Z.; Wang, L.; Liu, J.; Deng, Y.; Xiao, Z.; Fang, J.; Liang, Y. Green Preparation of Fluorescent Nitrogen-Doped Carbon Quantum Dots for Sensitive Detection of Oxytetracycline in Environmental Samples. Nanomaterials 2020, 10, 1561. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Bai, P.; Wang, X.; Zhao, G.; Feng, J.; Rend, H. Preparation of nitrogen-doped carbon quantum dots and its application as a fluorescent probe for Cr(vi) ion detection. New J. Chem. 2019, 43, 5488–5494. [Google Scholar] [CrossRef]
- Jing, S.; Zhao, Y.; Sun, R.-C.; Zhong, L.; Peng, X. Facile and High-Yield Synthesis of Carbon Quantum Dots from Biomass-Derived Carbons at Mild Condition. ACS Sustain. Chem. Eng. 2019, 7, 7833–7843. [Google Scholar] [CrossRef]
- Nguyen, H.A.; Srivastava, I.; Pan, D.; Gruebele, M. Unraveling the Fluorescence Mechanism of CQDs with Sub-Single-Particle Resolution. ACS Nano 2020, 14, 6127–6137. [Google Scholar] [CrossRef]
- Deng, Y.; Chen, M.; Chen, G.; Zou, W.; Zhao, Y.; Zhang, H.; Zhao, Q. Visible-Ultraviolet Upconversion Carbon Quantum Dots for Enhancement of the Photocatalytic Activity of Titanium Dioxide. ACS Omega 2021, 6, 4247–4254. [Google Scholar] [CrossRef]
- Papagiannouli, I.; Patanen, M.; Blanchet, V.; Bozek, J.D.; Villa, M.D.; Huttula, M.; Kokkonen, E.; Lamour, E.; Mevel, E.; Pelimanni, E.; et al. Depth Profiling of the Chemical Composition of Free-Standing Carbon Dots Using X‑ray Photoelectron Spectroscopy. J. Phys. Chem. C 2018, 122, 14889–14897. [Google Scholar] [CrossRef] [Green Version]
- Chawla, K.K. Carbon Fibers in Composite Materials, 2nd ed.; Springer: New York, NY, USA, 1998. [Google Scholar]
- Petersen, R. Carbon Fiber Biocompatibility for Implants. Fibers 2016, 4, 1. [Google Scholar] [CrossRef] [Green Version]
- Park, S.-J. Carbon Fibers; Springer Nature: Singapore, 2018. [Google Scholar]
- Al-Saleh, M.H.; Sundararaj, U. Review of the mechanical properties of carbon nanofiber/polymer composites. Compos. A 2011, 42, 2126–2142. [Google Scholar] [CrossRef]
- Shamsuddin, S.R.; Ho, K.K.C.; Lee, K.H.; Hodgkinson, J.M.; Bismarck, A. Carbon Fiber: Properties, Testing, and analysis. In Wiley Encyclopedia of Composites, 2nd ed.; Nicolais, L., Borzacchiello, A., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2012. [Google Scholar]
- Zou, G.; Zhang, D.; Dong, C.; Li, H.; Xiong, K.; Fei, L.; Qian, Y. Carbon nanofibers: Synthesis, characterization, and electrochemical properties. Carbon 2006, 44, 828–832. [Google Scholar] [CrossRef]
- Rodriguez, N.M. A review of catalytically grown carbon nanofibres. J. Mater. Res. 1993, 8, 3233–3250. [Google Scholar] [CrossRef]
- Manafi, S.A.; Badiee, S.H. Production of carbon nanofibers using a CVD method with lithium fluoride as a supported cobalt catalyst. Res. Lett. Mater. Sci. 2008, 2008, 850975. [Google Scholar] [CrossRef] [Green Version]
- Tibbets, G.G.; Gorkiewicz, D.W.; Alig, R.L. A new reactor for growing carbon-fibers from liquid-phase and vapor-phase hydrocarbons. Carbon 1993, 31, 809–814. [Google Scholar] [CrossRef]
- Ge, M.; Sattler, K. Observation of fullerene cones. Chem. Phys. Lett. 1994, 220, 192–196. [Google Scholar] [CrossRef]
- Zhao, T.; Kvande, I.; Yu, Y.; Ronning, M.; Holmen, A.; Chen, D. Synthesis of Platelet Carbon Nanofiber/Carbon Felt Composite on in Situ Generated Ni-Cu Nanoparticles. J. Phys. Chem. C 2011, 115, 1123–1133. [Google Scholar] [CrossRef]
- Kim, Y.A.; Hayashi, T.; Endo, M.; Dresselhaus, M.S. Carbon nanofibers. In Handbook of Nanomaterials; Vajtai, R., Ed.; Springer: Berlin/Heidelberg, Germany, 2013; pp. 233–262. [Google Scholar]
- Huang, H.; Kajiura, H.; Murakami, Y.; Ata, M. Metal sulfide catalyzed growth of carbon nanofibers and nanotubes. Carbon 2003, 41, 615–618. [Google Scholar] [CrossRef]
- Kajiura, H.; Huang, H.; Tsutsui, S.; Murakami, Y.; Miyakoshi, M. High purity fibrous carbon deposit on the anode surface in hydrogen DC arc-discharge. Carbon 2002, 40, 2423–2428. [Google Scholar] [CrossRef]
- Inagaki, M.; Yang, Y.; Kang, F. Carbon nanofibers prepared via electrospinning. Adv. Mater. 2012, 24, 2547–2566. [Google Scholar] [CrossRef]
- Zhang, L.; Aboagye, A.; Kelkar, A.; Lai, C.; Fong, H. A review: Carbon nanofibers from electrospun polyacrylonitrile and their applications. J. Mater. Sci. 2014, 49, 463–480. [Google Scholar] [CrossRef]
- Omoriyekomwan, J.E.; Tahmasebi, A.; Dou, J.; Wang, R. A review on the recent advances in the production of carbon nanotubes and carbon nanofibers via microwave-assisted pyrolysis of biomass. Fuel Proc. Technol. 2021, 214, 106686. [Google Scholar] [CrossRef]
- Zhang, J.; Tahmasebi, A.; Omoriyekomwan, J.E.; Yu, J. Direct synthesis of hollow carbon nanofibers on bio-char during microwave pyrolysis of pine nut shell. J. Anal. Appl. Pyrolysis 2018, 130, 142–148. [Google Scholar] [CrossRef]
- Bengtsson, A.; Bengtsson, J.; Sedin, M.; Sjoholm, E. Carbon Fibers from Lignin-Cellulose Precursors: Effect of Stabilization Conditions. ACS Sustain. Chem. Eng. 2019, 7, 8440–8448. [Google Scholar] [CrossRef] [Green Version]
- Nuryantini, A.Y.; Rahayu, F.; Mahen, E.C.S.; Sawitri, A.; Nuryadin, B.W. Synthesis of activated carbon fiber from pyrolyzed cotton for adsorption of fume pollutants. J. Phys. Conf. Ser. 2018, 1013, 012200. [Google Scholar] [CrossRef]
- Gardner, S.; Singamsetty, C.S.K.; Booth, G.L.; He, G.-R.; Pittman, J.C.U. Surface characterization of carbon fibers using angle-resolved XPS and ISS. Carbon 1995, 33, 587–595. [Google Scholar] [CrossRef]
- Akia, M.; Salinas, N.; Luna, S.; Medina, E.; Valdez, A.; Lopez, J.; Ayala, J.; Alcoutlabi, M.; Lozano, K. In situ synthesis of Fe3O4-reinforced carbon fiber composites as anodes in lithium-ion batteries. J. Mater. Sci. 2019, 54, 13479–13490. [Google Scholar] [CrossRef]
- He, G.; Song, Y.; Chen, S.; Wang, L. Porous carbon nanofiber mats from electrospun polyacrylonitrile/polymethylmethacrylate composite nanofibers for super-capacitor electrode materials. J. Mater. Sci. 2018, 53, 9721–9730. [Google Scholar] [CrossRef]
- Li, T.F.; Lv, Y.L.; Su, J.H.; Wang, Y.; Yang, Q.; Zhang, Y.W.; Zhou, J.C.; Xu, L.; Sun, D.M.; Tang, Y.W. Anchoring CoFe2O4 nanoparticles on N-doped carbon nanofibers for high-performance oxygen evolution reaction. Adv. Sci. 2017, 4, 1700226. [Google Scholar] [CrossRef]
- Chiang, Y.; Wu, C.; Chen, Y. Effects of activation on the properties of electrospun carbon nanofibers and their adsorption performance for carbon dioxide. Sep. Purif. Technol. 2020, 233, 116040. [Google Scholar] [CrossRef]
- Lindsay, B.; Abel, M.-L.; Watts, J.F. A study of electrochemically treated PAN based carbon fibres by IGC and XPS. Carbon 2007, 45, 2433–2444. [Google Scholar] [CrossRef]
- Boukhvalov, D.W.; Zhidkov, I.S.; Kiryakov, A.; Menendez, J.L.; Garcia, L.F.; Kukharenko, A.I.; Cholakh, S.O.; Zatsepin, A.F.; Kurmaev, E.Z. Unveiling the Atomic and Electronic Structure of StackedCup Carbon Nanofibers. Nanoscale Res. Lett. 2021, 16, 153. [Google Scholar] [CrossRef]
- Yadava, D.; Aminib, F.; Ehrmann, A. Recent advances in carbon nanofibers and their applications–A review. Eur. Polym. J. 2020, 138, 109963. [Google Scholar] [CrossRef]
- Guo, Q.; Liu, D.; Zhang, X.; Li, L.; Hou, H.; Niwa, O.; You, T. Pd-Ni Alloy Nanoparticle/Carbon Nanofiber Composites:Preparation, Structure, and Superior Electrocatalytic Properties for Sugar Analysis. Anal. Chem. 2014, 86, 5898–5905. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Wan, K.; Liu, B.; Wang, Y.; Wei, G. Recent advance in the fabrication of carbon nanofiber-based composite materials for wearable devices. Nanotechnology 2021, 32, 442001. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Wang, Y.; Gong, C.; Liu, B.; Wei, G. Production, structural design, functional control, and broad applications of carbon nanofiber-based nanomaterials: A comprehensive review. Chem. Eng. J. 2020, 402, 126189. [Google Scholar] [CrossRef]





















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
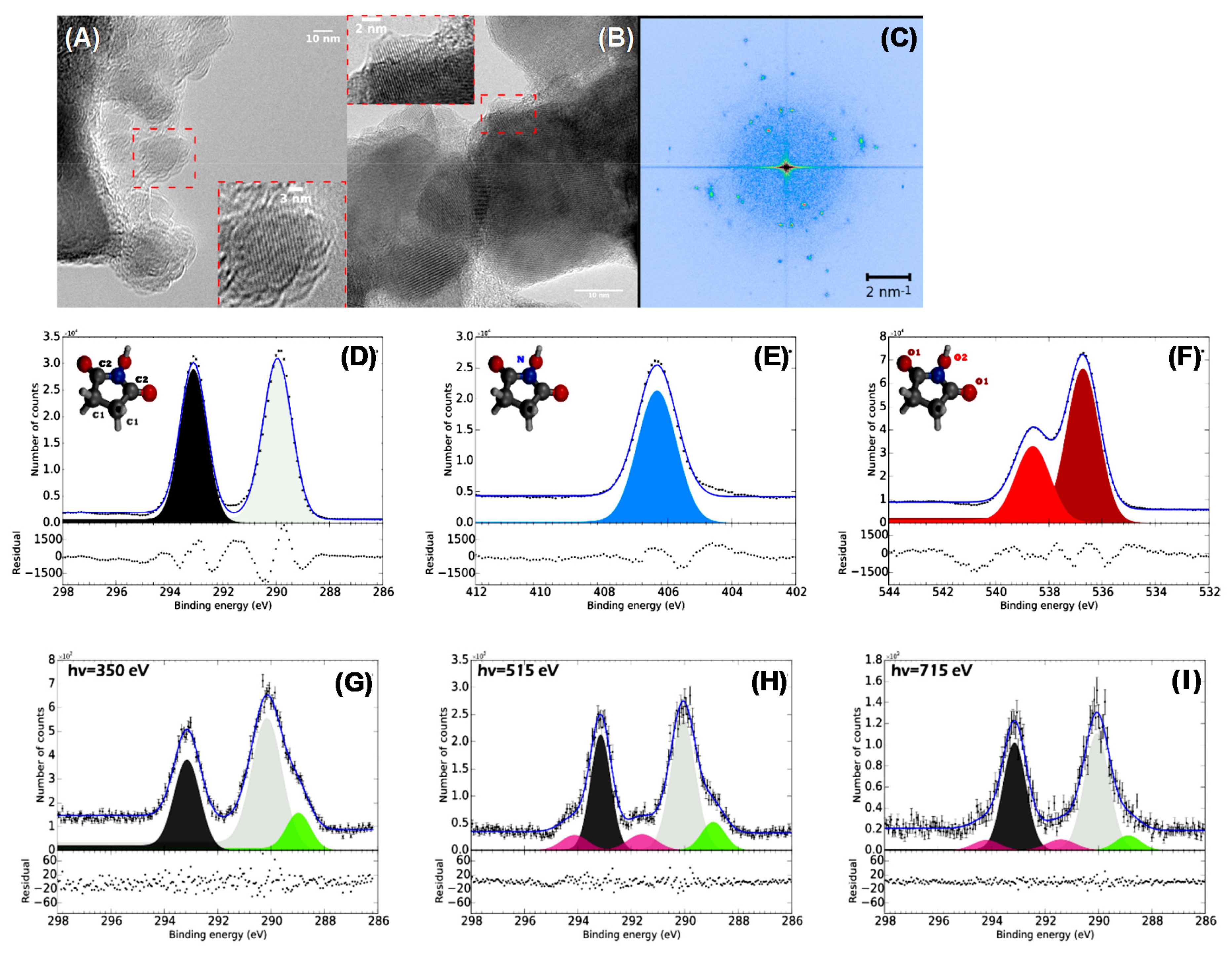{kind=link}
{kind=link}
| XPS | UPS: HeI | HeII | UV Synchrotron | HAXPES | ||
|---|---|---|---|---|---|---|
| Energy (eV) | 1486.6 (Al Kα) | 1253.6 (Mg Kα) | 20.2 | 40.8 | 10–102 | 102–104 eV |
| Δλ (eV) | 0.25 | 0.7 | 0.003 | 0.017 | 10−4 | ~0.01 eV |
| Energy resolution (eV) | 0.28 | 0.9–1 | <0.1 | 0.01 | 0.6 | |
| Lateral resolution (μm) | 3 | 2 | - | Submicron | ||
| Sampling depth (nm) | 3–10 | 0–1.5 | 0–2 | 0–20 | ||
| Angle-resolved | Yes | Yes | Yes | Yes | ||
| X diffraction | // | // | Yes | Yes | ||
| Characterization | ||||||
| Electronic structure | XPS VB and DOS | UV DOS | UV DOS | XPS VB and DOS | ||
| Quantum confinement | BE shift of core lines | Work function shift, energy gaps | Work function shift, energy gaps | BE shift of core lines | ||
| Structural information | NP size estimation | Presence of curvature | Presence of curvature | NP size estimation | ||
| Chemical information | Chemical shift | VB orbital bands | VB orbital bands | Chemical shift | ||
| Different hybridization | Core lines, VB and KLL Auger spectra | VB analysis | VB analysis | Core lines, VB and KLL Auger spectra | ||
| Band structure | Angle-resolved VB | Angle-resolved VB | Angle-resolved VB | Angle-resolved VB | ||
| Microscopy | - | - | - | Submicron structure | ||
| Property | VGCNF | SWCNT | MWCNT | CNF |
|---|---|---|---|---|
| Diameter (nm) | 50–200 | 0.6–0.8 | 5–50 | 7300 |
| Aspect ratio | 250–2000 | 100–10,000 | 100–10,000 | 440 |
| Density (g/cm3) | 2 | ~1.3 | ~1.75 | 1.74 |
| Thermal conductivity (W/mK) | 1950 | 3000–6000 | 3000–6000 | 20 |
| Electrical resistivity (Ω/cm) | 1 × 10−4 | 1 × 10−3–1 × 10−4 | 2 × 10−3–1 × 10−4 | 1.7 × 10−3 |
| Tensile strength (GPa) | 2.92 | 50–500 | 10–60 | 3.8 |
| Tensile modulus (GPa) | 240 | 1500 | 1000 | 227 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Speranza, G. Characterization of Carbon Nanostructures by Photoelectron Spectroscopies. Materials 2022, 15, 4434. https://doi.org/10.3390/ma15134434
Speranza G. Characterization of Carbon Nanostructures by Photoelectron Spectroscopies. Materials. 2022; 15(13):4434. https://doi.org/10.3390/ma15134434
Chicago/Turabian StyleSperanza, Giorgio. 2022. "Characterization of Carbon Nanostructures by Photoelectron Spectroscopies" Materials 15, no. 13: 4434. https://doi.org/10.3390/ma15134434
APA StyleSperanza, G. (2022). Characterization of Carbon Nanostructures by Photoelectron Spectroscopies. Materials, 15(13), 4434. https://doi.org/10.3390/ma15134434