
Synthesis and Crystal Structure of the Zintl Phases NaSrSb, NaBaSb and NaEuSb


Abstract
:1. Introduction
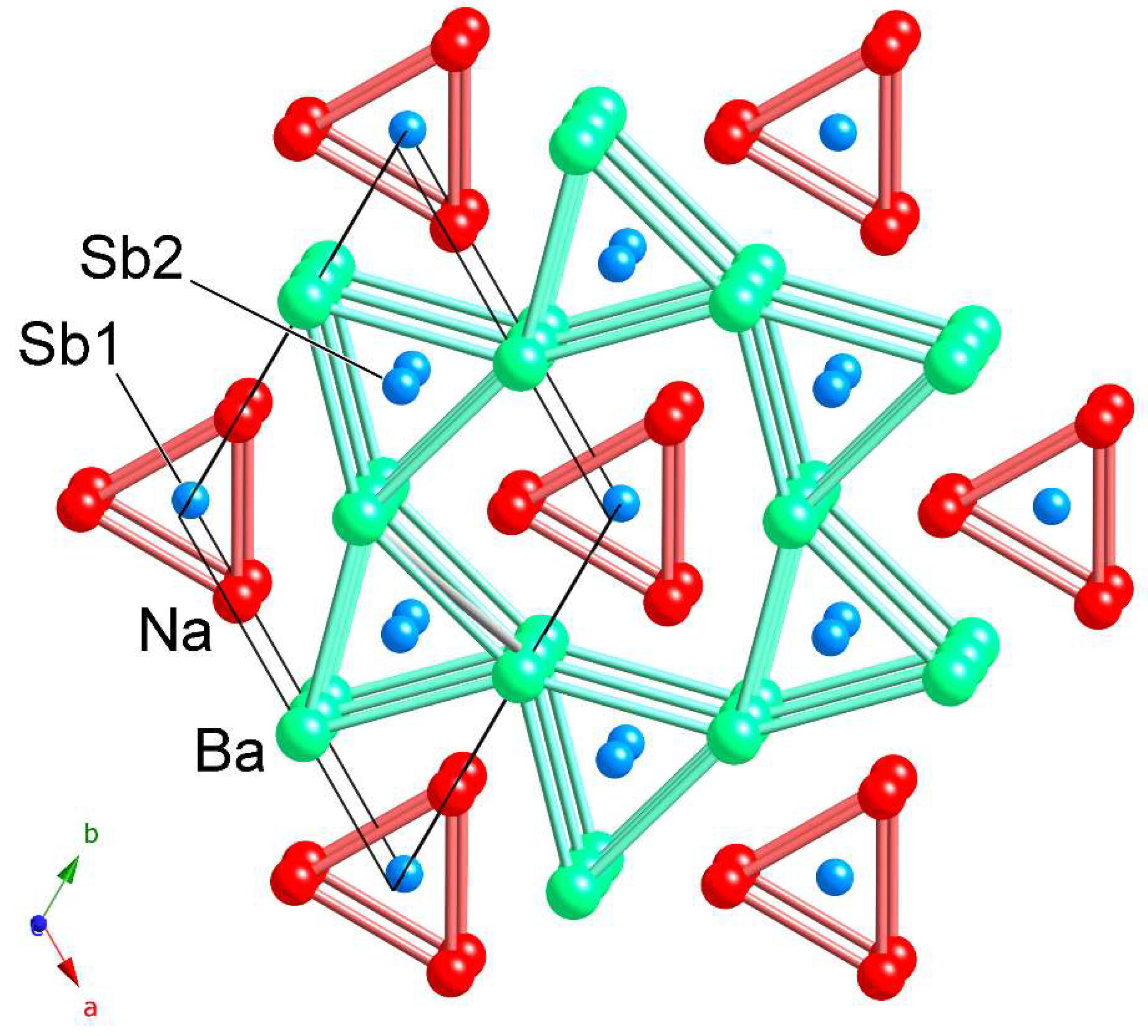
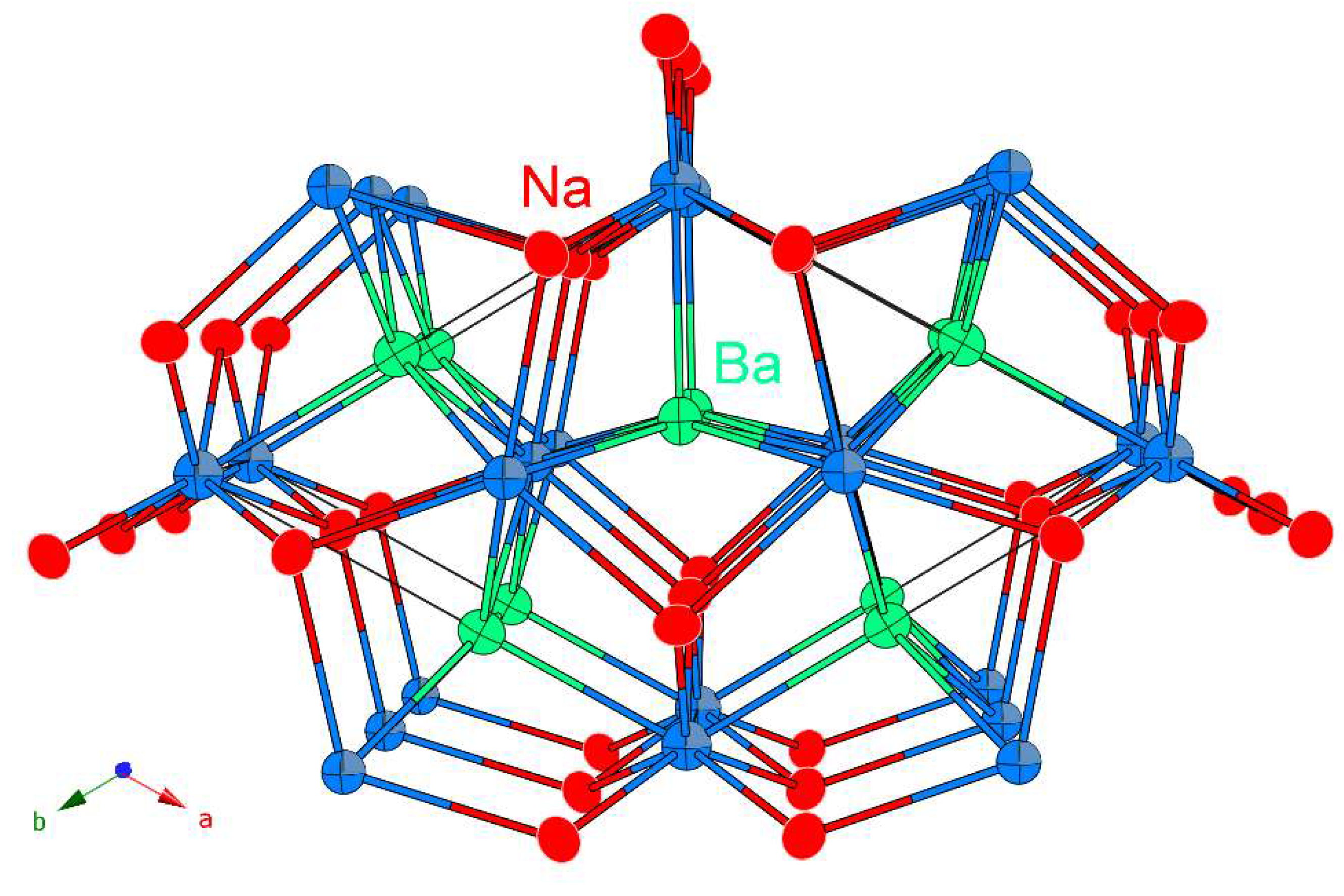
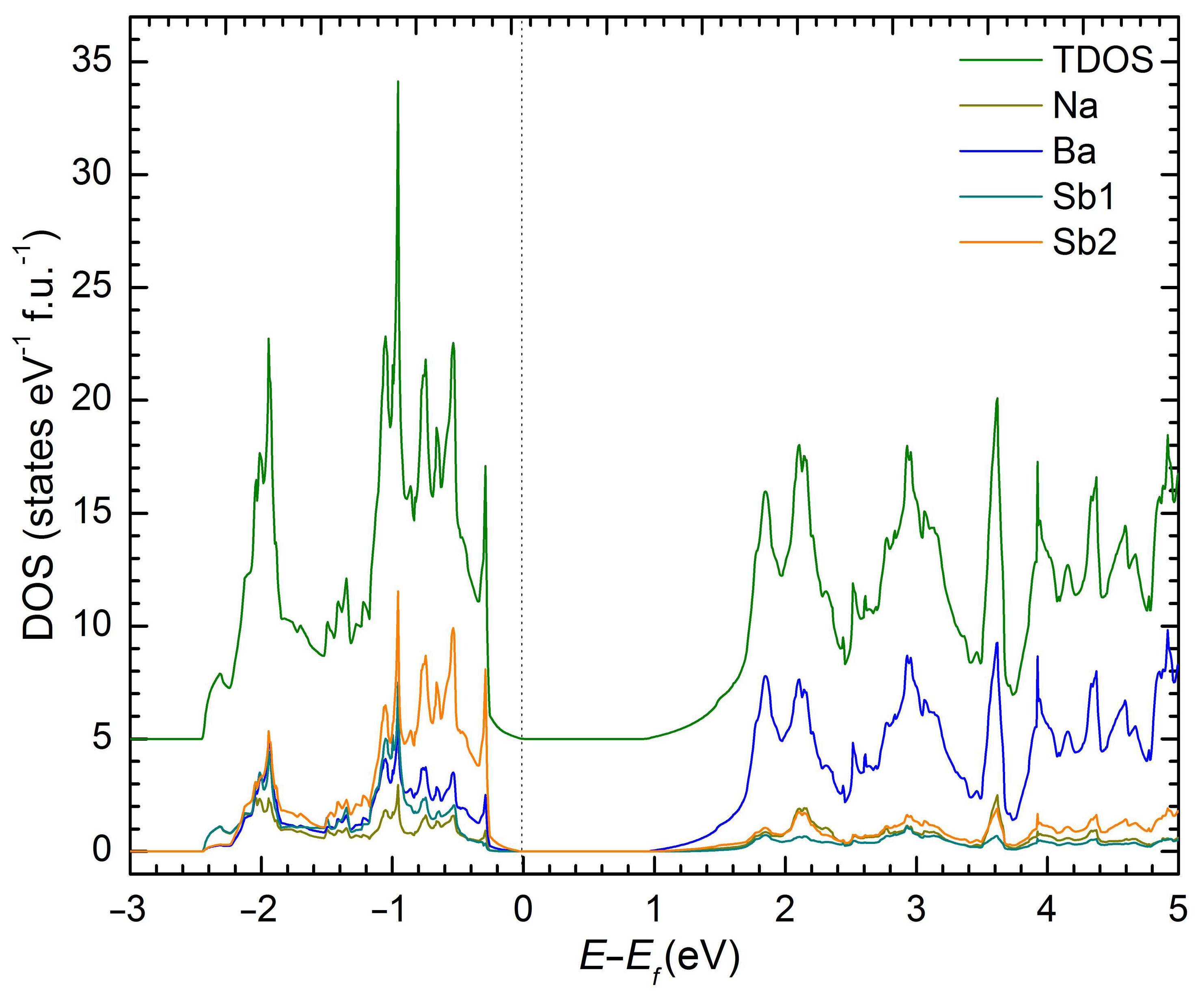
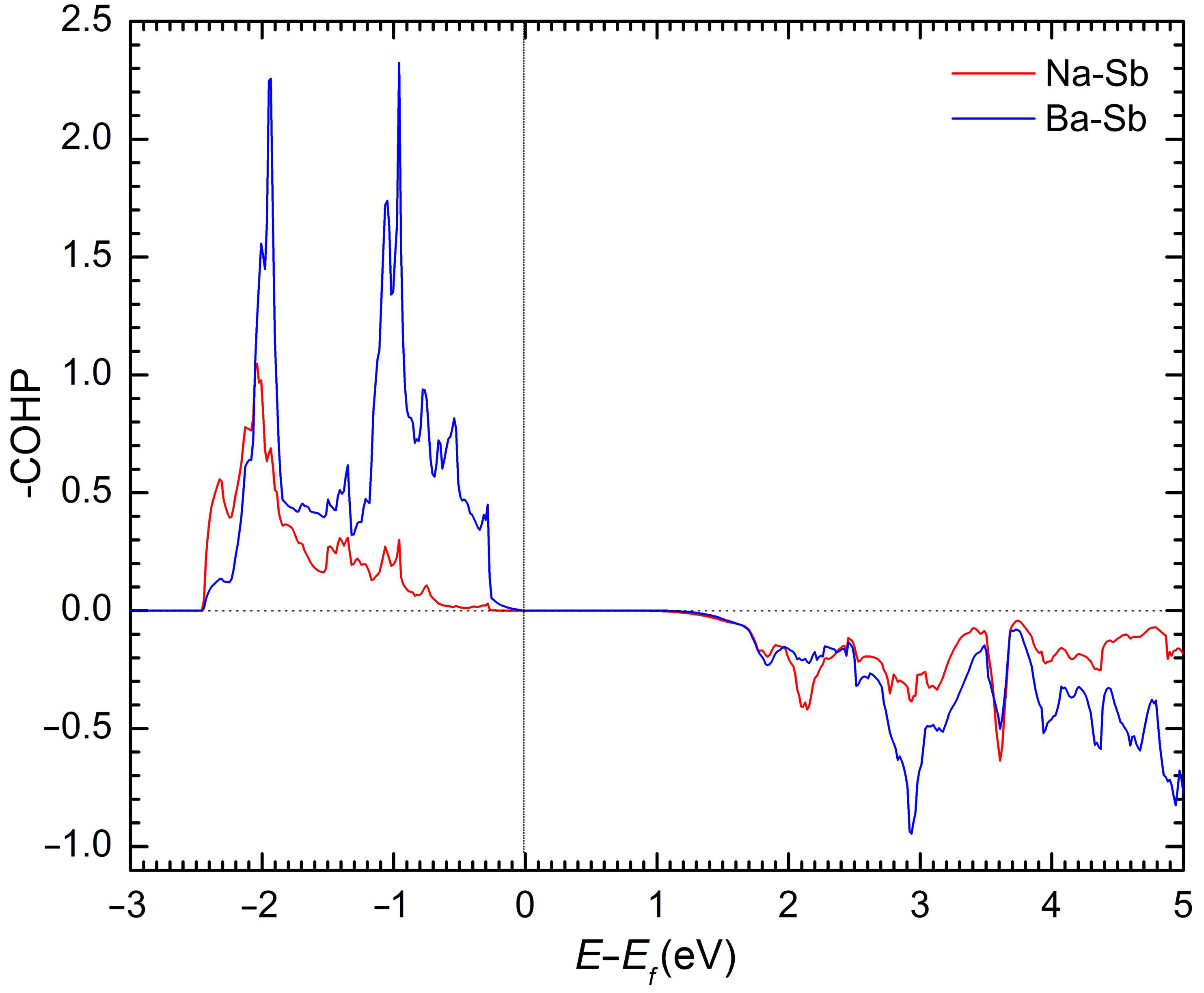
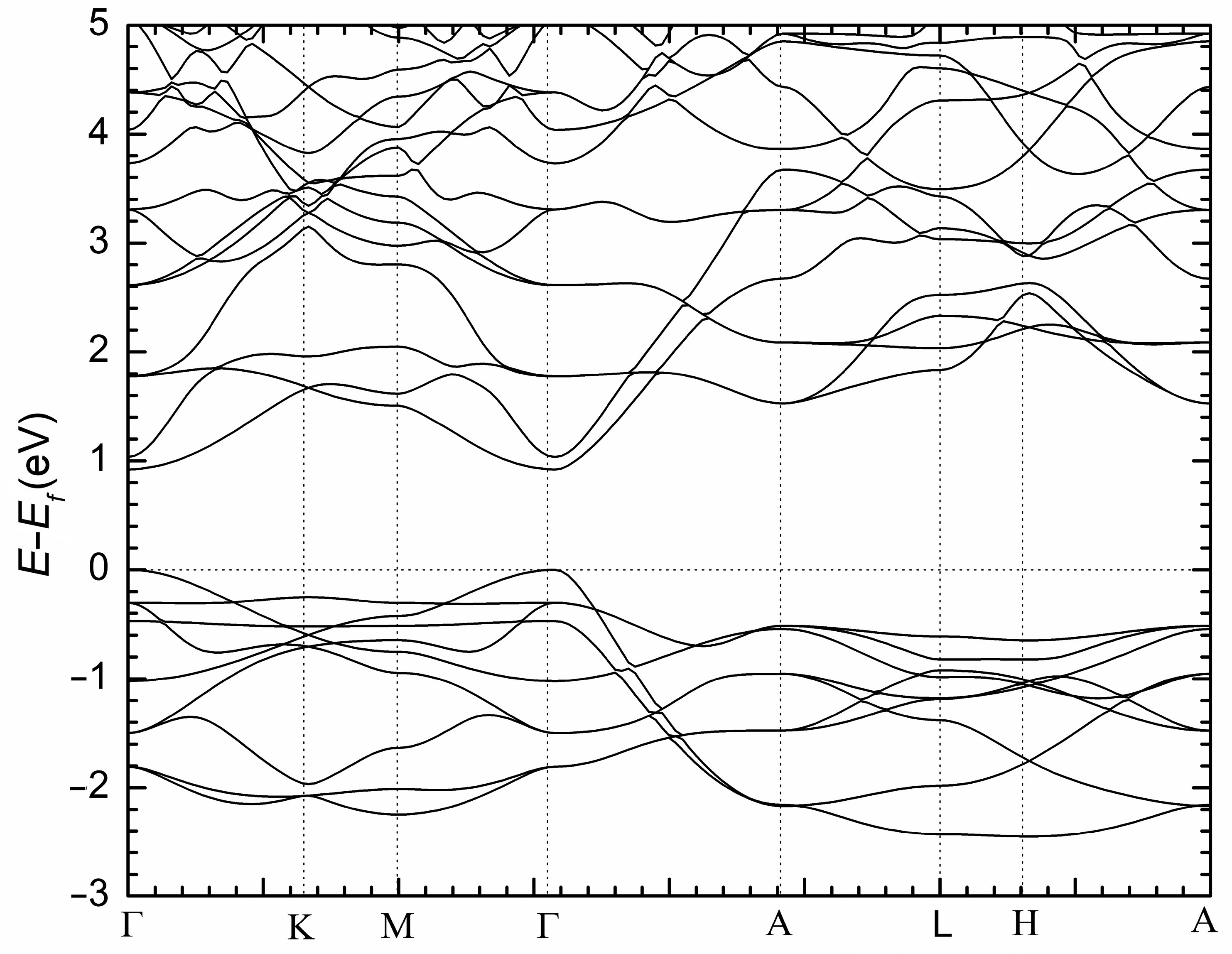
2. Results and Discussion
3. Materials and Methods
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Villars, P.; Calvert, L.D. (Eds.) Pearson’s Handbook of Crystallographic Data for Intermetallic Compounds, 2nd ed.; American Society for Metals: Materials Park, OH, USA, 1991. [Google Scholar]
- El Maslout, A.; Motte, J.P.; Gleitzer, C.; Aubry, J. Preparation et proprietes d’un nouveau compose dans la serie LiMP: Le phosphure de lithium-cadmium LiCdP. Comptes Rendus Seances L’academie Sci. Ser. C Sci. Chim. 1971, 273, 707–710. [Google Scholar]
- Tiburtius, C.; Schuster, H.U. LiBeSb und LiZnBi, ternaere Verbindungen mit Wurtzitgeruest. Z. Naturforsch. 1978, 33b, 35–38. [Google Scholar] [CrossRef]
- Krenkel, B.; Schuster, H.U. NaMgAs(Sb)—Ternaere Verbindungen mit modifizierter Cu2Sb-Struktur. Z. Naturforsch. 1978, 33b, 1080–1082. [Google Scholar]
- Hirt, H.; Deiseroth, H.J. Crystal structure of potassium calcium bismuthide, KCaBi. Z. Kristallogr. 2003, 218, 5. [Google Scholar]
- Tiburtius, C.; Schuster, H.U. NaBeAs(Sb)—Ternaere Phasen im ‘aufgefuellten’ NiAs (Ni2In)-Typ. Z. Naturforsch. 1978, 32b, 1133–1138. [Google Scholar]
- Dong, Y.K.; DiSalvo, F.J. Synthesis and single crystal structures of ternary phosphides Li4SrP2 and AAeP (A = Li, Na; Ae = Sr, Ba). J. Solid State Chem. 2007, 180, 432–439. [Google Scholar]
- Eisenmann, B.; Liebrich, O.; Schäfer, H.; Weiss, A. Darstellung und Kristallstruktur von CaLiSb (Ternaere E-Phasen von Hauptgruppenelementen II). Z. Naturforsch. 1969, 24b, 1344–1345. [Google Scholar] [CrossRef]
- Feng, X.J.; Prots, Y.; Schmidt, M.P.; Hoffmann, S.; Schnelle, W.; Burkhardt, U.; Zhao, J.-T.; Grin, Y. Synthesis, structure, and properties of two Zintl phases around the composition SrLiAs. Inorg. Chem. 2013, 52, 8971–8978. [Google Scholar] [CrossRef]
- Albering, J.H.; Ebel, T.; Jeitschko, W. Praeparation, Kristallstruktur und magnetische Eigenschaften der Verbindungen LiAX (A = Ca, Sr, Ba, Eu, Yb; X = P, As, Sb, Bi). Z. Kristallogr. Suppl. Issue 1997, 12, 242. [Google Scholar]
- Carrillo Cabrera, W.; Somer, M.; Peters, E.M.; Peters, K.; von Schnering, H.-G. Crystal structure of sodium strontium arsenide, NaSrAs. Z. Kristallogr. 1997, 212, 252. [Google Scholar] [CrossRef]
- Carrillo Cabrera, W.; Somer, M.; Peters, E.M.; Peters, K.; von Schnering, H.-G. Crystal structure of sodium barium phosphide, NaBaP. Z. Kristallogr. 1997, 212, 191. [Google Scholar] [CrossRef]
- Hirt, H.; Deiseroth, H.J. The new polar intermetallic compound NaBaBi. Z. Anorg. Allg. Chem. 2004, 630, 1357–1359. [Google Scholar] [CrossRef]
- Ovchinnikov, A.; Bobev, S. Zintl phases with group 15 elements and the transition metals: A brief overview of pnictides with diverse and complex structures. J. Solid State Chem. 2019, 270, 346. [Google Scholar]
- Baranets, S.; Ovchinnikov, A.; Bobev, S. Chapter 322: Structural diversity of the Zintl pnictides with rare-earth metals. In Handbook of Chemistry and Physics of the Rare Earths; Elsevier: Amsterdam, The Netherlands, 2021; Volume 60, pp. 227–324. [Google Scholar]
- Schäfer, M.C.; Suen, N.-T.; Bobev, S. Synthesis and crystal chemistry of new ternary pnictides containing lithium—Adding structural complexity one step at a time. Dalton Trans. 2014, 43, 1688. [Google Scholar]
- Makongo, J.P.A.; You, T.-S.; He, H.; Suen, N.-T.; Bobev, S. New lithium-containing pnictides with 1-D infinite chains of super-tetrahedral clusters. Synthesis, crystal and electronic structure of Ba4Li2Cd3Pn6 (Pn = P, As and Sb). Eur. J. Inorg. Chem. 2014, 2014, 5113. [Google Scholar] [CrossRef]
- Ojwang, D.O.; Bobev, S. Synthesis and structural characterization of Ba7Li11Bi10 and AE4(Li,Tr)7Pn6 (AE = Sr, Ba, Eu; Tr = Ga, In; Pn = Sb, Bi). Inorganics 2018, 6, 109. [Google Scholar]
- Wang, Y.; Suen, N.-T.; Kunene, T.; Stoyko, S.; Bobev, S. Synthesis and structural characterization of the Zintl phases Na3Ca3TrPn4, Na3Sr3TrPn4, and Na3Eu3TrPn4 (Tr = Al, Ga, In; Pn = P, As, Sb). J. Solid State Chem. 2017, 249, 160. [Google Scholar] [CrossRef]
- Wang, Y.; Stoyko, S.; Bobev, S. Quaternary pnictides with complex, non-centrosymmetric structures. Synthesis and structural characterization of the new Zintl phases Na11Ca2Al3Sb8, Na4CaGaSb3 and Na15Ca3In5Sb12. Inorg. Chem. 2015, 54, 1931. [Google Scholar]
- Saparov, B.; Bobev, S. Synthesis and crystal structure of the Zintl phases Na2CaCdSb2, Na2SrCdSb2 and Na2EuCdSb2. Inorganics 2022, 10, 265. [Google Scholar]
- Nesper, R. The Zintl-Klemm concept—A historical survey. Z. Anorg. Allg. Chem. 2014, 640, 2639–2648. [Google Scholar]
- Hoffmann, R.-D.; Pöttgen, R. AlB2-related intermetallic compounds—A comprehensive view based on group-subgroup relations. Z. Kristallogr. 2001, 216, 127–145. [Google Scholar]
- Oliynyk, A.O.; Adutwum, L.A.; Rudyk, B.W.; Pisavadia, H.; Lofti, S.; Hlukhyy, V.; Harynuk, J.J.; Mar, A.; Brgoch, J. Disentangling structural confusion through machine learning: Structure prediction and polymorphism of equiatomic ternary phases ABC. J. Am. Chem. Soc. 2017, 139, 17870–17881. [Google Scholar] [CrossRef] [PubMed]
- Shannon, R.D. Effective ionic radii in oxides and fluorides. Acta Crystallogr. 1969, B25, 925–946. [Google Scholar]
- Xiong, J.; Kushwaha, S.K.; Liang, T.; Krizan, J.W.; Hirschberger, M.; Wang, W.; Cava, R.J.; Ong, N.P. Evidence for the chiral anomaly in the Dirac semimetal Na3Bi. Science 2015, 350, 413–416. [Google Scholar] [CrossRef] [Green Version]
- Narayan, A.; DiSante, D.; Picozzi, S.; Sanvito, S. Topological tuning in three-dimensional Dirac semimetals. Phys. Rev. Lett. 2014, 113, 256403. [Google Scholar] [CrossRef]
- Zhou, W.; Zhang, S.; Guo, S.; Qu, H.; Cai, B.; Chen, Z.; Zeng, H. High-performance monolayer Na3Sb shrinking transistors: A DFT-NEGF study. Nanoscale 2020, 12, 18931. [Google Scholar]
- Andersen, O.K. Linear Methods in Band Theory. Phys. Rev. B 1975, 12, 3060–3083. [Google Scholar]
- Andersen, O.K.; Jepsen, O. Explicit, First-Principles Tight-Binding Theory. Phys. Rev. Lett. 1984, 53, 2571. [Google Scholar] [CrossRef]
- Steinberg, S.; Dronskowski, R. The crystal orbital Hamilton population (COHP) method as a tool to visualize and analyze chemical bonding in intermetallic compounds. Crystals 2018, 8, 225. [Google Scholar]
- Sun, Y.; Wang, Q.-Z.; Wu, S.-C.; Felser, C.; Liu, C.-X.; Yan, B. Pressure-induced topological insulator in NaBaBi with right-handed surface spin texture. Phys. Rev. B 2016, 93, 205303. [Google Scholar] [CrossRef]
- Toberer, E.S.; May, A.F.; Snyder, G.J. Zintl chemistry for designing high efficiency thermoelectric materials. Chem. Mater. 2010, 22, 624. [Google Scholar]
- Kauzlarich, S.M.; Brown, S.R.; Snyder, G.J. Zintl Phases for thermoelectric devices. Dalton Trans. 2007, 21, 2099. [Google Scholar]
- Liu, Z.K.; Zhou, B.; Zhang, Y.; Wang, Z.J.; Weng, H.M.; Prabhakaran, D.; Mo, S.-K.; Shen, Z.X.; Fang, Z.; Dai, X.; et al. Discovery of a Three-Dimensional Topological Dirac Semimetal, Na3Bi. Science 2014, 343, 864–867. [Google Scholar] [PubMed]
- Ogunbunmi, M.O.; Baranets, S.; Childs, A.B.; Bobev, S. The Zintl phases AIn2As2 (A = Ca, Sr, Ba): New topological insulators and thermoelectric material candidates. Dalton Trans. 2021, 50, 9173–9184. [Google Scholar] [PubMed]
- Guo, W.-T.; Huang, Z.; Zhang, J.-M. The Zintl phase compounds AEIn2As2 (AE = Ca, Sr, Ba): Topological phase transition under pressure. Phys. Chem. Chem. Phys. 2022, 24, 17337–17347. [Google Scholar] [CrossRef] [PubMed]
- Schindler, F.; Cook, A.M.; Vergniory, M.G.; Wang, Z.; Parkin, S.S.P.; Bernevig, B.A.; Neupert, T. Higher-order topological insulators. Sci. Adv. 2018, 4, eaat0346. [Google Scholar] [PubMed]
- Baranets, S.; He, H.; Bobev, S. Niobium-bearing arsenides and germanides from elemental mixtures not involving niobium: A new twist to an old problem in solid-state synthesis. Acta Crystallogr. C 2018, 74, 623. [Google Scholar]
- He, H.; Tyson, C.; Bobev, S. Synthesis and crystal structures of the quaternary Zintl phases RbNa8Ga3Pn6 (Pn = P, As) and Na10NbGaAs6. Crystals 2012, 2, 213–223. [Google Scholar] [CrossRef]
- SMART, version 2.10; Bruker Analytical X-ray Systems, Inc.: Madison, WI, USA, 2003.
- SAINT, version 6.45; Bruker Analytical X-ray Systems, Inc.: Madison, WI, USA, 2003.
- SADABS, version 2.10; Bruker Analytical X-ray Systems, Inc.: Madison, WI, USA, 2003.
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. C 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Tank, R.; Jepsen, O.; Burkhardt, A.; Andersen, O. TB-LMTO-ASA Program; Max-Planck-Institut für Festkörperforschung: Stuttgart, Germany, 1994. [Google Scholar]
- von Barth, U.; Hedin, L. A local exchange-correlation potential for the spin polarized case. J. Phys. C Solid State Phys. 1972, 5, 1629. [Google Scholar]
- Yamada, T.; Matsuo, N.; Enoki, M.; Yamane, H. A novel ternary bismuthide, NaMgBi: Crystal amd electronic structure and electrical properties. Z. Naturforsch. 2021, 76b, 789–795. [Google Scholar]
- Vogel, R.; Schuster, H.U. Neue elektrovalente ternare Verbindungen des Kaliums mit Magnesium unde Elementen der 5 Hauptgruppe. Z. Naturforsch. 1979, 34b, 1719–1721. [Google Scholar] [CrossRef]
- Cardoso, G.; Caroca-Canalesr, N.; Hönle, W.; von Schnering, H.-G. Crystal structure of rubidium calcium arsenide, RbCaAs, and rubidium calcium antimonide, RbCaSb. Z. Kristallogr. NCS 2003, 218, 455–456. [Google Scholar] [CrossRef]
- Nowotny, H.M.; Holub, F. Untersuchungen an metalischen Systemen mit Flussspatphasen. Monatsh. Chem. 1960, 91, 877–887. [Google Scholar]
- Monconduit, L.; Belin, C. A new ternary antimonide phase, LiBaSb. Acta Cryst. E 2001, 57, 17–18. [Google Scholar]
- Gupta, S.; Ganguli, A.K. Synthesis, structure and properties of a new Zintl phase: SrLiSb. J. Soild State Chem. 2006, 179, 1318–1322. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Empirical Formula | NaSrSb | NaBaSb | NaEuSb |
|---|---|---|---|
| Formula weight | 232.36 | 282.08 | 296.70 |
| Temperature (K) | 200(2) | 200(2) | 200(2) |
| Space group, Z | P2m, 3 | P2m, 3 | P2m, 3 |
| a (Å) | 8.2183(3) | 8.4779(4) | 8.1514(4) |
| c (Å) | 4.8475(4) | 5.0338(5) | 4.8154(5) |
| V (Å3) | 283.54(3) | 313.33(4) | 277.09(3) |
| c/a | 0.590 | 0.594 | 0.591 |
| ρcal (g/cm3) | 4.08 | 4.49 | 5.33 |
| μ (cm–1) | 210.9 | 156.9 | 239.7 |
| Goodness-of-fit on F2 | 1.11 | 1.12 | 1.02 |
| Unique reflections | 329 | 383 | 355 |
| Refined parameters | 15 | 15 | 15 |
| R1 (I > 2σI) a | 0.0153 | 0.0173 | 0.0187 |
| wR2 (I > 2σI) a | 0.0330 | 0.0368 | 0.0361 |
| R1 (all data) a | 0.0157 | 0.0179 | 0.0197 |
| wR2 (all data) a | 0.0331 | 0.0369 | 0.0364 |
| Largest peak and hole difference (e–/Å3) | 0.84 and –0.85 | 0.61 and –0.89 | 0.88 and –0.82 |
| CCDC deposition no. | 2237818 | 2237817 | 2237816 |
| Atom | Site | x | y | z | Ueq (Å2) |
|---|---|---|---|---|---|
| NaSrSb | |||||
| Na | 3g | 0.2424(3) | 0 | 1/2 | 0.0132(5) |
| Sr | 3f | 0.58090(8) | 0 | 0 | 0.0146(2) |
| Sb2 | 2d | 1/3 | 2/3 | 1/2 | 0.0119(1) |
| Sb1 | 1a | 0 | 0 | 0 | 0.0151(2) |
| NaBaSb | |||||
| Na | 3g | 0.2400(4) | 0 | 1/2 | 0.0173(8) |
| Ba | 3f | 0.58118(6) | 0 | 0 | 0.0122(1) |
| Sb2 | 2d | 1/3 | 2/3 | 1/2 | 0.0100(2) |
| Sb1 | 1a | 0 | 0 | 0 | 0.0114(2) |
| NaEuSb | |||||
| Na | 3g | 0.2416(5) | 0 | 1/2 | 0.0171(9) |
| Eu | 3f | 0.58180(7) | 0 | 0 | 0.0142(1) |
| Sb2 | 2d | 1/3 | 2/3 | 1/2 | 0.0125(2) |
| Sb1 | 1a | 0 | 0 | 0 | 0.0132(2) |
| Atom Pair | Distance (Å) | Atom Pair | Distance (Å) | Atom Pair | Distance (Å) |
|---|---|---|---|---|---|
| NaSrSb | NaBaSb | NaEuSb | |||
| Na–Sb1 (×2) | 3.138(2) | Na–Sb1 (×2) | 3.237(2) | Na–Sb1 (×2) | 3.111(3) |
| Na–Sb2 (×2) | 3.180(2) | Na–Sb2 (×2) | 3.293(2) | Na–Sb2 (×2) | 3.158(3) |
| Na–Sr (×2) | 3.690(2) | Na–Ba (×2) | 3.834(3) | Na–Eu (×2) | 3.672(3) |
| Na–Sr (×4) | 3.8529(5) | Na–Ba (×4) | 3.9822(5) | Na–Eu (×4) | 3.8187(5) |
| Na–Na (×2) | 3.451(4) | Na–Na (×2) | 3.525(6) | Na–Na (×2) | 3.412(7) |
| Sr–Sb1 | 3.4443(6) | Ba–Sb1 | 3.5508(6) | Eu–Sb1 | 3.4089(6) |
| Sr–Sb2 (×4) | 3.4562(2) | Ba–Sb2 (×4) | 3.5774(2) | Eu–Sb2 (×4) | 3.4320(2) |
| Sr–Na (×2) | 3.690(2) | Ba–Na (×2) | 3.834(3) | Eu–Na (×2) | 3.672(3) |
| Sr–Na (×4) | 3.8529(5) | Ba–Na (×4) | 3.9822(5) | Eu–Na (×4) | 3.8187(5) |
| Sb1–Na (×6) | 3.138(2) | Sb1–Na (×6) | 3.237(2) | Sb1–Na (×6) | 3.111(3) |
| Sb1–Sr (×3) | 3.4443(6) | Sb1–Ba (×3) | 3.5508(6) | Sb1–Eu (×3) | 3.4089(6) |
| Sb2–Na (×3) | 3.180(2) | Sb2–Na (×3) | 3.293(2) | Sb2–Na (×3) | 3.158(3) |
| Sb2–Sr (×6) | 3.4562(2) | Sb2–Ba (×6) | 3.5774(2) | Sb2–Eu (×6) | 3.4320(2) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Bobev, S. Synthesis and Crystal Structure of the Zintl Phases NaSrSb, NaBaSb and NaEuSb. Materials 2023, 16, 1428. https://doi.org/10.3390/ma16041428
Wang Y, Bobev S. Synthesis and Crystal Structure of the Zintl Phases NaSrSb, NaBaSb and NaEuSb. Materials. 2023; 16(4):1428. https://doi.org/10.3390/ma16041428
Chicago/Turabian StyleWang, Yi, and Svilen Bobev. 2023. "Synthesis and Crystal Structure of the Zintl Phases NaSrSb, NaBaSb and NaEuSb" Materials 16, no. 4: 1428. https://doi.org/10.3390/ma16041428
APA StyleWang, Y., & Bobev, S. (2023). Synthesis and Crystal Structure of the Zintl Phases NaSrSb, NaBaSb and NaEuSb. Materials, 16(4), 1428. https://doi.org/10.3390/ma16041428