
Dissolution of Portlandite in Pure Water: Part 2 Atomistic Kinetic Monte Carlo (KMC) Approach

 ,
,  ,
,  ,
, 
Abstract
:1. Introduction
2. Methods and Computational Models
2.1. Quantum Chemistry Computations
2.2. Atomistic Kinetic Monte Carlo
2.3. The MATLAB Code Implementation by Employing the KMC Upscaling Approach
3. Results and Discussions
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Salah Uddin, K.M.; Izadifar, M.; Ukrainczyk, N.; Koenders, E.; Middendorf, B. Dissolution of Portlandite in Pure Water: Part 1 Molecular Dynamics (MD) Approach. Materials 2022, 15, 1404. [Google Scholar] [CrossRef]
- Chatterjee, A.; Vlachos, D.G. An overview of spatial microscopic and accelerated kinetic Monte Carlo methods. J. Comput. Mater. Des. 2007, 14, 253–308. [Google Scholar] [CrossRef]
- Elts, E.; Greiner, M.; Briesen, H. Predicting Dissolution Kinetics for Active Pharmaceutical Ingredients on the Basis of Their Molecular Structures. Cryst. Growth Des. 2016, 16, 4154–4164. [Google Scholar] [CrossRef]
- Piana, S.; Reyhani, M.; Gale, J.D. Simulating micrometre-scale crystal growth from solution. Nature 2005, 438, 70–73. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Wei, S.; Wang, Q.; Tang, M.; Shen, X.; Zou, X.; Shen, Y.; Ma, B. Morphology prediction of portlandite: Atomistic simulations and experimental research. Appl. Surf. Sci. 2020, 502, 144296. [Google Scholar] [CrossRef]
- Martin, P.; Manzano, H.; Dolado, J.S. Mechanisms and dynamics of mineral dissolution: A new kinetic Monte Carlo model. Adv. Theory Simul. 2019, 2, 1900114. [Google Scholar] [CrossRef] [Green Version]
- Martin, P.; Gaitero, J.J.; Dolado, J.S.; Manzano, H. New Kinetic Monte Carlo Model to Study the Dissolution of Quartz. ACS Earth Space Chem. 2021, 5, 516–524. [Google Scholar] [CrossRef]
- Perko, J.; Ukrainczyk, N.; Šavija, B.; Phung, Q.T.; Koenders, E.A.B. Influence of Micro-Pore Connectivity and Micro-Fractures on Calcium Leaching of Cement Pastes—A Coupled Simulation Approach. Materials 2020, 13, 2697. [Google Scholar] [CrossRef]
- Yang, T.; Keller, B.; Magyari, E.; Hametner, K.; Günther, D. Direct observation of the carbonation process on the surface of calcium hydroxide crystals in hardened cement paste using an Atomic Force Microscope. J. Mater. Sci. 2003, 38, 1909–1916. [Google Scholar] [CrossRef]
- Czechowski, F.; Marcinkowski, T. Primary sludge stabilisation with calcium hydroxide. Environ. Chem. Lett. 2006, 4, 11–14. [Google Scholar] [CrossRef]
- Ma, X.; Ye, J.; Jiang, L.; Sheng, L.; Liu, J.; Li, Y.-Y.; Xu, Z.P. Alkaline fermentation of waste activated sludge with calcium hydroxide to improve short-chain fatty acids production and extraction efficiency via layered double hydroxides. Bioresour. Technol. 2019, 279, 117–123. [Google Scholar] [CrossRef]
- Wang, X.-Y. Effect of fly ash on properties evolution of cement based materials. Constr. Build. Mater. 2014, 69, 32–40. [Google Scholar] [CrossRef]
- Yu, Z.; Ye, G. New perspective of service life prediction of fly ash concrete. Constr. Build. Mater. 2013, 48, 764–771. [Google Scholar] [CrossRef]
- Glass, G.K.; Reddy, B.; Buenfeld, N.R. Corrosion inhibition in concrete arising from its acid neutralisation capacity. Corros. Sci. 2000, 42, 1587–1598. [Google Scholar] [CrossRef]
- Sato, T.; Beaudoin, J.J.; Ramachandran, V.S.; Mitchell, L.D.; Tumidajski, P.J. Thermal decomposition of nanoparticulate Ca(OH)2-anomalous effects. Adv. Cem. Res. 2007, 19, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Sun, Z.; Chi, H.; Fan, L.-S. Physical and Chemical Mechanism for Increased Surface Area and Pore Volume of CaO in Water Hydration. Ind. Eng. Chem. Res. 2012, 51, 10793–10799. [Google Scholar] [CrossRef]
- Ukrainczyk, N.; Muthu, M.; Vogt, O.; Koenders, E. Geopolymer, Calcium Aluminate, and Portland Cement-Based Mortars: Comparing Degradation Using Acetic Acid. Materials 2019, 12, 3115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drugă, B.; Ukrainczyk, N.; Weise, K.; Koenders, E.; Lackner, S. Interaction between wastewater microorganisms and geopolymer or cementitious materials: Biofilm characterization and deterioration characteristics of mortars. Int. Biodeterior. Biodegrad. 2018, 134, 58–67. [Google Scholar] [CrossRef]
- Regnault, O.; Lagneau, V.; Catalette, H.; Schneider, H. Experimental study of pure mineral phases/supercritical CO2 reactivity. Implications for geological CO2 sequestration. C. R. Geosci. 2005, 337, 1331–1339. [Google Scholar] [CrossRef]
- Gu, W.; Bousfield, D.W.; Tripp, C.P. Formation of calcium carbonate particles by direct contact of Ca(OH)2 powders with supercritical CO2. J. Mater. Chem. 2006, 16, 3312–3317. [Google Scholar] [CrossRef]
- Boualleg, S.; Bencheikh, M.; Belagraa, L.; Daoudi, A.; Chikouche, M.A. The Combined Effect of the Initial Cure and the Type of Cement on the Natural Carbonation, the Portlandite Content, and Nonevaporable Water in Blended Cement. Adv. Mater. Sci. Eng. 2017, 2017, 5634713. [Google Scholar] [CrossRef] [Green Version]
- Galmarini, S.; Aimable, A.; Ruffray, N.; Bowen, P. Changes in portlandite morphology with solvent composition: Atomistic simulations and experiment. Cem. Concr. Res. 2011, 41, 1330–1338. [Google Scholar] [CrossRef] [Green Version]
- Jam, A.N.; Jam, N.N.; Izadifar, M.; Rabczuk, T. Molecular dynamics study on the crack propagation in carbon doped polycrystalline boron-nitride nanosheets. Comput. Mater. Sci. 2022, 203, 111066. [Google Scholar] [CrossRef]
- Izadifar, M.; Thissen, P.; Abadi, R.; Jam, A.N.; Gohari, S.; Burvill, C.; Rabczuk, T. Fracture toughness of various percentage of doping of boron atoms on the mechanical properties of polycrystalline graphene: A molecular dynamics study. Phys. E Low-Dimens. Syst. Nanostruct. 2019, 114, 113614. [Google Scholar] [CrossRef]
- Kohn, W.; Sham, L.J. Self-Consistent Equations Including Exchange and Correlation Effects. Phys. Rev. 1965, 140, A1133–A1138. [Google Scholar] [CrossRef] [Green Version]
- Izadifar, M.; Dolado, J.S.; Thissen, P.; Ayuela, A. Interactions between Reduced Graphene Oxide with Monomers of (Calcium) Silicate Hydrates: A First-Principles Study. Nanomaterials 2021, 11, 2248. [Google Scholar] [CrossRef]
- Izadifar, M.; Königer, F.; Gerdes, A.; Wöll, C.; Thissen, P. Correlation between Composition and Mechanical Properties of Calcium Silicate Hydrates Identified by Infrared Spectroscopy and Density Functional Theory. J. Phys. Chem. C 2019, 123, 10868–10873. [Google Scholar] [CrossRef]
- Ren, K.; Zheng, R.; Xu, P.; Cheng, D.; Huo, W.; Yu, J.; Zhang, Z.; Sun, Q. Electronic and Optical Properties of Atomic-Scale Heterostructure Based on MXene and MN (M = Al, Ga): A DFT Investigation. Nanomaterials 2021, 11, 2236. [Google Scholar] [CrossRef]
- Ren, K.; Shu, H.; Huo, W.; Cui, Z.; Yu, J.; Xu, Y. Mechanical, electronic and optical properties of a novel B2P6 monolayer: Ultrahigh carrier mobility and strong optical absorption. Phys. Chem. Chem. Phys. 2021, 23, 24915–24921. [Google Scholar] [CrossRef]
- Izadifar, M.; Thissen, P.; Steudel, A.; Kleeberg, R.; Kaufhold, S.; Kaltenbach, J.; Schuhmann, R.; Dehn, F.; Emmerich, K. Comprehensive Examination of Dehydroxylation of Kaolinite, Disordered Kaolinite, and Dickite: Experimental Studies and Density Functional Theory. Clays Clay Miner. 2020, 68, 319–333. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B 1993, 47, 558–561. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [Green Version]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Momma, K.; Izumi, F. VESTA: A three-dimensional visualization system for electronic and structural analysis. J. Appl. Cryst. 2008, 41, 653–658. [Google Scholar] [CrossRef]
- Sheppard, D.; Xiao, P.; Chemelewski, W.; Johnson, D.D.; Henkelman, G. A generalized solid-state nudged elastic band method. J. Chem. Phys. 2012, 136, 074103. [Google Scholar] [CrossRef]
- Sheppard, D.; Terrell, R.; Henkelman, G. Optimization methods for finding minimum energy paths. J. Chem. Phys. 2008, 128, 134106. [Google Scholar] [CrossRef] [Green Version]
- Jansen, A.P.J. An Introduction to Kinetic Monte Carlo Simulations of Surface Reactions; Springer: New York, NY, USA, 2012. [Google Scholar]
- Wang, J.U.N.; Keener, T.I.M.C.; Li, G.; Khang, S.-J. The Dissolution Rate of Ca(OH)2 in Aqueous Solutions. Chem. Eng. Commun. 1998, 169, 167–184. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
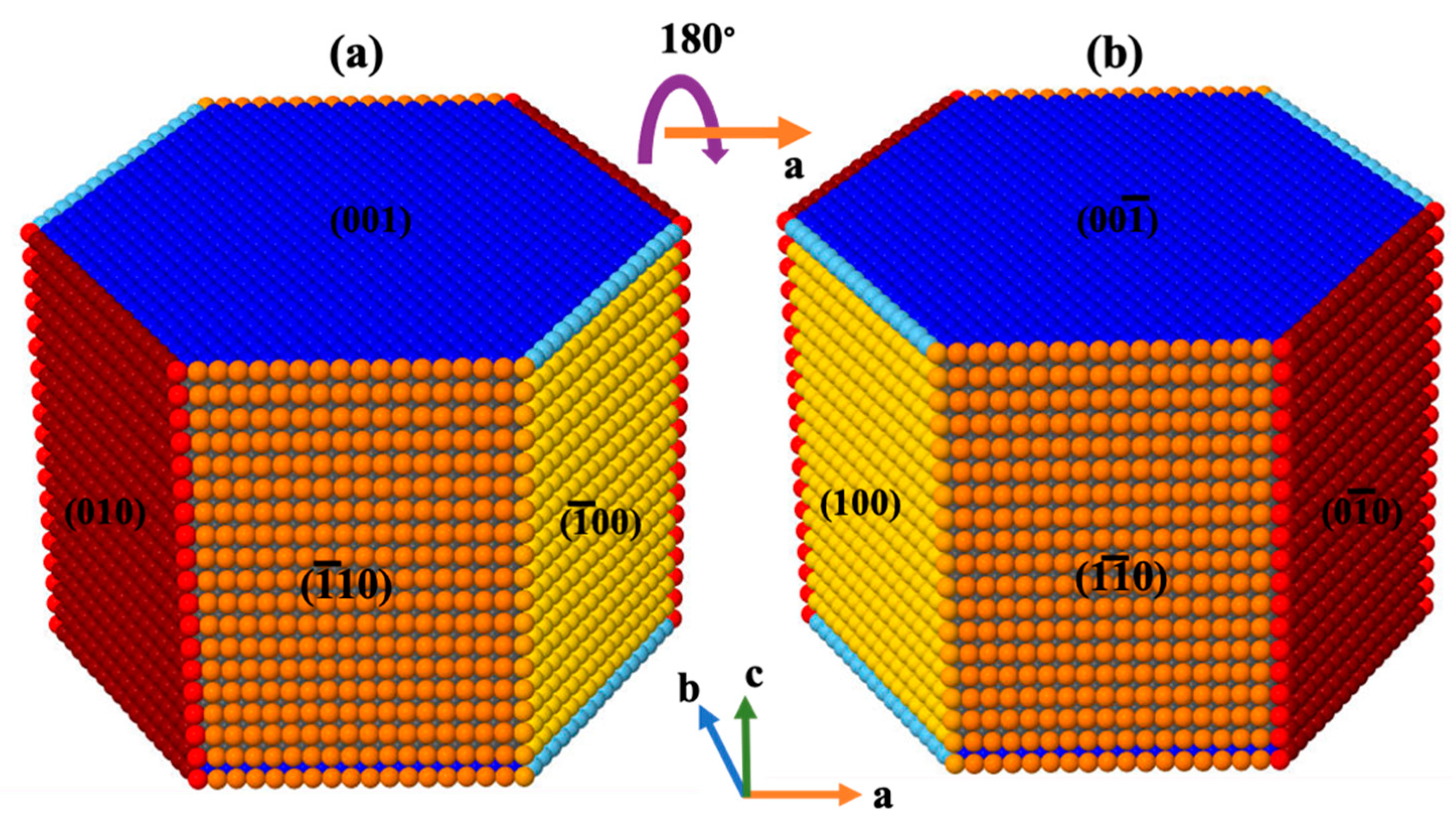{kind=link}
{kind=link}
{kind=link}
{kind=link}
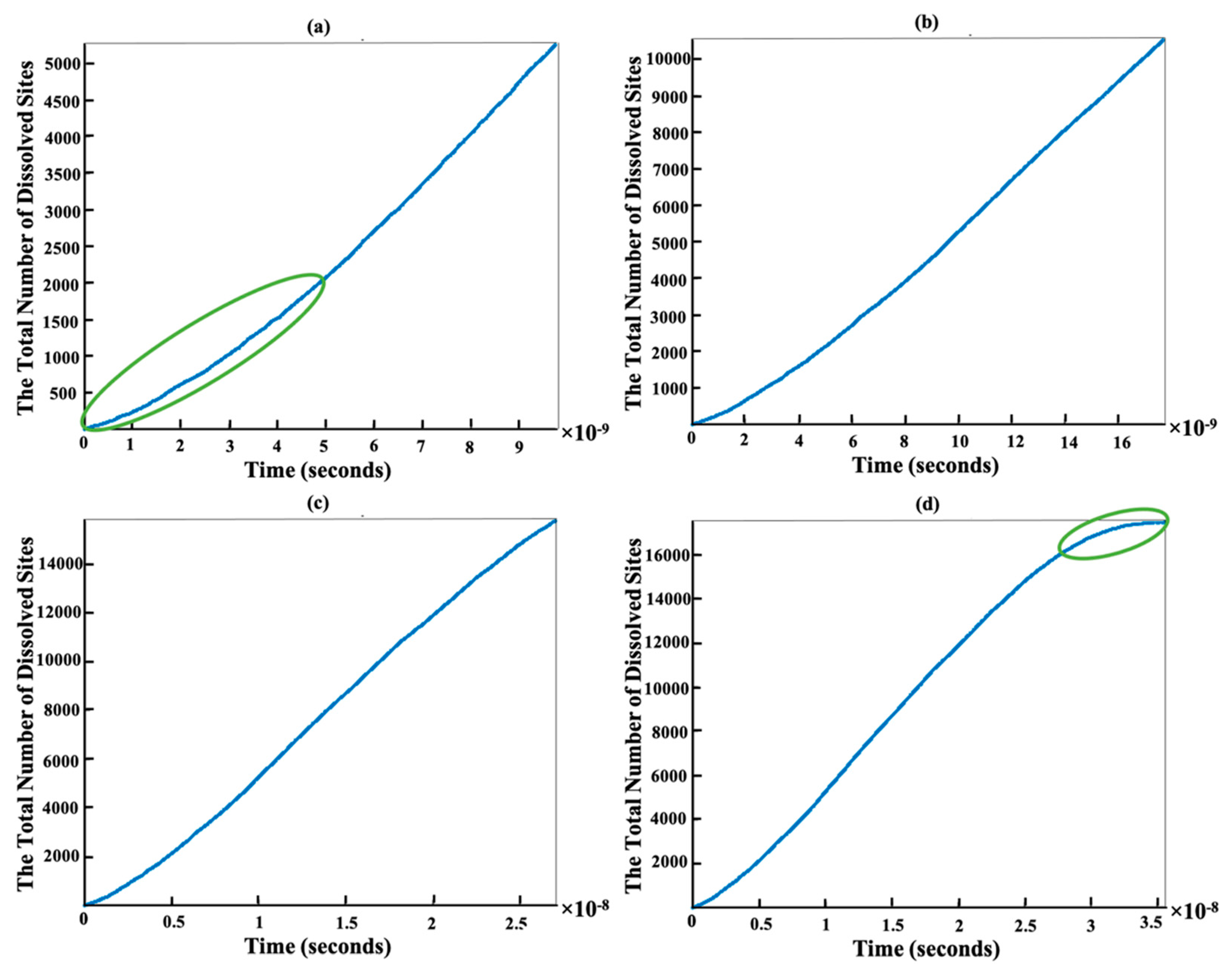{kind=link}
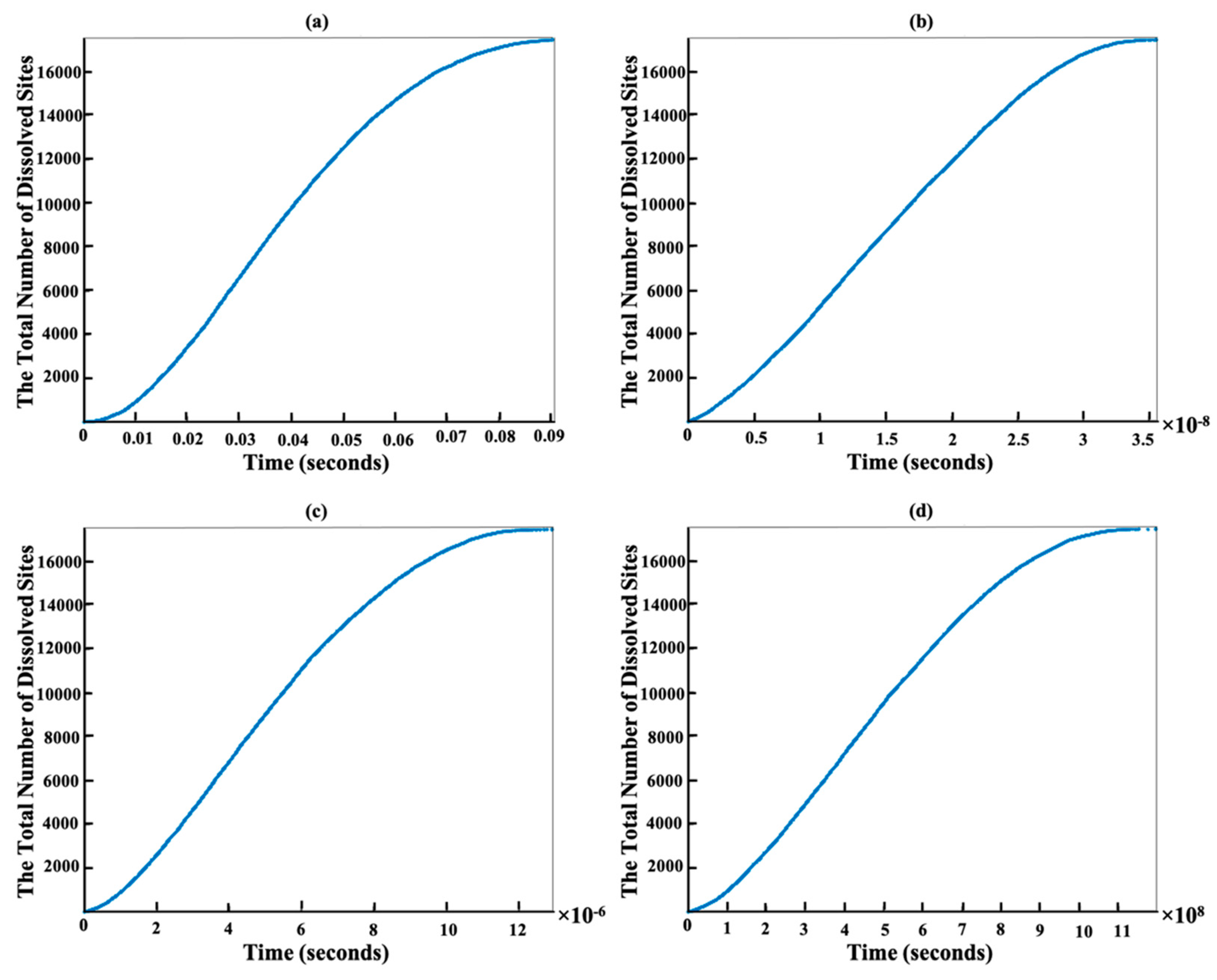{kind=link}
| Figure 2 | (a) | (b) | (c) | (d) | (e) | (f) | (g) |
|---|---|---|---|---|---|---|---|
| ΔG* (kJ/mol) | 352.00 | 199.10 | 175.40 | 56.14 | 55.80 | 54.90 | 25.90 |
| ΔHa (kJ/mol) | 242.18 | 140.87 | 120.12 | 41.43 | 40.85 | 39.87 | 18.32 |
| k (s−1) | 1.243 × 10−49 | 7.849 × 10−23 | 1.119 × 10−18 | 0.897 × 103 | 1.029 × 103 | 1.479 × 103 | 1.791 × 108 |
| Figure 3 | (a) | (b) | (c) |
|---|---|---|---|
| ΔG* (kJ/mol) | 195.30 | 114.60 | 70.00 |
| k (s−1) | 3.638 × 10−22 | 5.081 × 10−8 | 3.337 |
| Figure 3 | (a) | (b) | (c) |
|---|---|---|---|
| ΔG* (kJ/mol) | 29.90 | 20.55 | 7.1 |
| k (s−1) | 3.565 × 107 | 1.552 × 109 | 3.536 × 1011 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Izadifar, M.; Ukrainczyk, N.; Salah Uddin, K.M.; Middendorf, B.; Koenders, E. Dissolution of Portlandite in Pure Water: Part 2 Atomistic Kinetic Monte Carlo (KMC) Approach. Materials 2022, 15, 1442. https://doi.org/10.3390/ma15041442
Izadifar M, Ukrainczyk N, Salah Uddin KM, Middendorf B, Koenders E. Dissolution of Portlandite in Pure Water: Part 2 Atomistic Kinetic Monte Carlo (KMC) Approach. Materials. 2022; 15(4):1442. https://doi.org/10.3390/ma15041442
Chicago/Turabian StyleIzadifar, Mohammadreza, Neven Ukrainczyk, Khondakar Mohammad Salah Uddin, Bernhard Middendorf, and Eduardus Koenders. 2022. "Dissolution of Portlandite in Pure Water: Part 2 Atomistic Kinetic Monte Carlo (KMC) Approach" Materials 15, no. 4: 1442. https://doi.org/10.3390/ma15041442
APA StyleIzadifar, M., Ukrainczyk, N., Salah Uddin, K. M., Middendorf, B., & Koenders, E. (2022). Dissolution of Portlandite in Pure Water: Part 2 Atomistic Kinetic Monte Carlo (KMC) Approach. Materials, 15(4), 1442. https://doi.org/10.3390/ma15041442