Polymer Layered Silicate Nanocomposites: A Review

Abstract
:1. Introduction
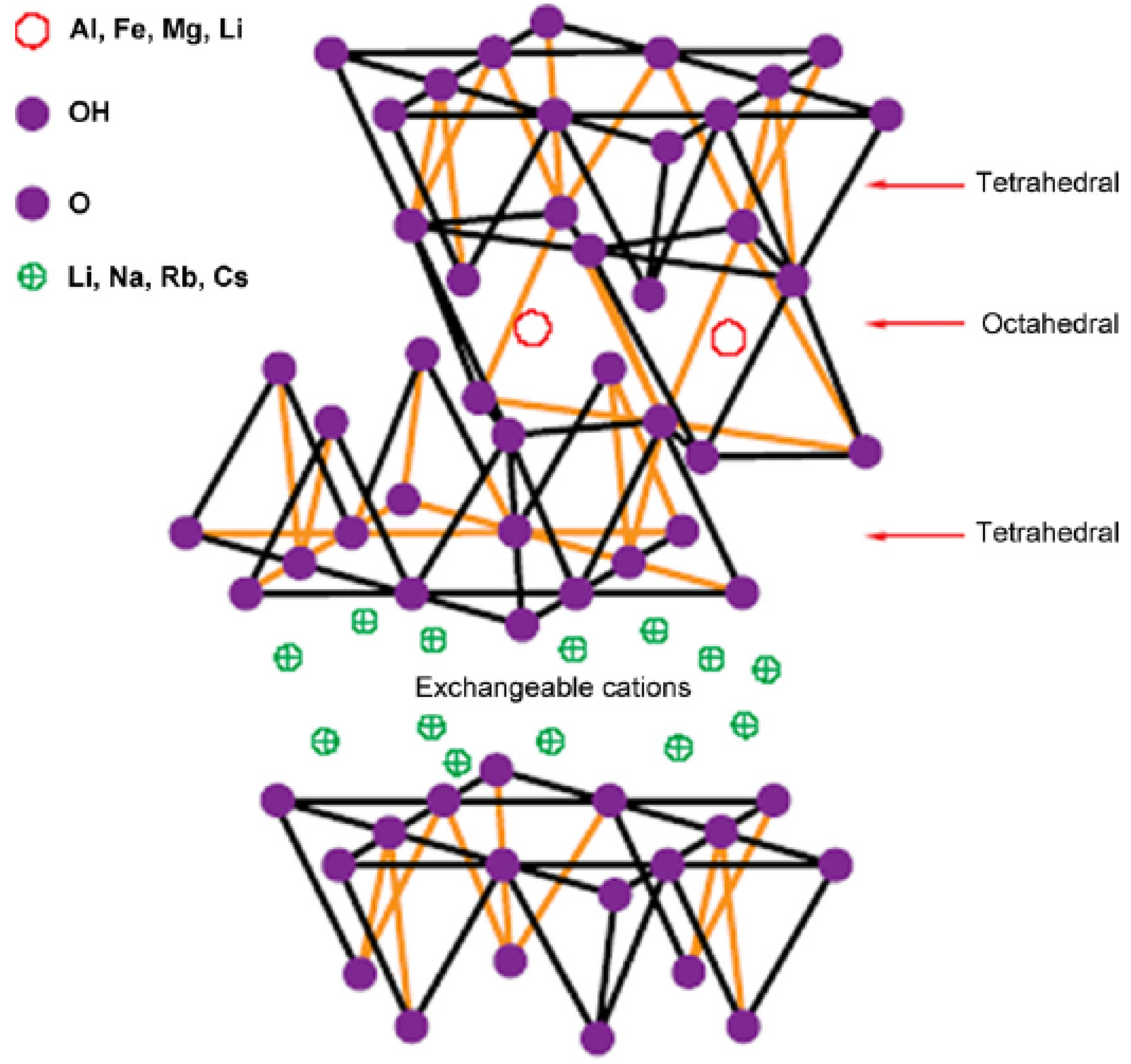
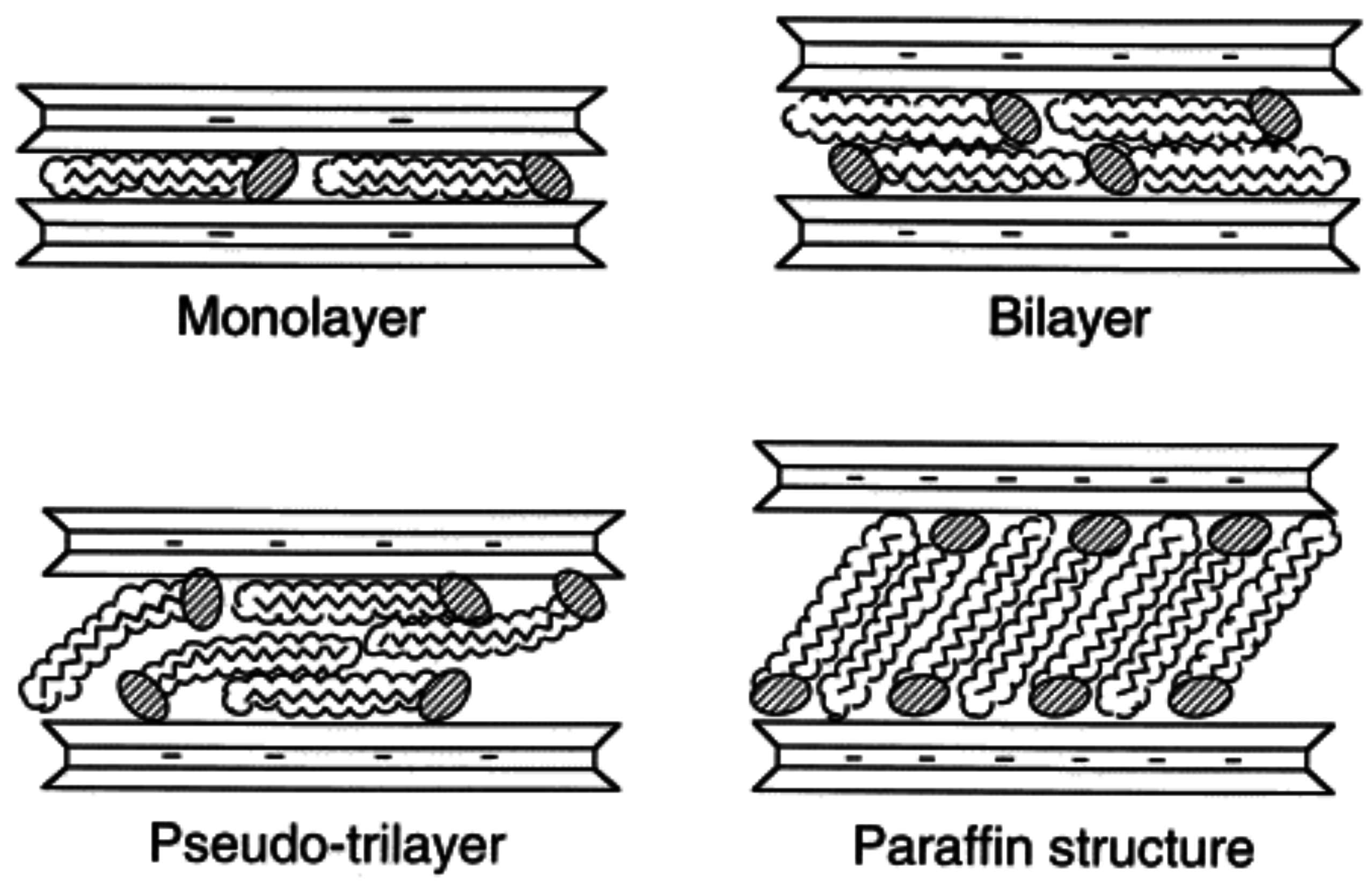
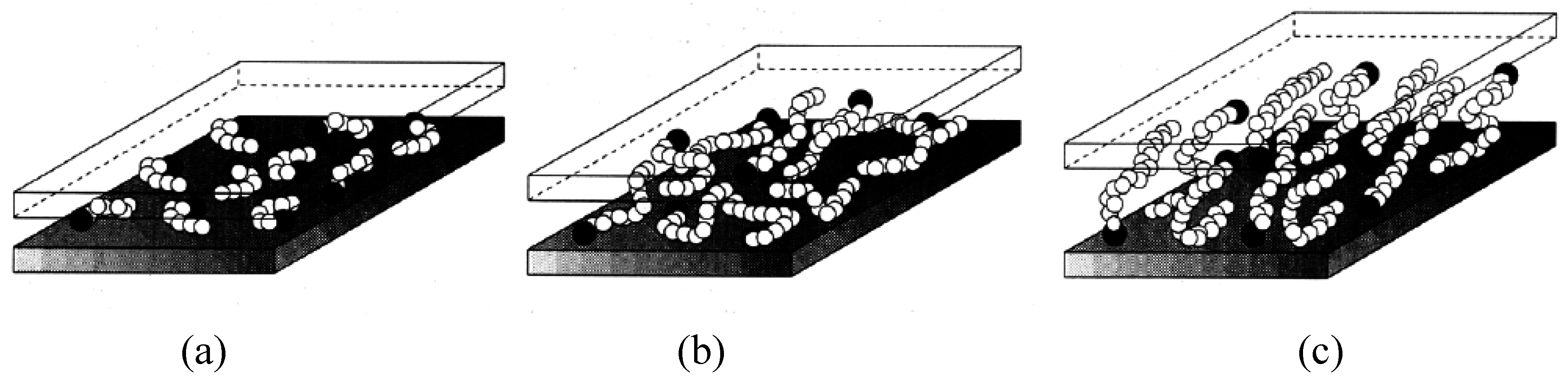
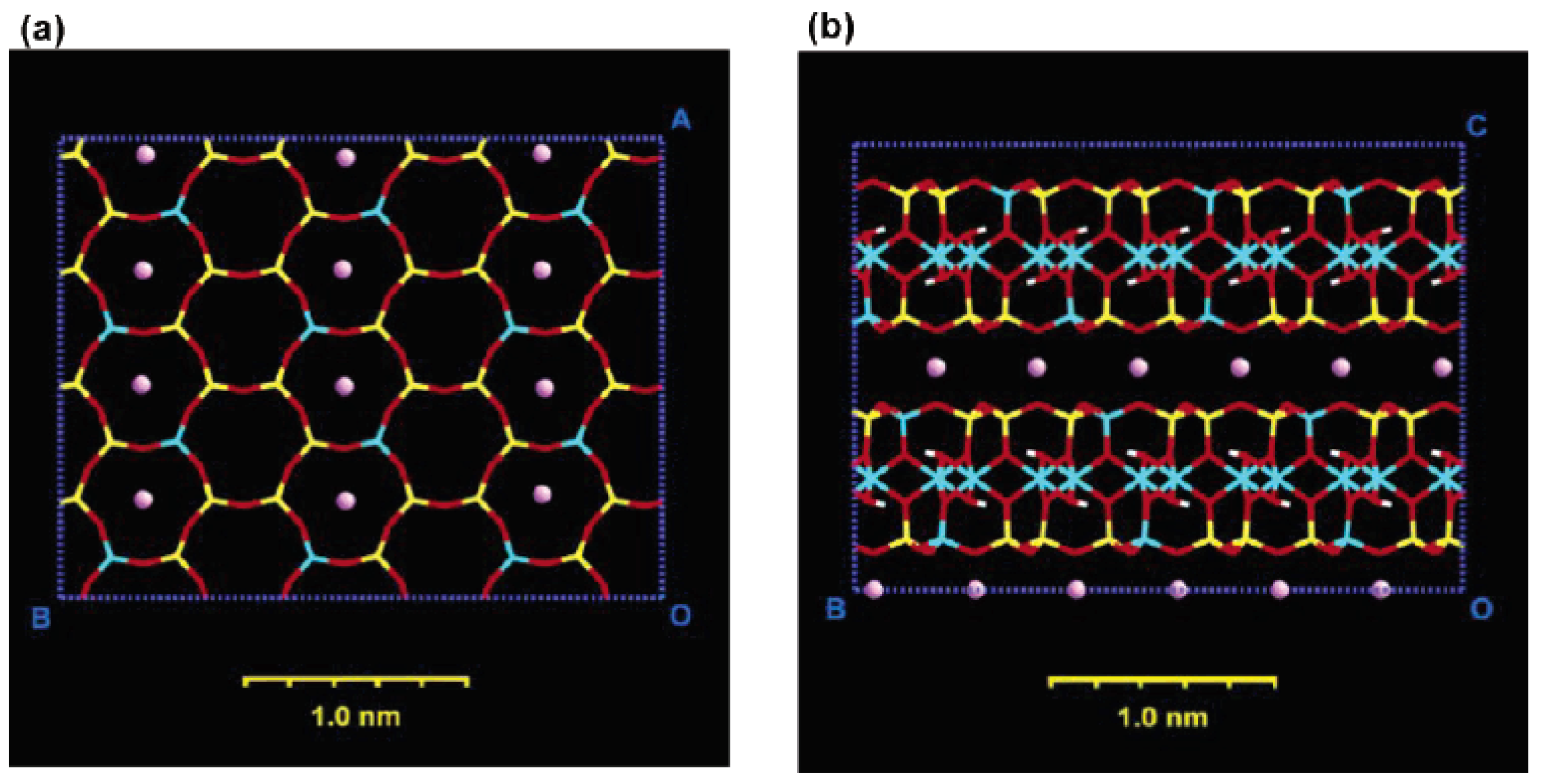
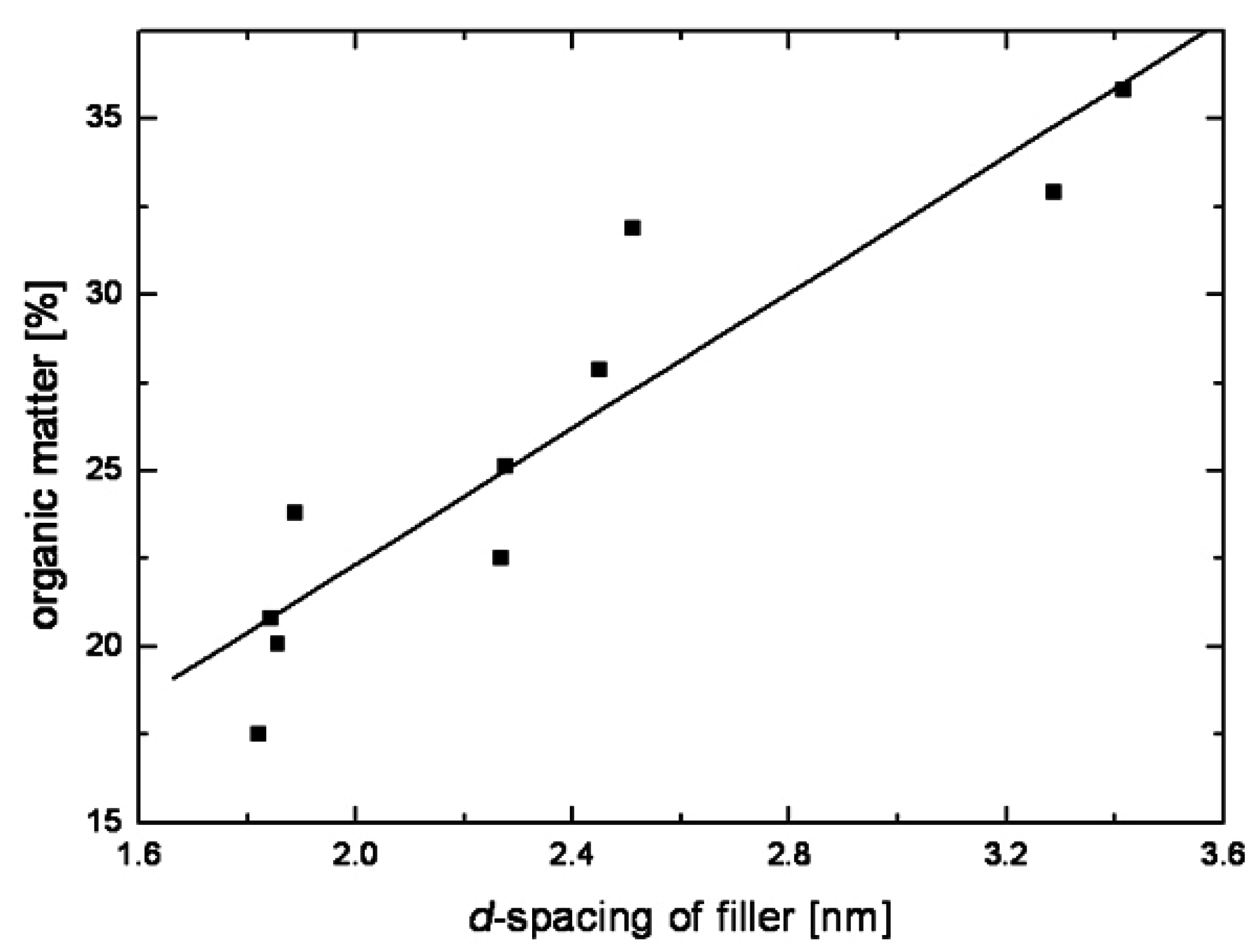
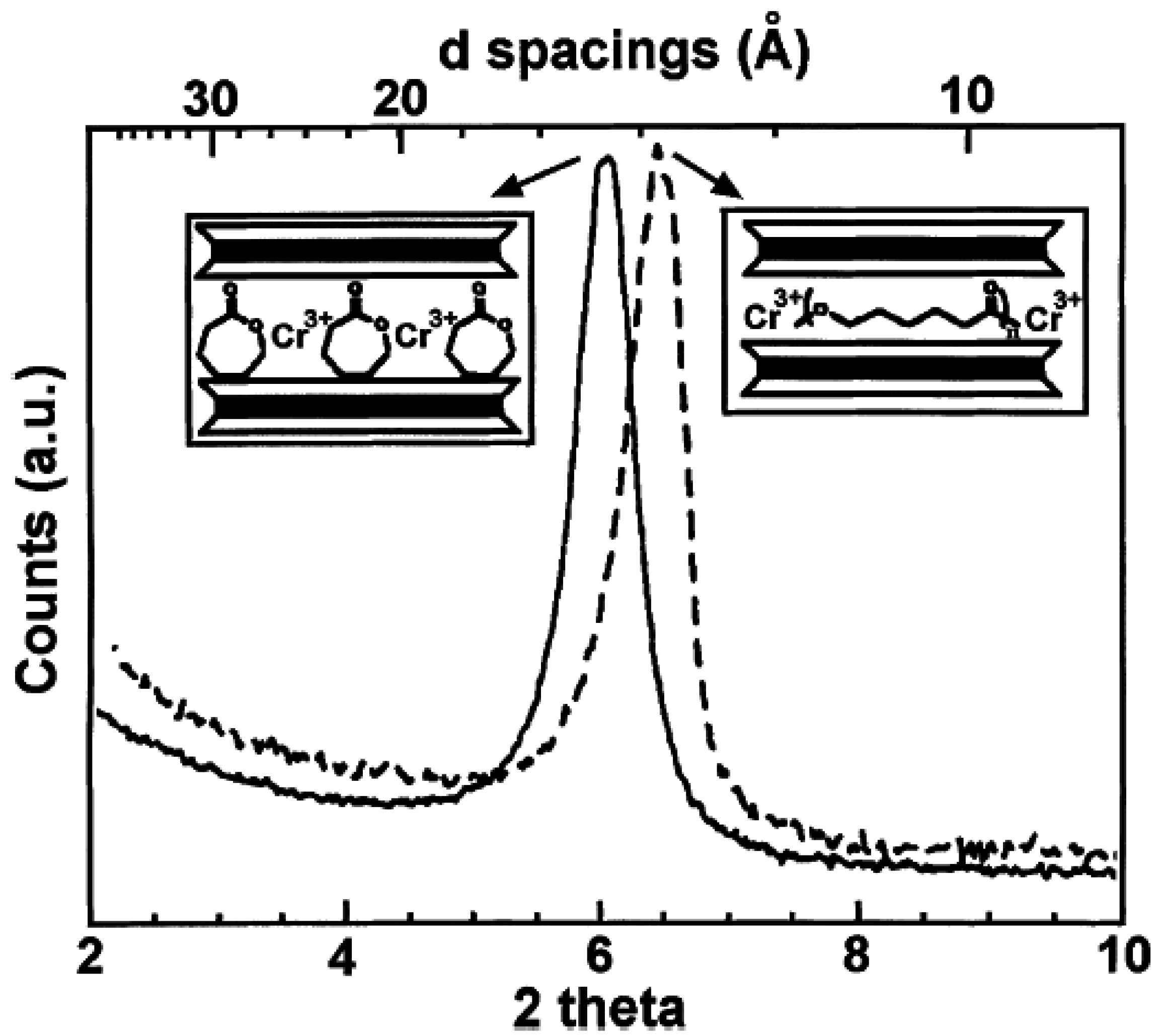
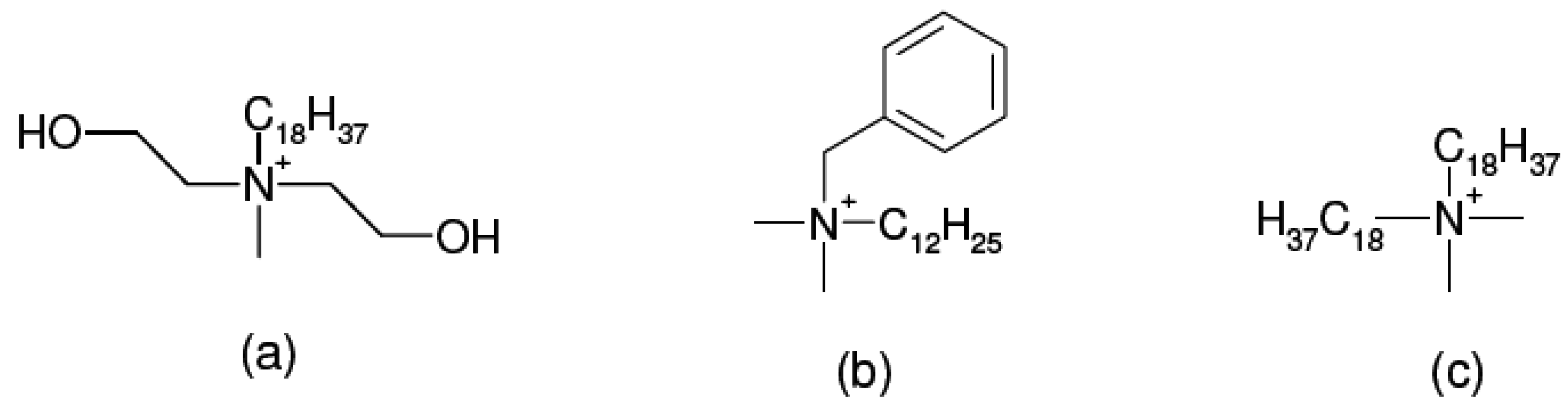
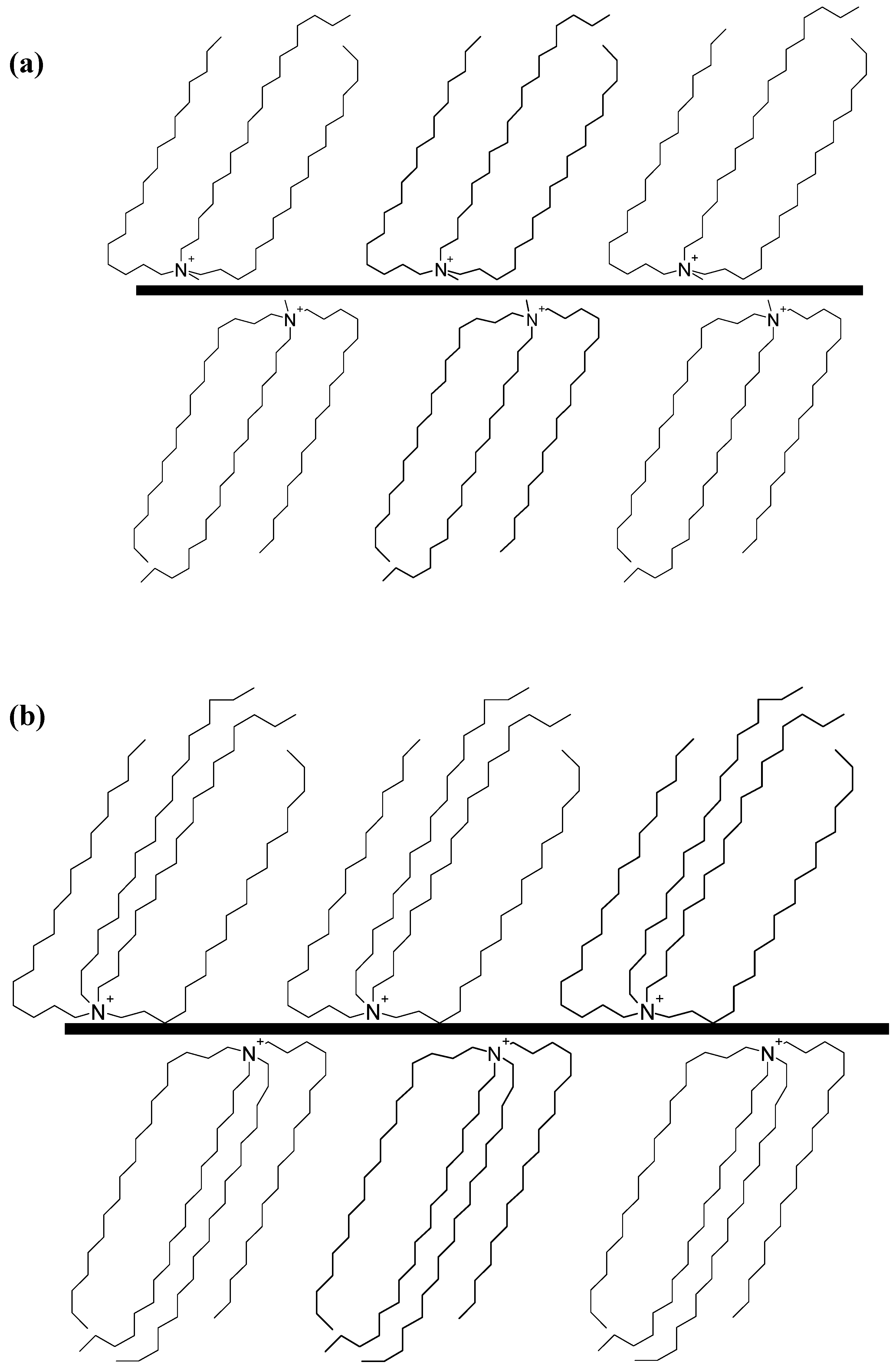
2. Layered Silicates






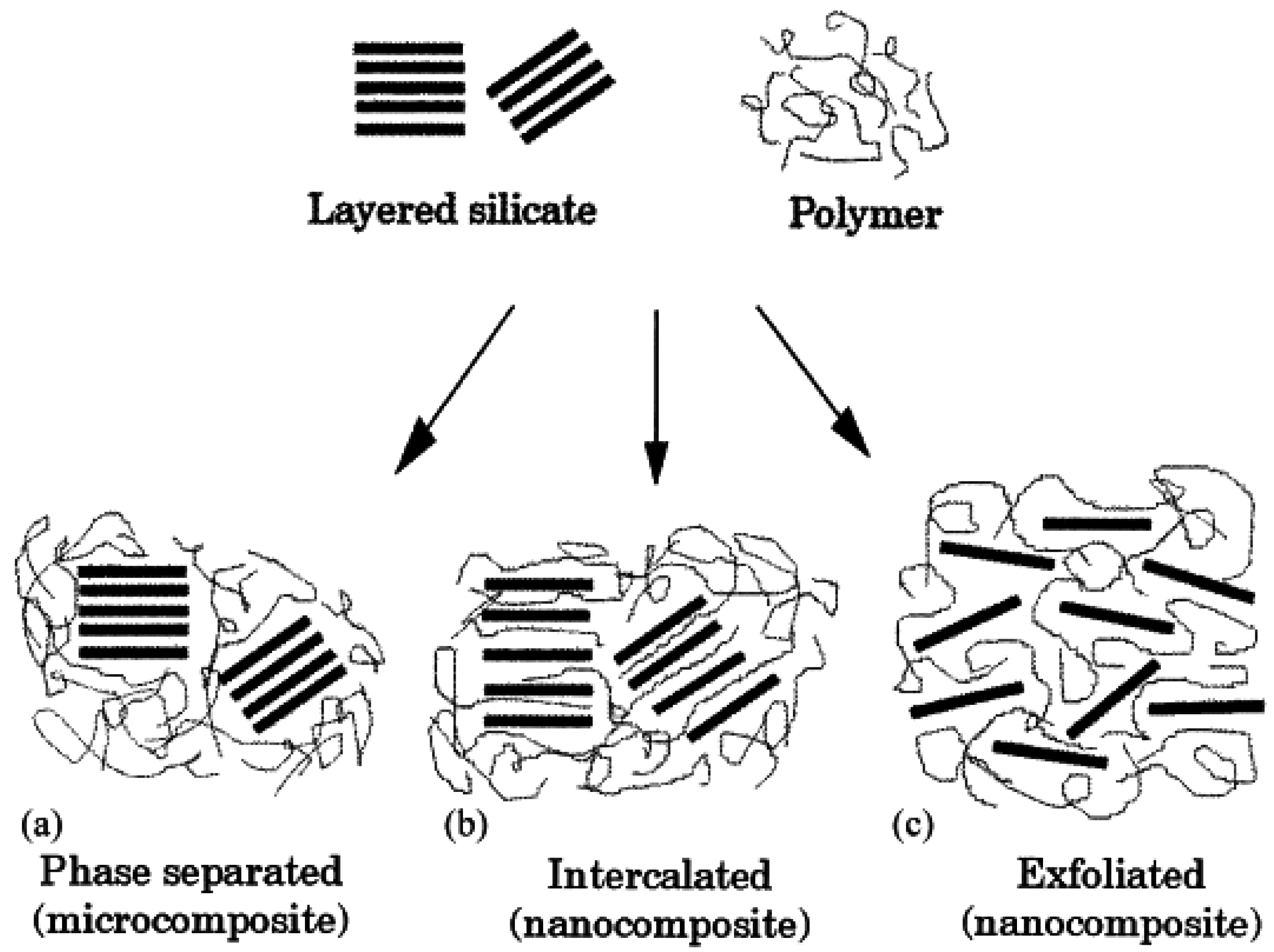
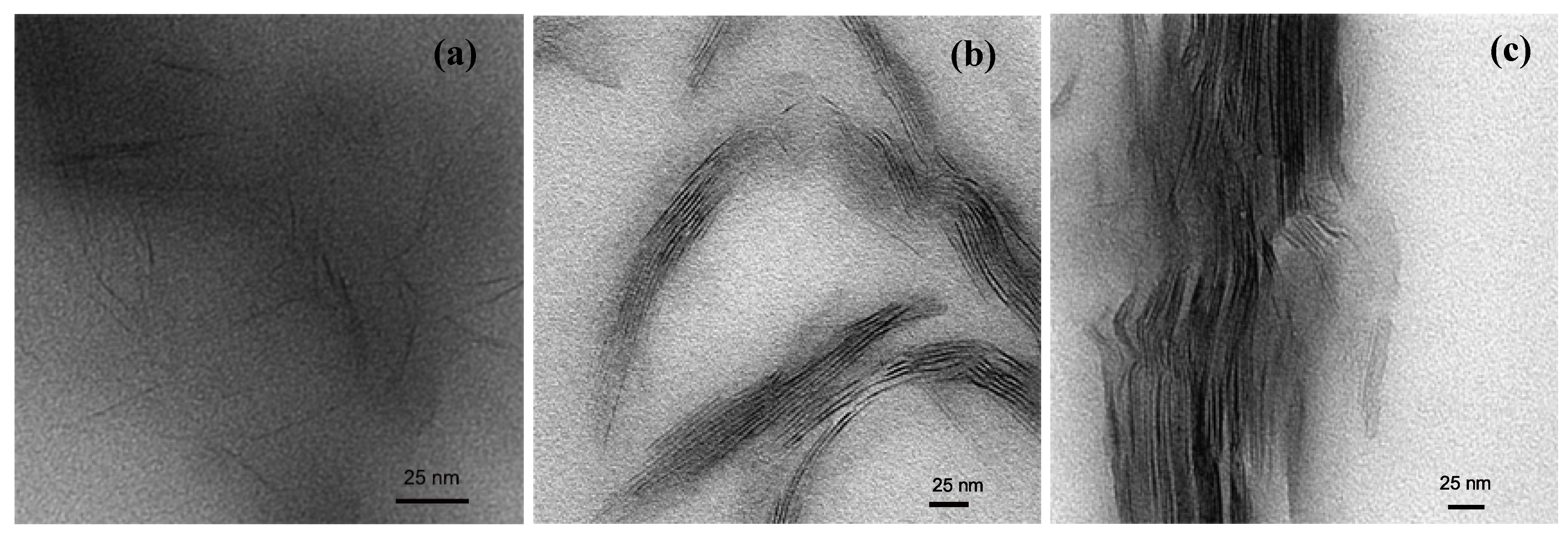
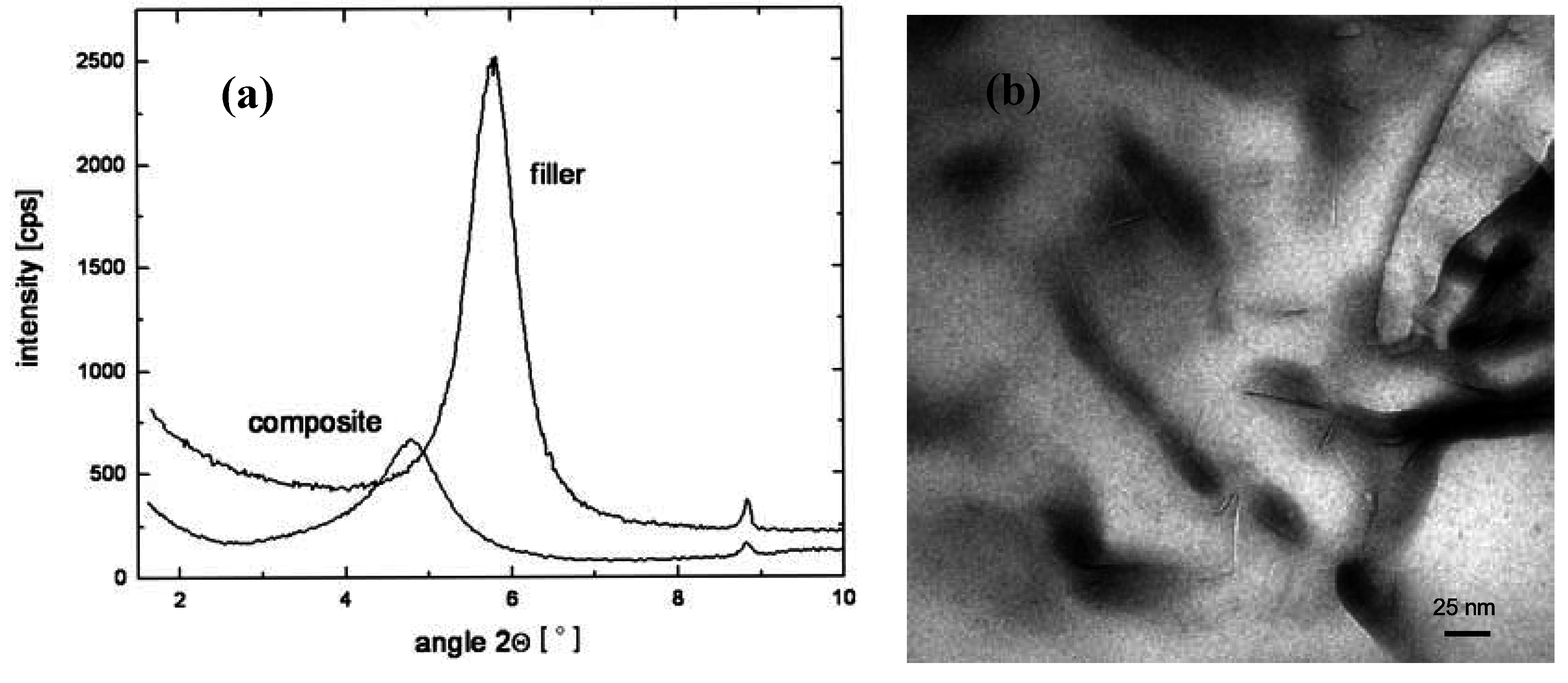
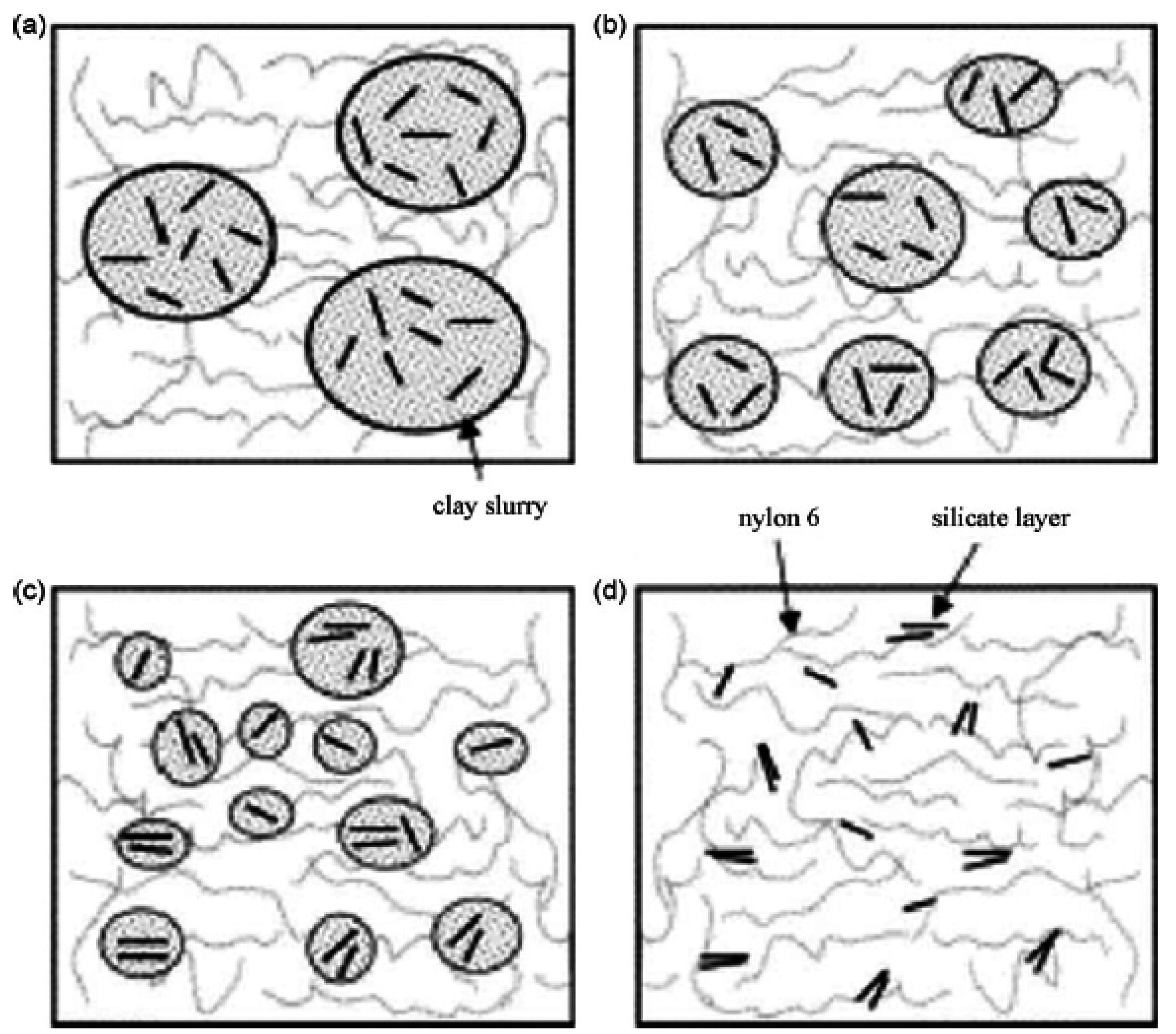
3. Nanocomposite Structures and Characterization








4. Nanocomposite Preparation
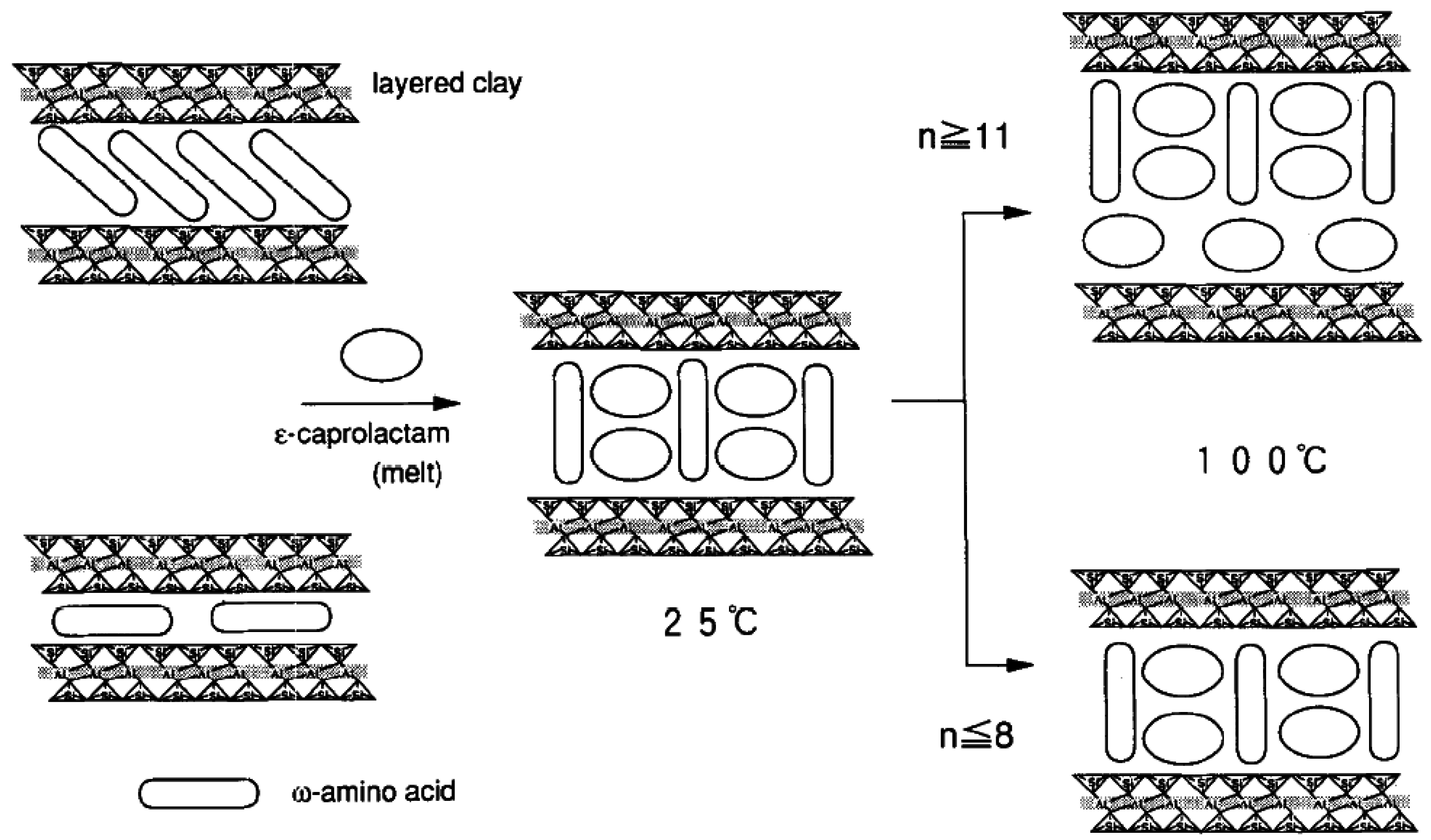
4.1. Template Synthesis
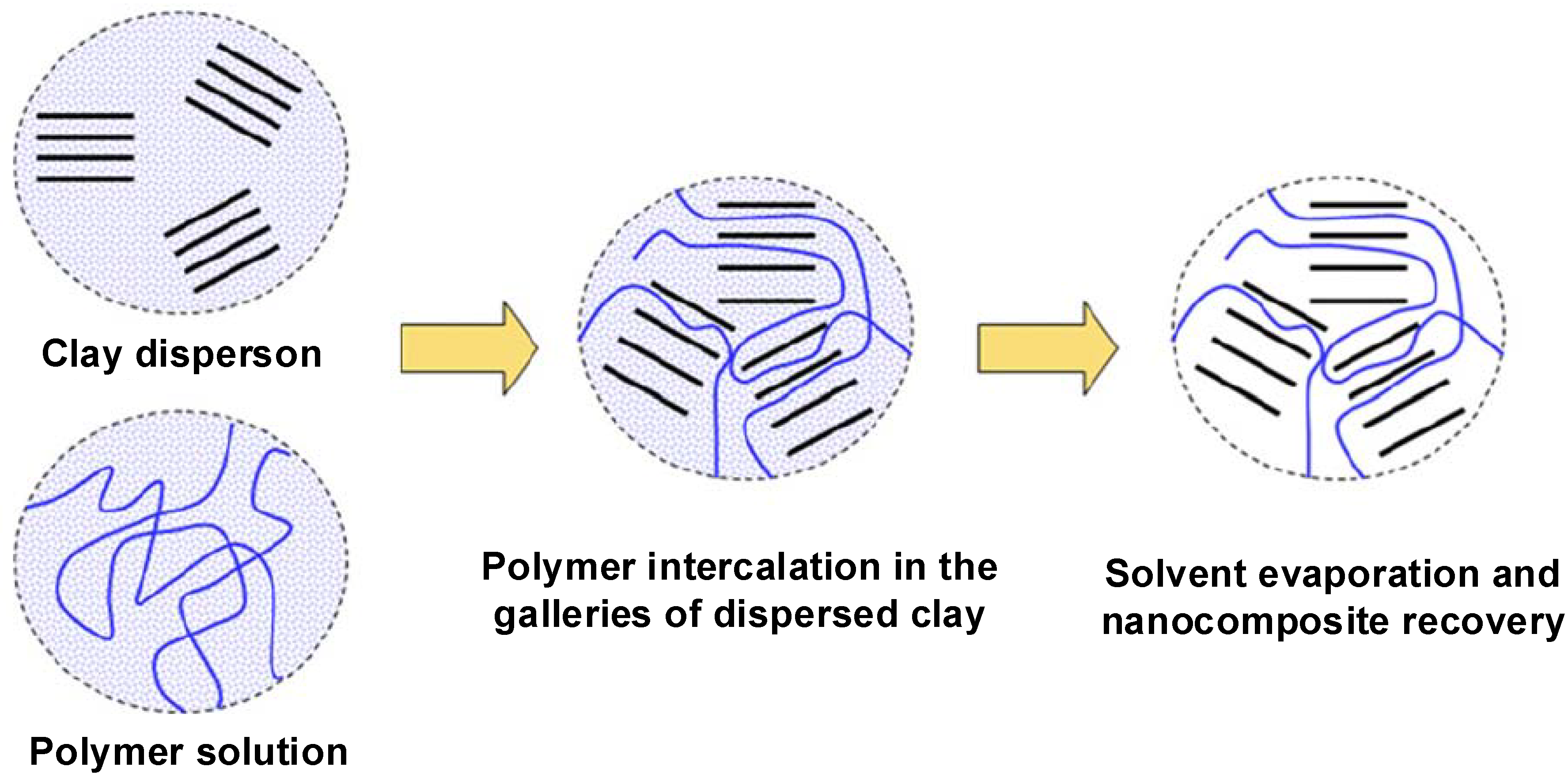
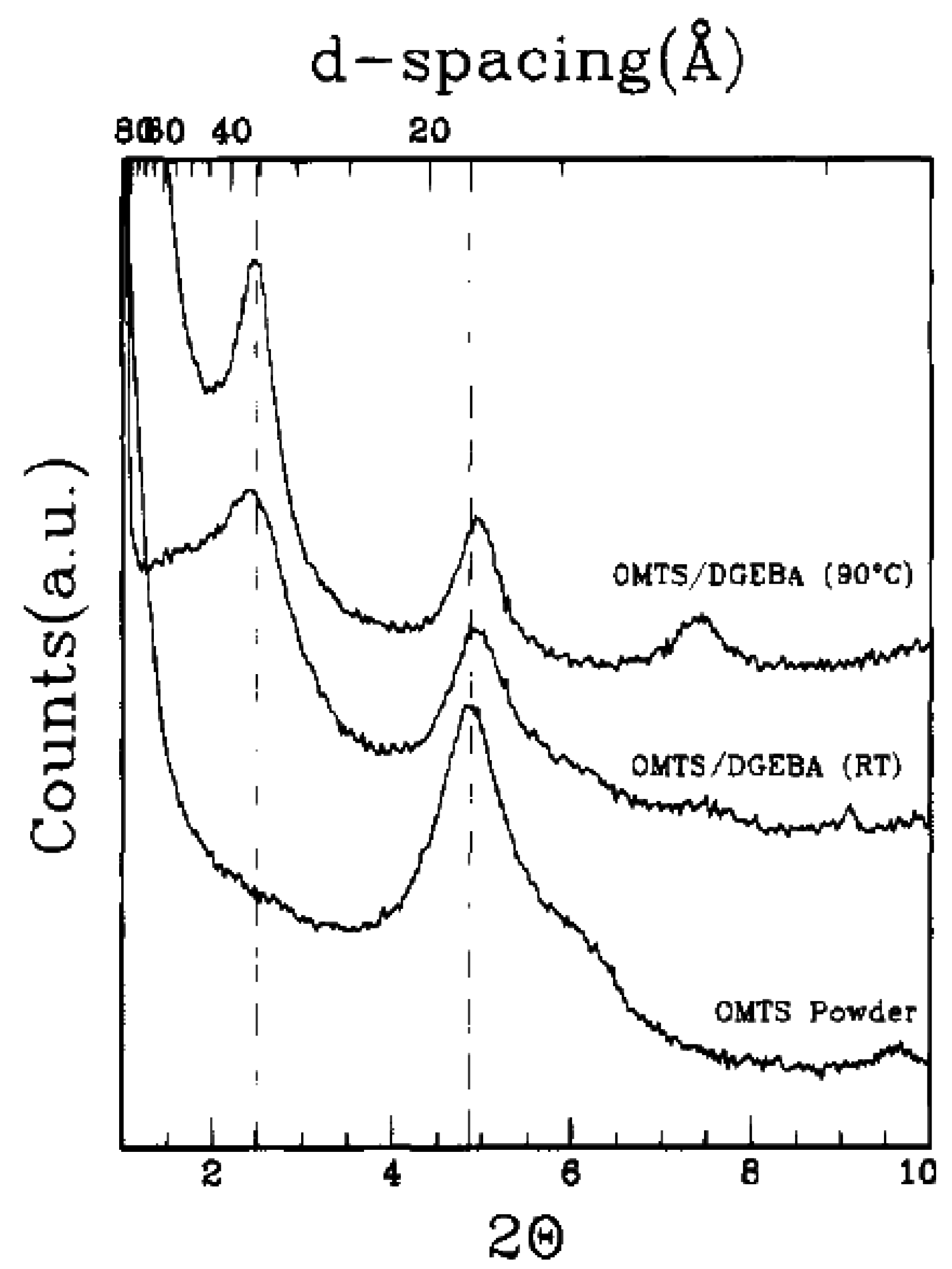
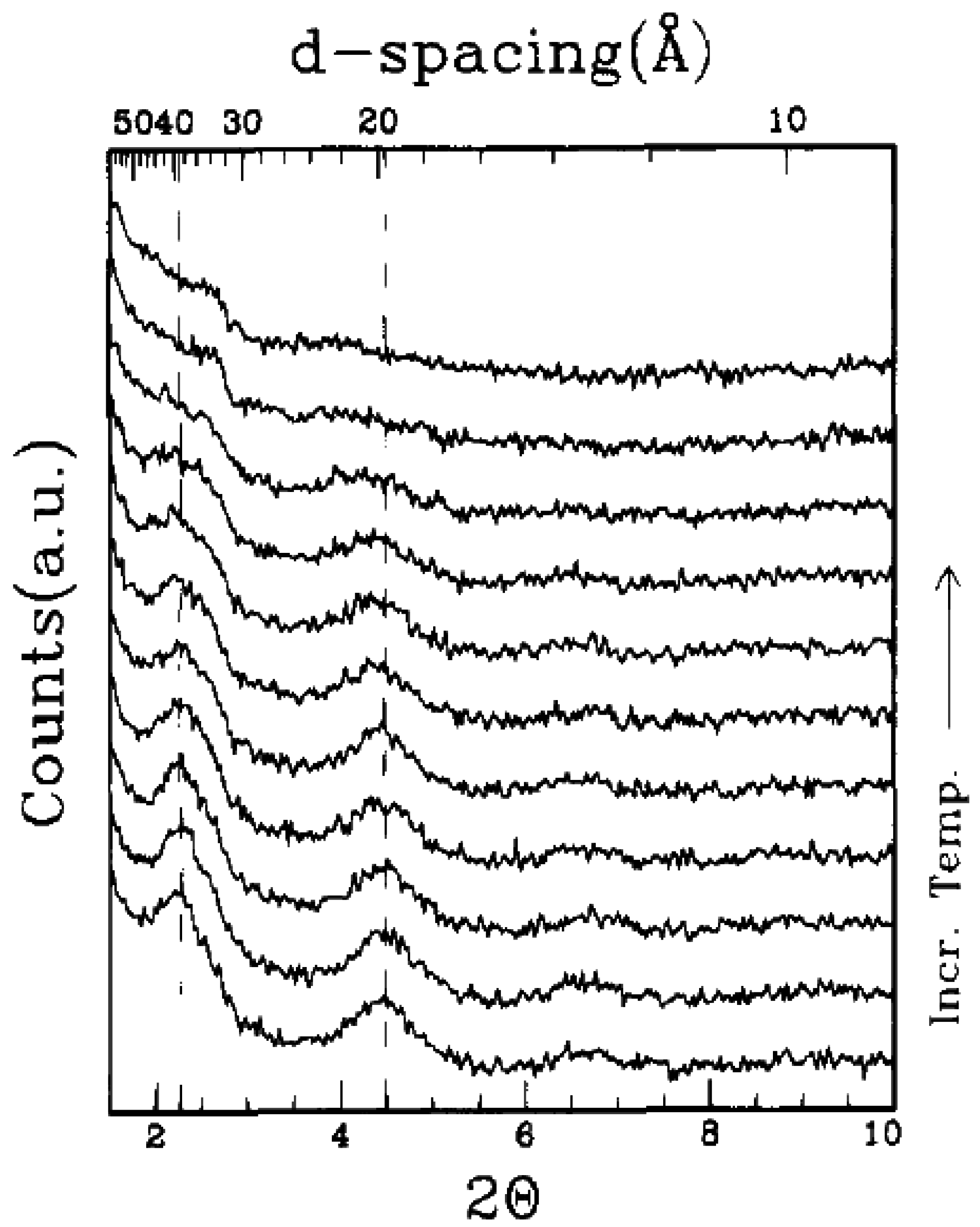
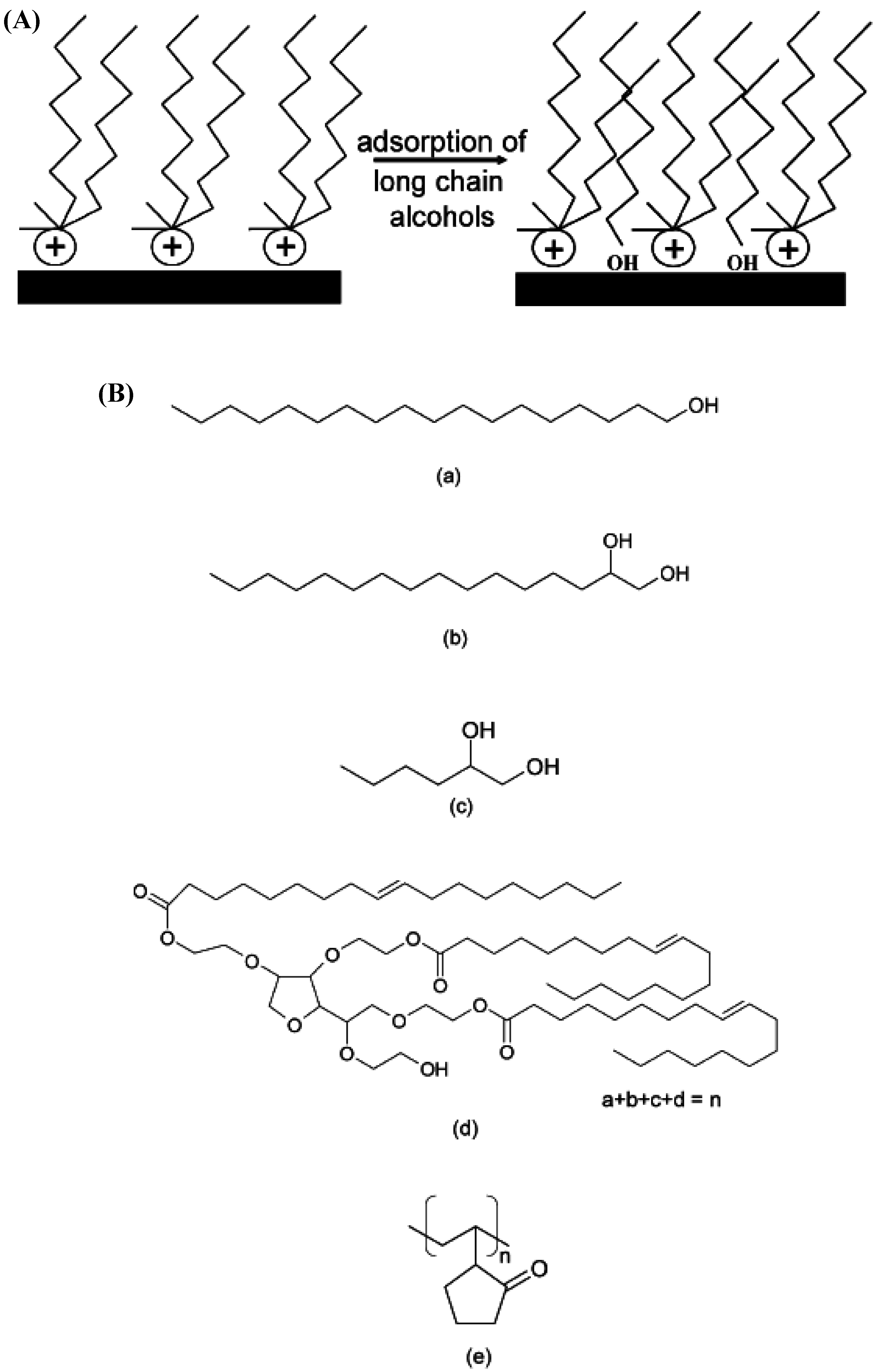
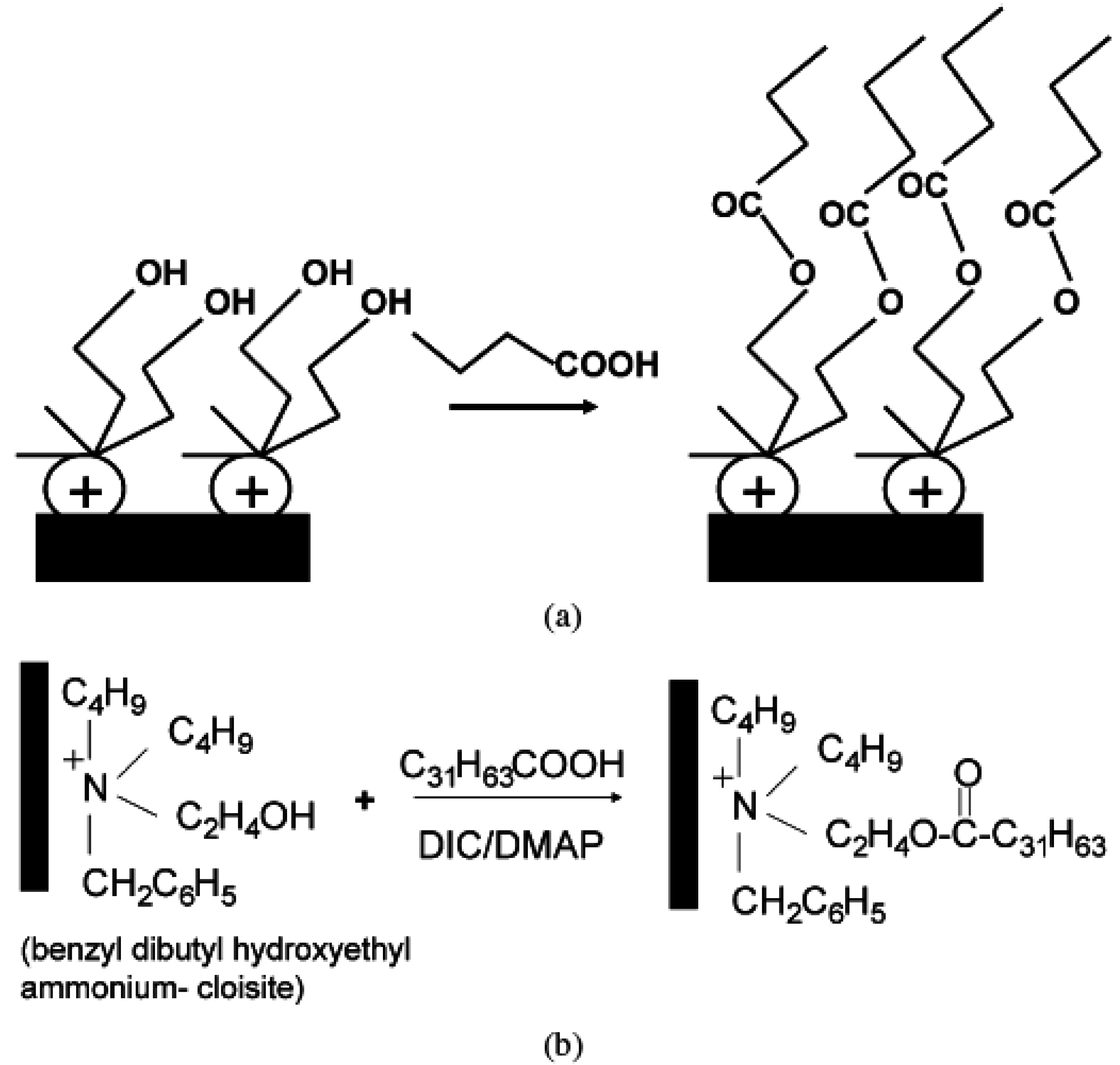
4.2. Intercalation of Polymer or Prepolymer from Solution

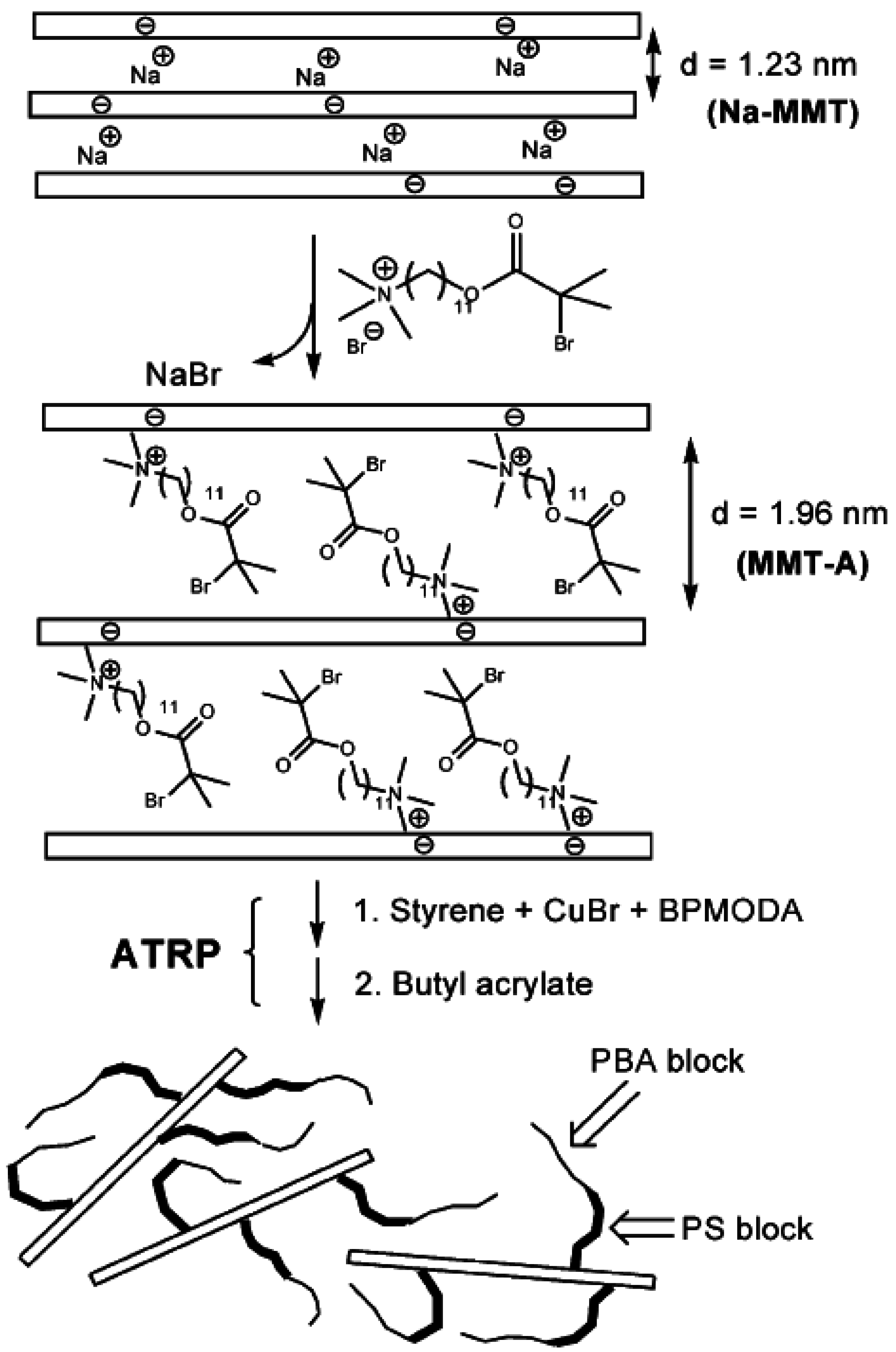
4.3. In-Situ Intercalative Polymerization







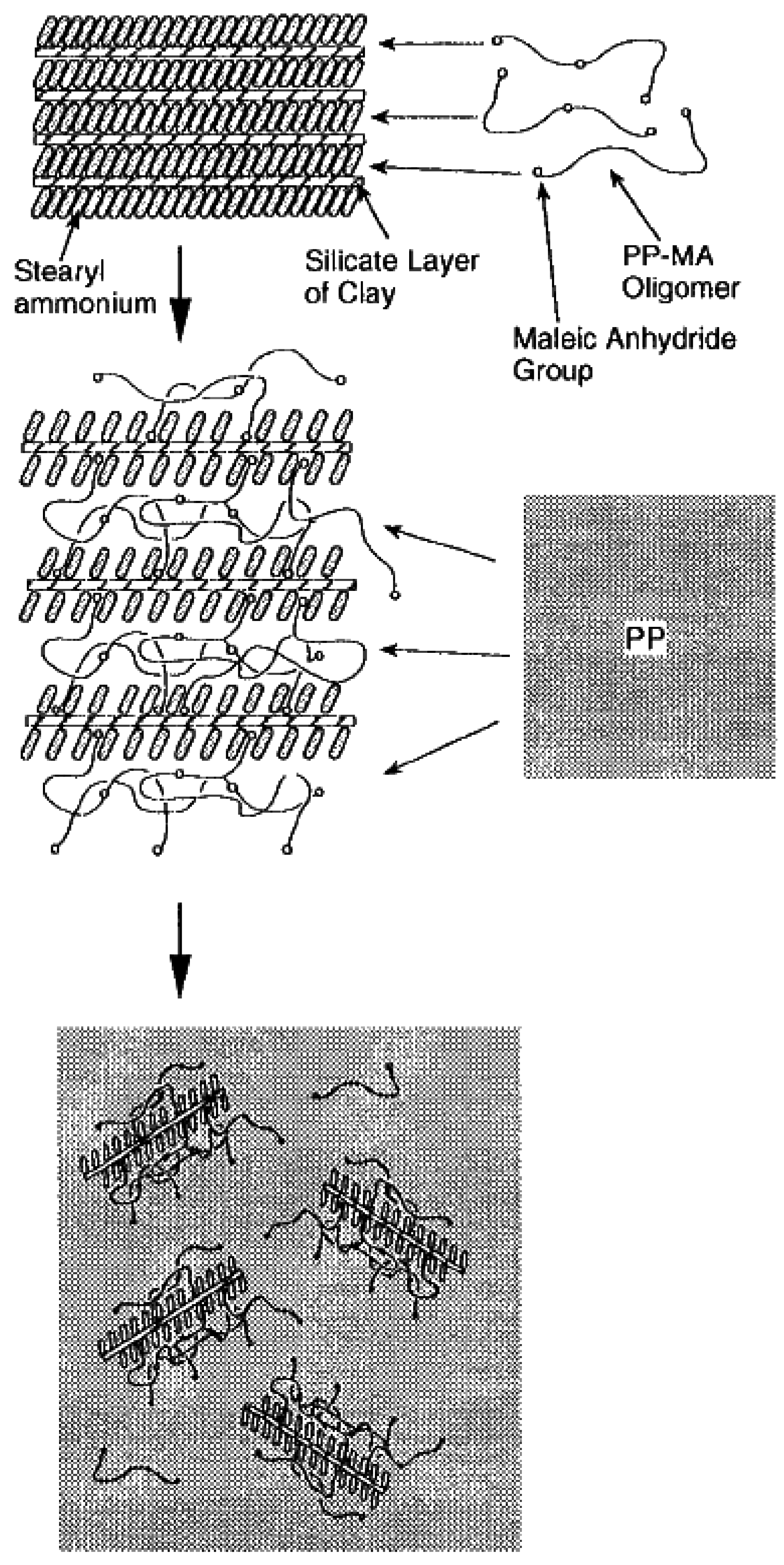
4.4. Melt Intercalation




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Modification | Substrate, CECμ.eq/g-1 | Basal spacing, nm |
|---|---|---|
| octadecyltrimethylammonium | montmorillonite, 880 | 1.84 |
| octadecyltrimethylammonium | montmorillonite, 680 | 1.82 |
| octadecyltrimethylammonium | montmorillonite, 900 | 1.85 |
| octadecyltrimethylammonium | montmorillonite, 1000 | 2.14 |
| dioctadecyldimethylammonium | montmorillonite, 880 | 2.51 |
| dioctadecyldimethylammonium | montmorillonite, 680 | 2.45 |
| trioctadecylmethylammonium | montmorillonite, 880 | 3.42 |
| trioctadecylmethylammonium | montmorillonite, 680 | 3.29 |
| benzylhexadecyldimethylammonium | montmorillonite, 880 | 1.88 |
| benzylhexadecyldimethylammonium | montmorillonite, 680 | 1.85 |
| docosyltriethylammonium | montmorillonite, 880 | 1.93 |
| decylmethyloctadecylimidazolium | montmorillonite, 880 | 2.24 |
| didocyldimethylammonium/dioctadecyldimethylammonium | montmorillonite, 880 | 2.28 |
| didocyldimethylammonium/dioctadecyldimethylammonium | montmorillonite, 680 | 2.27 |
| benzylhydroxyethylmethyloctadecyl ammonium | montmorillonite, 880 | 2.06 |
| benzldibutylhydroxyethylammonium | montmorillonite, 880 | 1.52 |
| benzyldi(hydroxyethyl)butyl ammonium | montmorillonite, 880 | 1.50 |
| benzyltriethanolammonium | montmorillonite, 880 | 1.52 |
| benzylhydroxyethylmethyloctadecyl ammonium | vermiculite, 1400 | 3.40 |
| benzylhexadecyldimethylammonium | vermiculite, 1400 | 3.25 |


5. Nanocomposite Properties
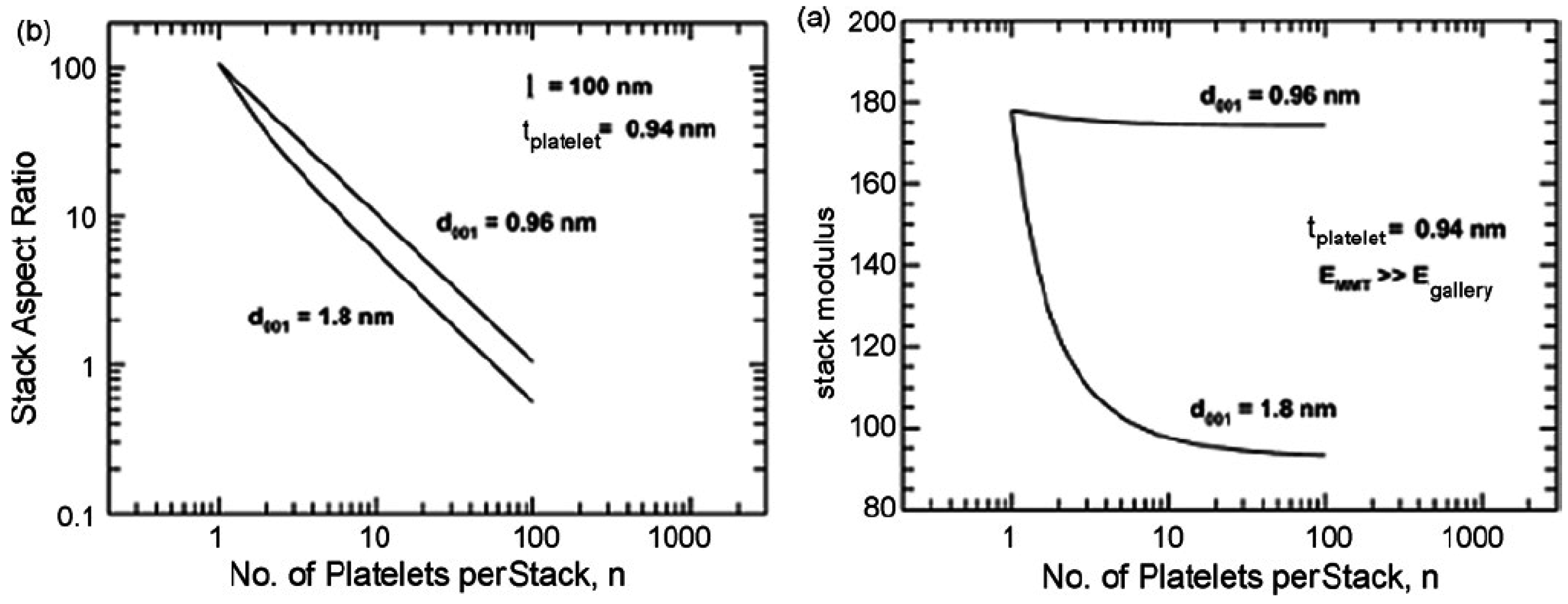
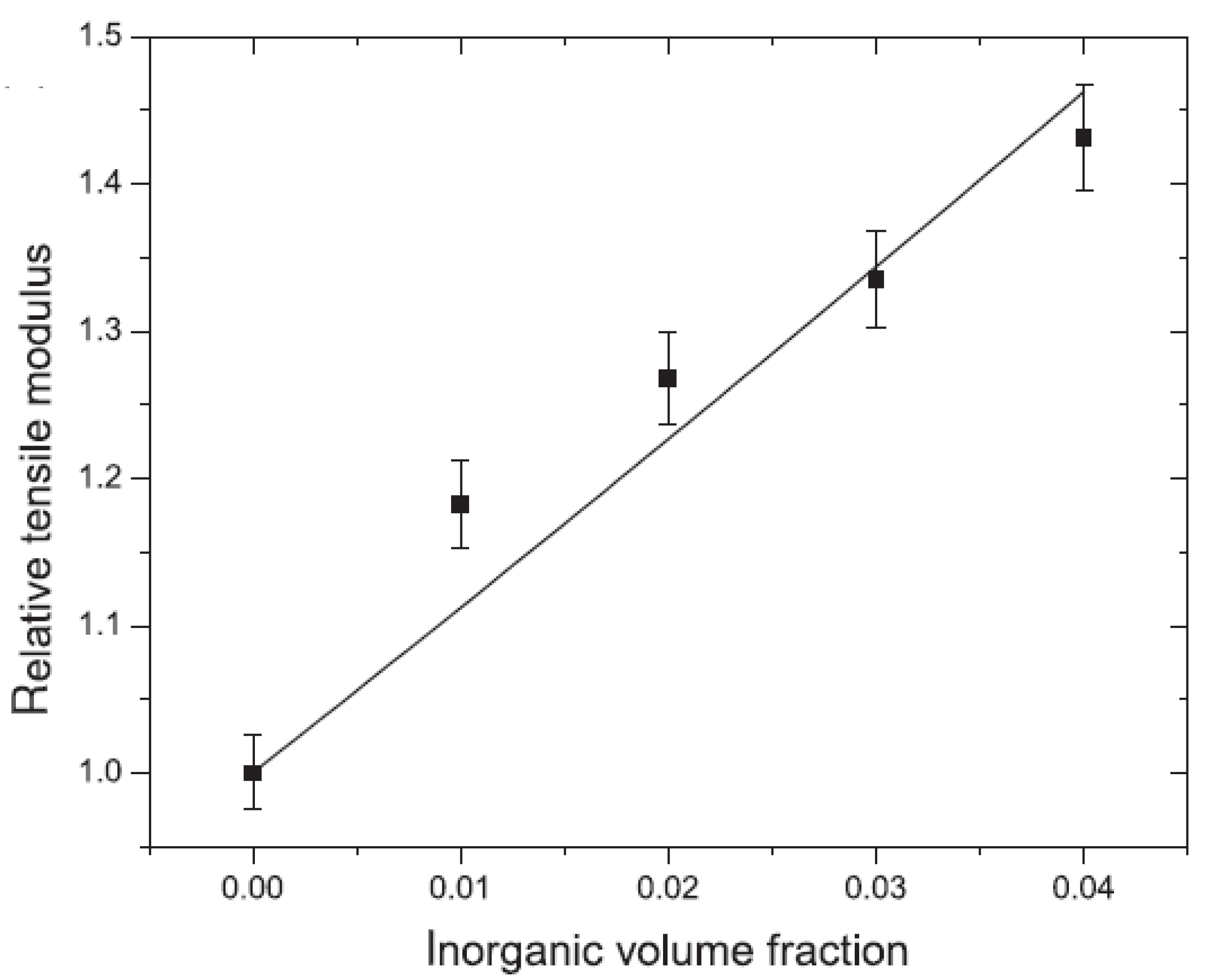
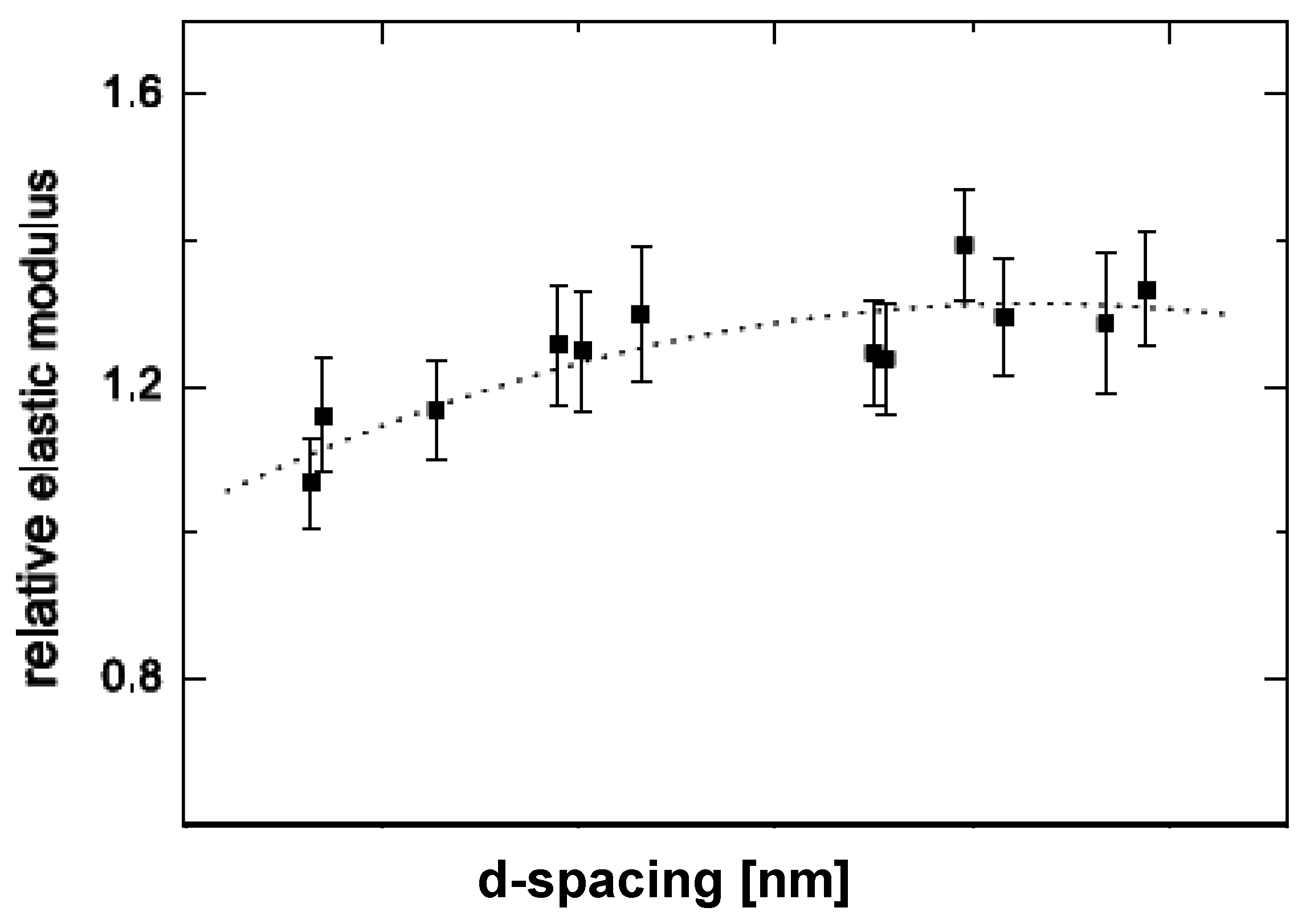
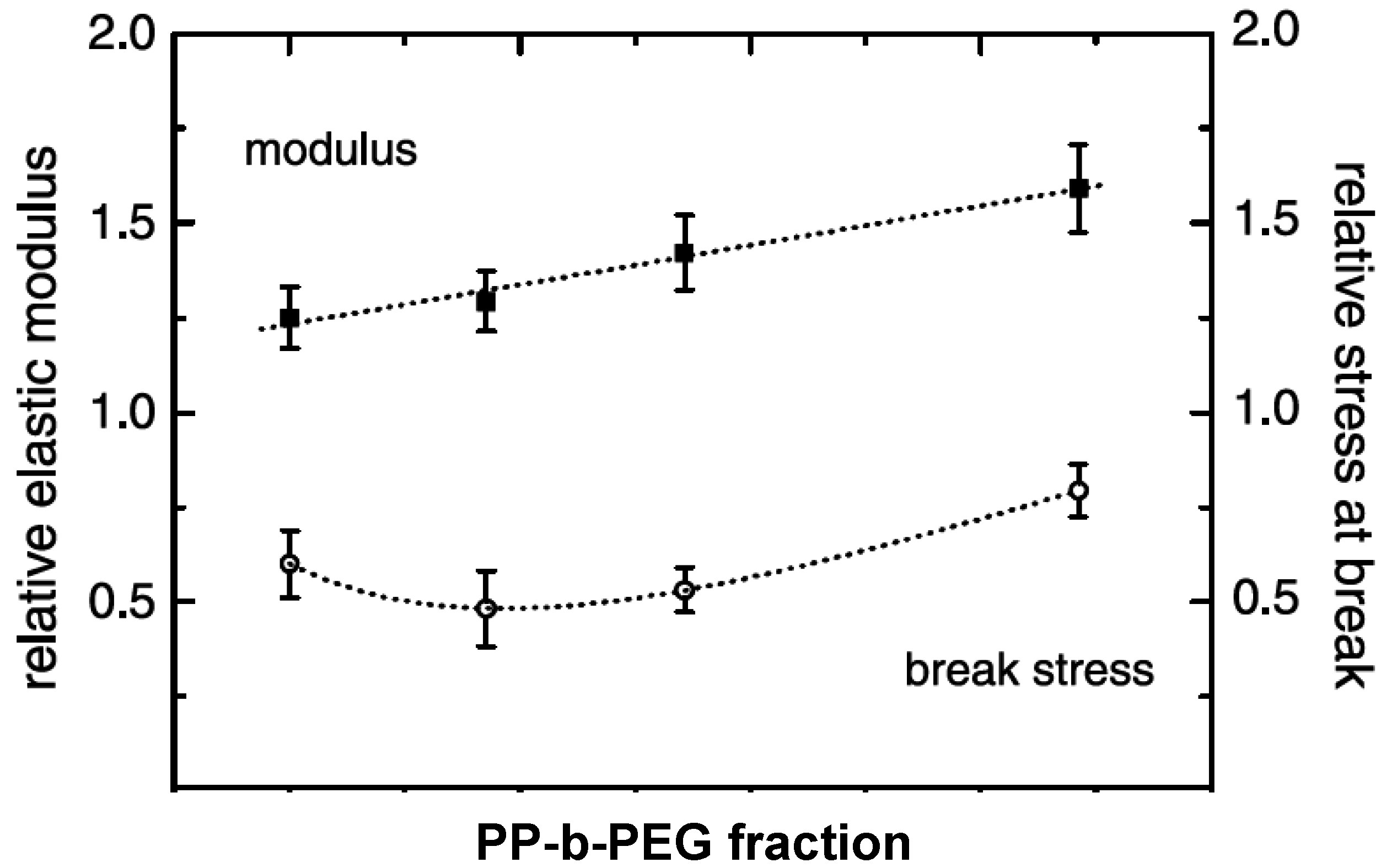
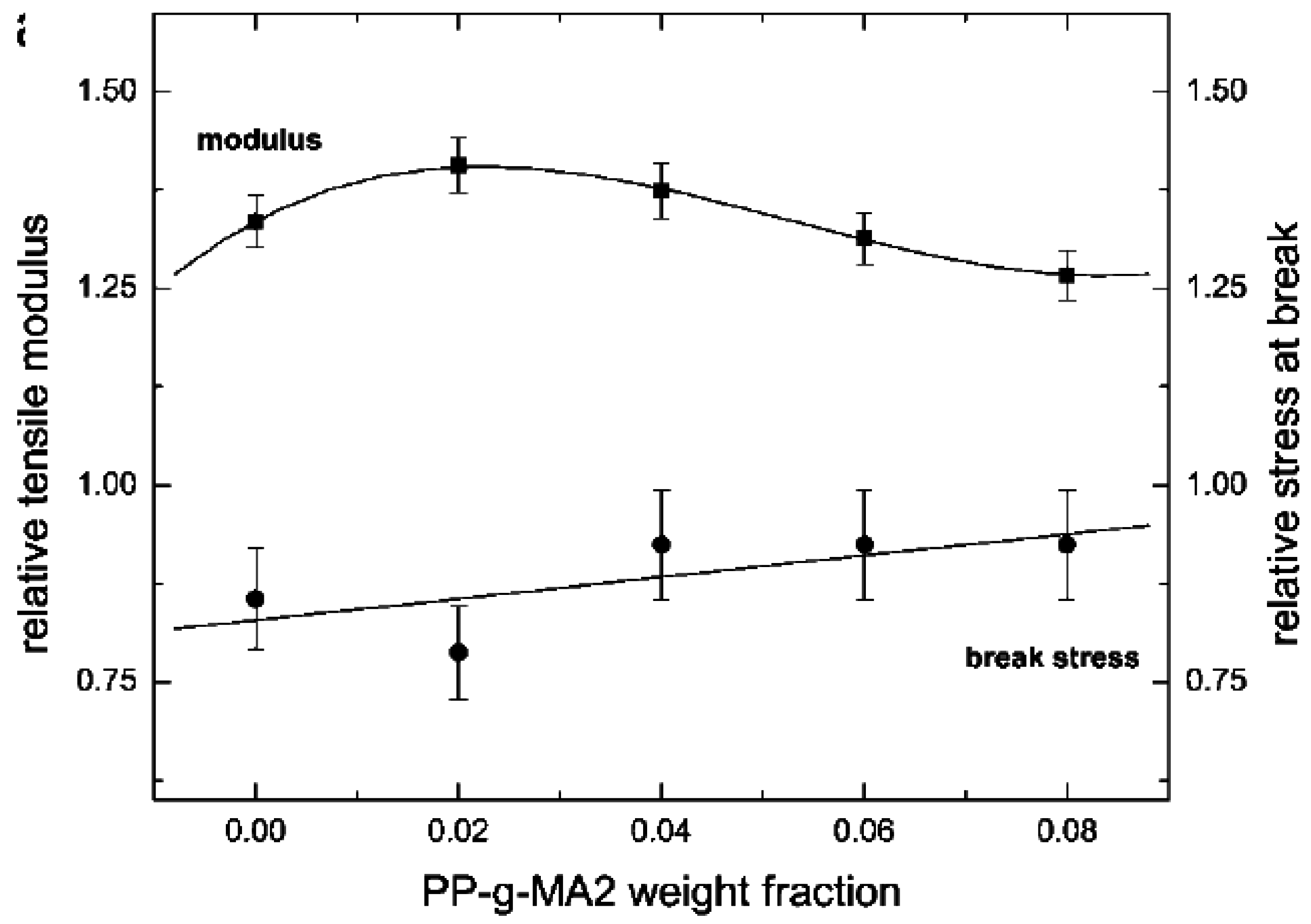
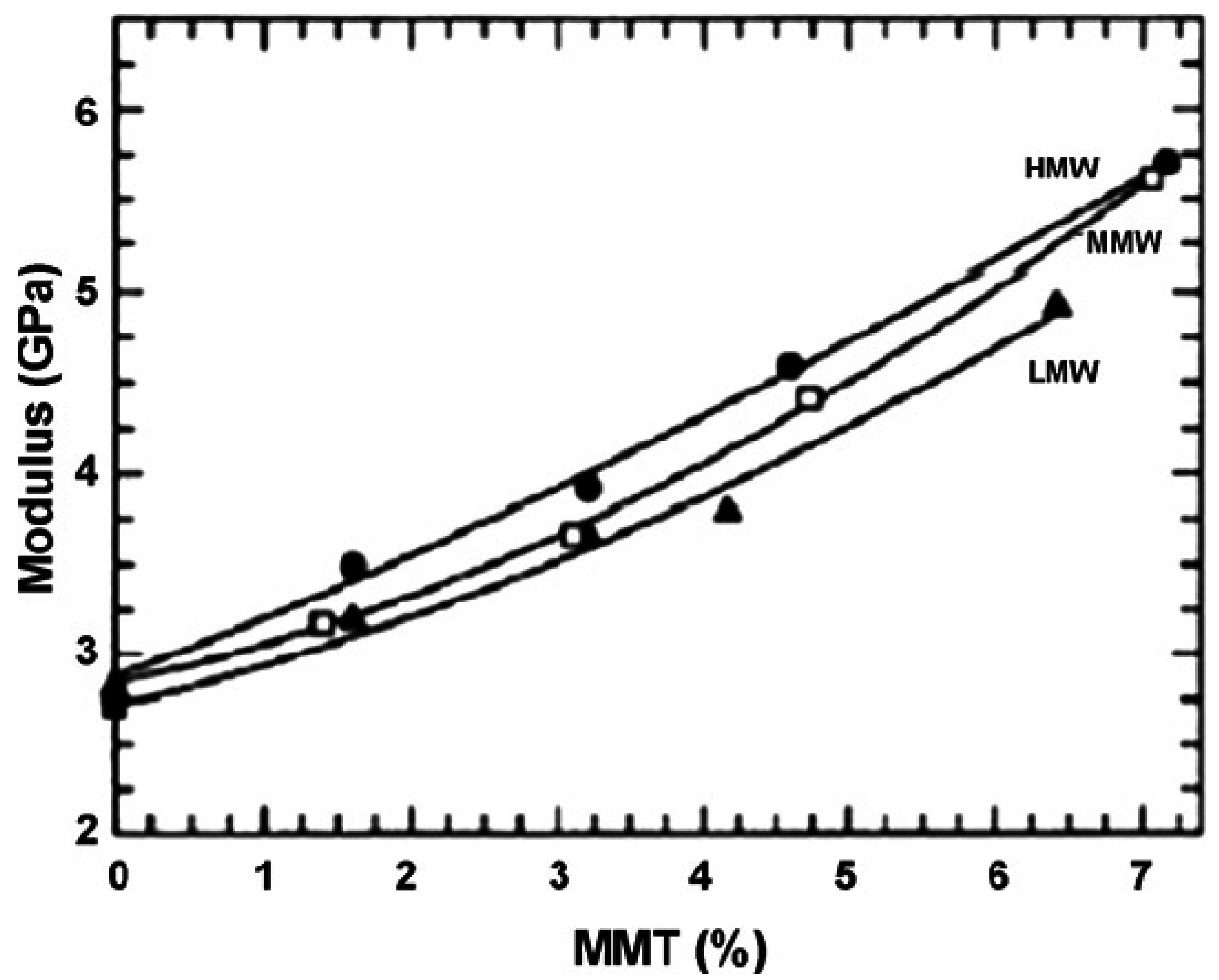
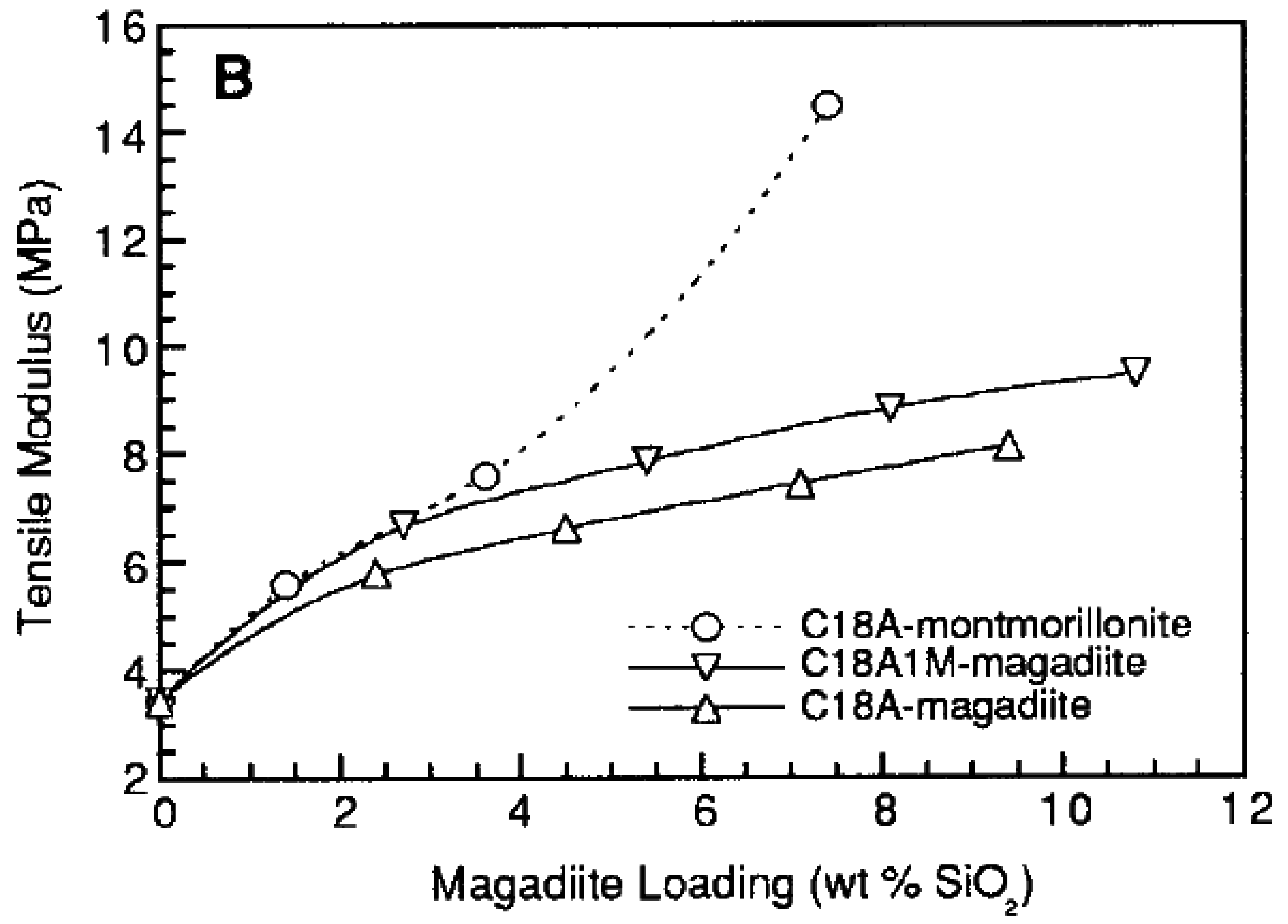
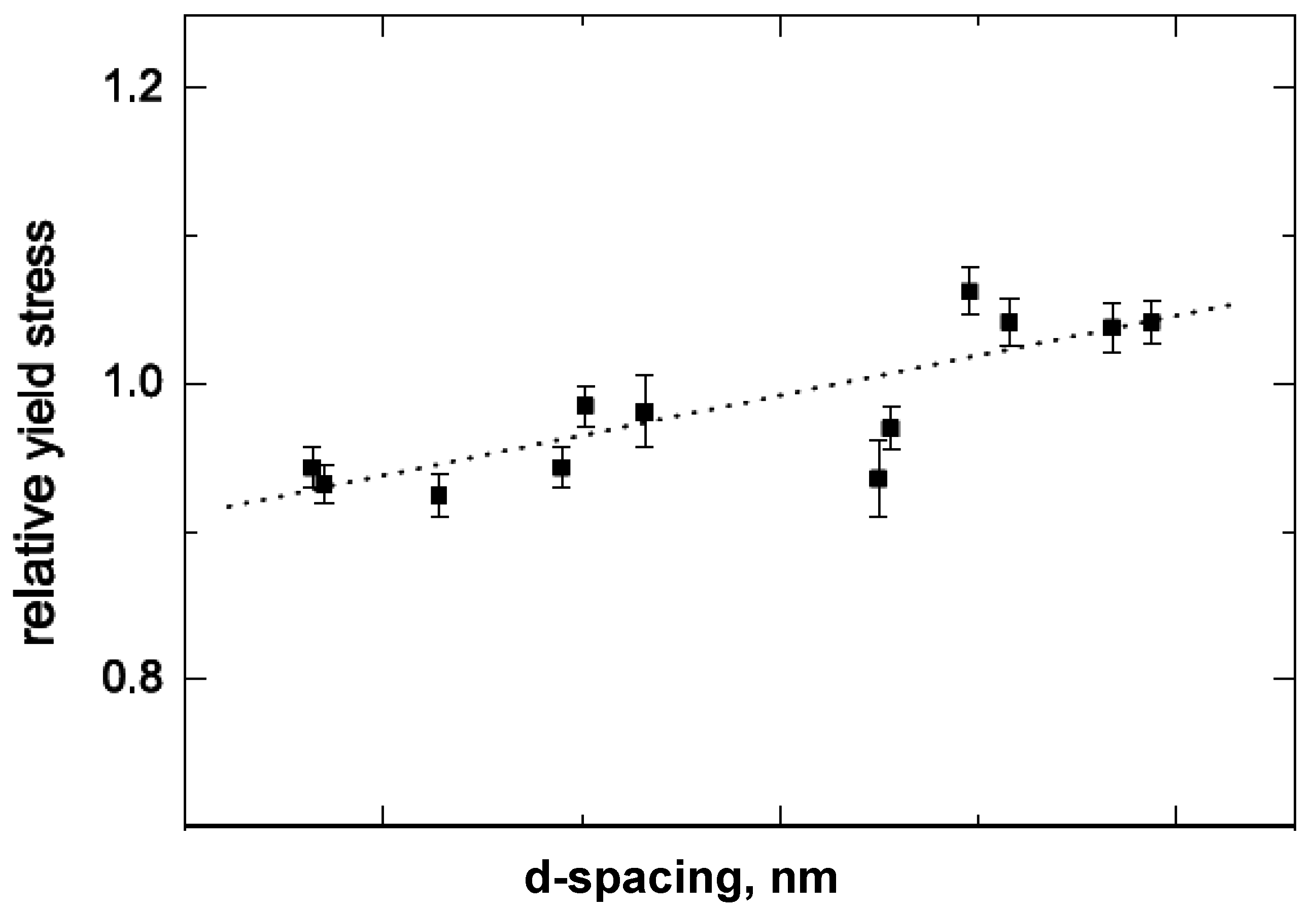
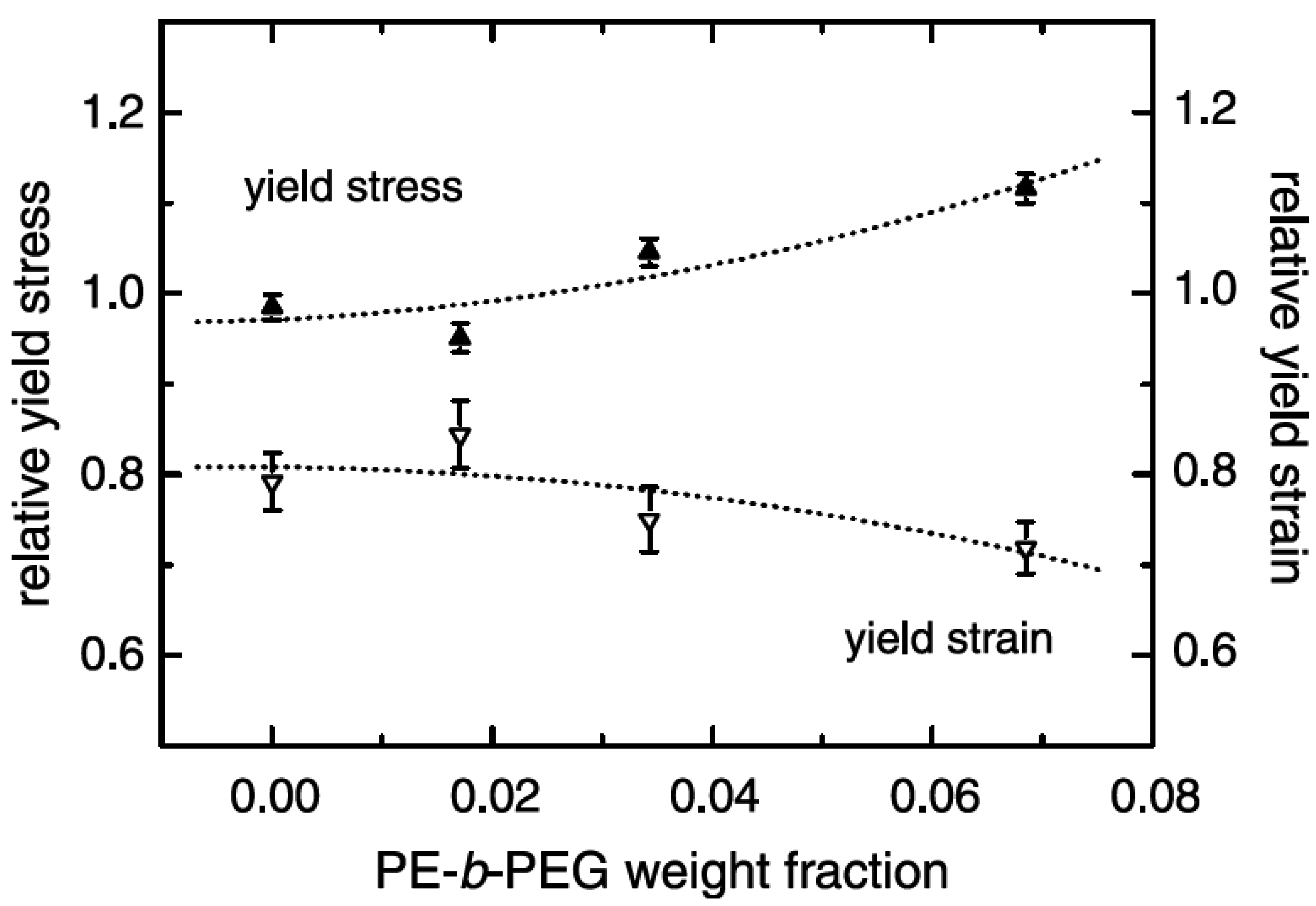
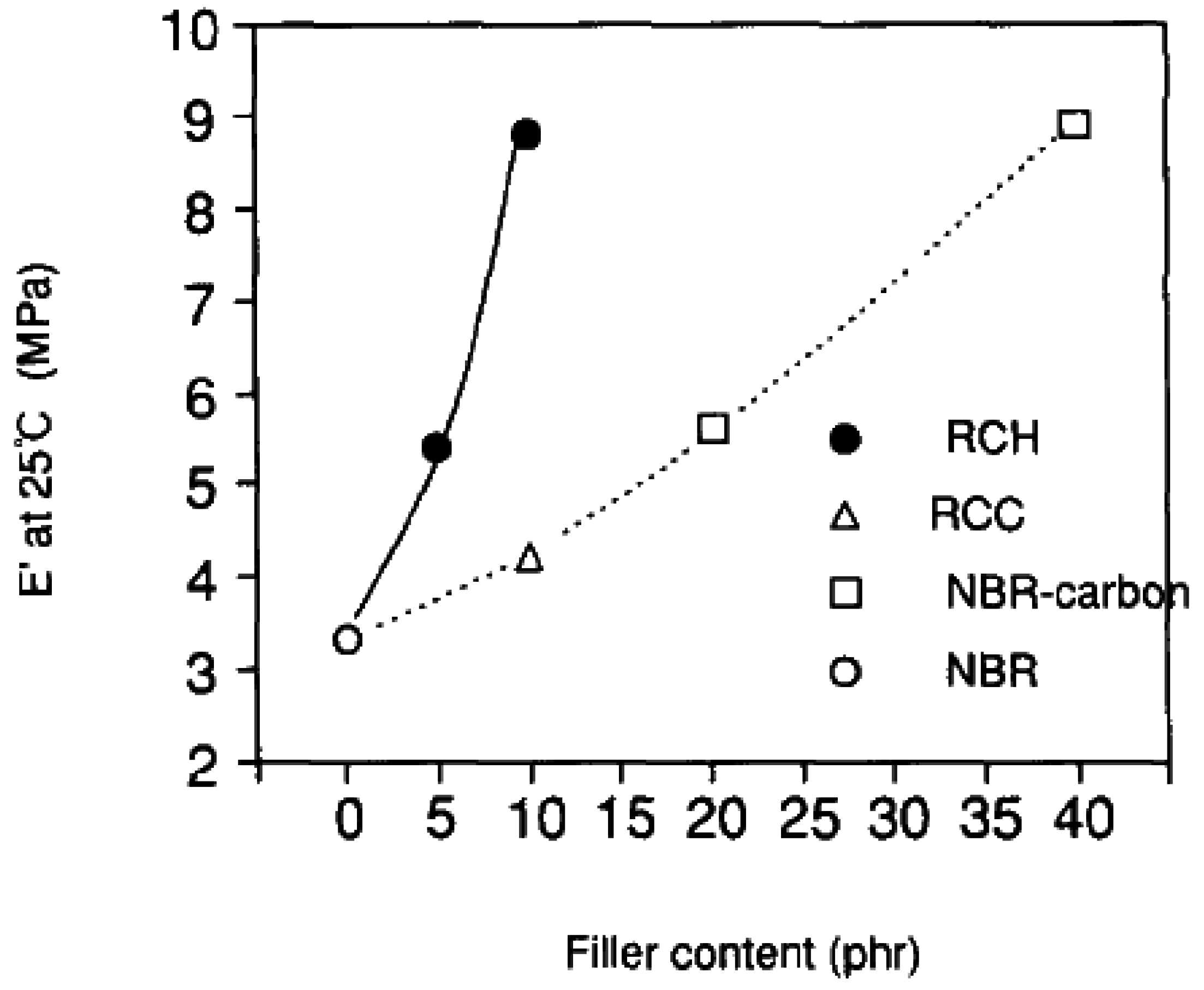
5.1. Mechanical Properties

| Nanocomposite | Clay Content (wt.%) | Young modulus (GPa) |
|---|---|---|
| PA6/MMT (in situ polymerization) | 0 | 1.11 |
| 4.7 | 1.87 | |
| 5.3 | 2.04 | |
| PA6(LMW)/MMT (melt intercalation) | 0 | 2.82 |
| 3.2 | 3.65 | |
| 6.4 | 4.92 | |
| PA6(MMW)/MMT (melt intercalation) | 0 | 2.71 |
| 3.1 | 3.66 | |
| 7.1 | 5.61 | |
| PA6(HMW)/MMT (melt intercalation) | 0 | 2.75 |
| 3.2 | 3.92 | |
| 7.2 | 5.70 | |
| PP(7.2% PP-g-MA)/OMLS | 0 | 0.714 |
| 7.2 | 0.838 | |
| PP(21.6% PP-g-MA)/OMLS | 0 | 0.760 |
| 7.2 | 1.010 | |
| EVA/Cloisite Na | 0 | 0.0122 |
| 3 | 0.0135 | |
| EVA/Cloisite 20A | 0 | 0.0122 |
| 3 | 0.0249 | |
| EVA/Cloisite 25A | 0 | 0.0122 |
| 3 | 0.0220 | |
| EVA/Cloisite 30B | 0 | 0.0122 |
| 3 | 0.0228 | |
| EVA/Nanofil 757 | 0 | 0.0122 |
| 3 | 0.0116 | |
| EVA/Nanofil 15 | 0 | 0.0122 |
| 3 | 0.0240 | |
| EVA/Somasif ME100 | 0 | 0.0122 |
| 3 | 0.0124 | |
| EVA/Somasif MAE | 0 | 0.0122 |
| 3 | 0.021 | |
| Soft PU/30B (solution intercalation) | 0 | 0.0075 |
| 3 | 0.0138 | |
| 7 | 0.024 | |
| Soft PU/30B (melt intercalation) | 0 | 0.0072 |
| 3 | 0.0114 | |
| 7 | 0.0193 | |
| Hard PU/30B (solution intercalation) | 0 | 0.050 |
| 3 | 0.086 | |
| 7 | 0.134 | |
| Hard PU/30B (melt intercalation) | 0 | 0.061 |
| 3 | 0.081 | |
| 7 | 0.119 | |
| HDPE/o-MMT | 0 | 1.020 |
| 0.9 | 1.060 | |
| 1.8 | 1.250 | |
| 2.8 | 1.380 | |
| 4.8 | 1.360 |






| Nanocomposite | Clay Content (wt.%) | Tensile Strength (MPa) |
|---|---|---|
| PA6/MMT (in situ polymerization) | 0 | 68.6 |
| 4.7 | 97.2 | |
| 5.3 | 97.3 | |
| PA6(LMW)/MMT (melt intercalation) | 0 | 69.2 |
| 3.2 | 78.9 | |
| 6.4 | 83.6 | |
| PA6(MMW)/MMT (melt intercalation) | 0 | 70.2 |
| 3.1 | 86.6 | |
| 7.1 | 95.2 | |
| PA6(HMW)/MMT (melt intercalation) | 0 | 69.7 |
| 3.2 | 84.9 | |
| 7.2 | 97.6 | |
| PMMA/OMLS | 0 | 53.9 |
| 12.6 | 62.0 | |
| PS/OMLS | 0 | 28.7 |
| 17.2 | 23.4 | |
| 24.6 | 16.6 | |
| EVA | 0 | 28.4 |
| EVA/Cloisite Na | 3 | 25.9 |
| EVA/Cloisite 20A | 3 | 25.8 |
| EVA/Cloisite 25A | 3 | 26.2 |
| EVA/Cloisite 30B | 3 | 30.7 |
| EVA/Nanofil 757 | 3 | 27.6 |
| EVA/Nanofil 15 | 3 | 26.7 |
| EVA/Somasif ME100 | 3 | 24.5 |
| EVA/Somasif MAE | 3 | 25.1 |
| Soft PU/30B (solution intercalation) | 0 | 45 |
| 3 | 31 | |
| 7 | 21 | |
| Hard PU/30B (solution intercalation) | 0 | 58 |
| 3 | 44 | |
| 7 | 34 | |
| PU/MMT | 0 | 5.9 |
| 5 | 6.2 | |
| 10 | 6.5 | |
| 21.5 | 8.3 | |
| PE/JS | 0 | 22 |
| 5 | 25 | |
| 10 | 27 | |
| 15 | 28 | |
| PE/DM | 0 | 22 |
| 5 | 21 | |
| 10 | 23 | |
| 15 | 24 | |
| HDPE/o-MMT | 0 | 27 |
| 0.9 | 26 | |
| 1.8 | 26 | |
| 2.8 | 26 | |
| 4.0 | 25 |




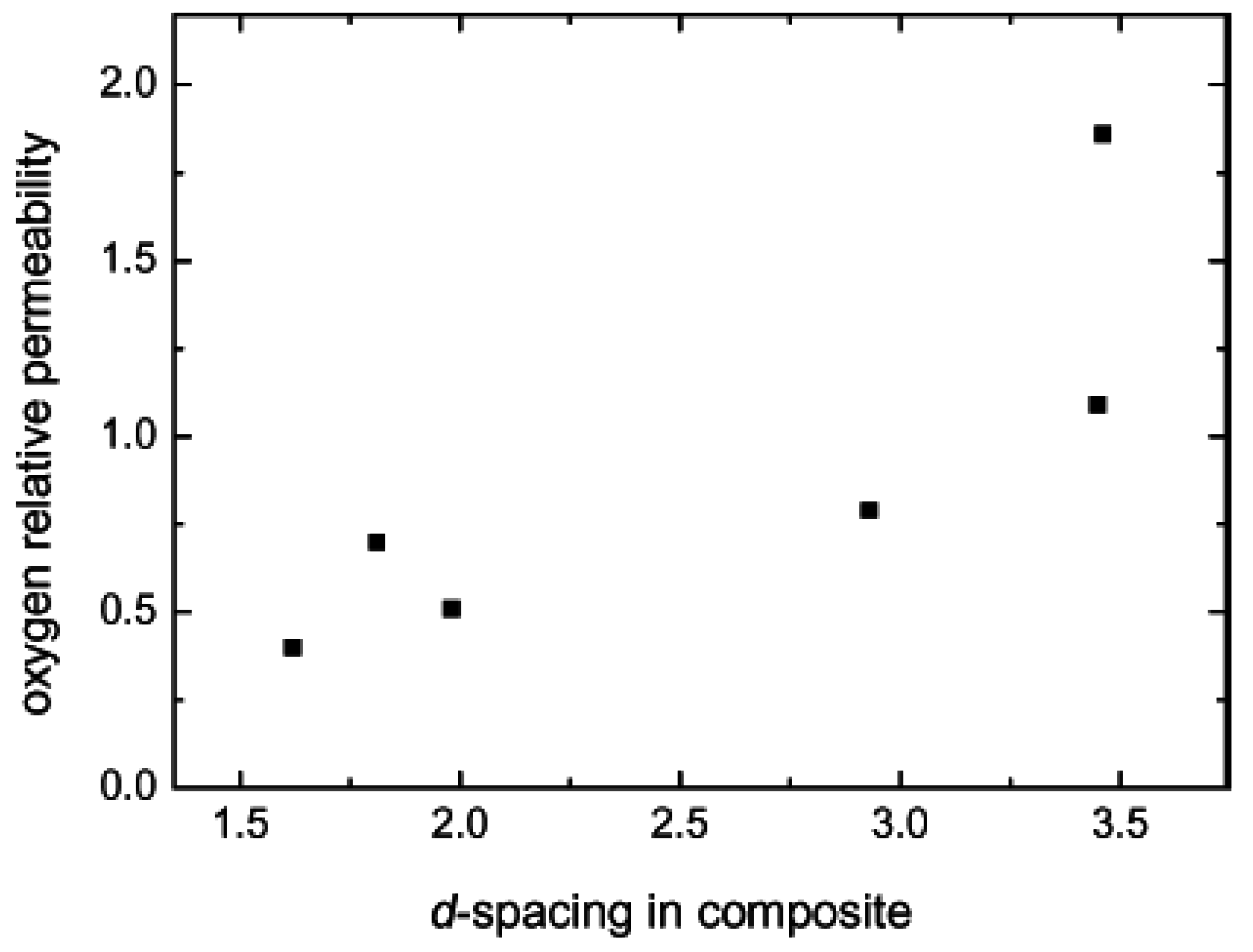
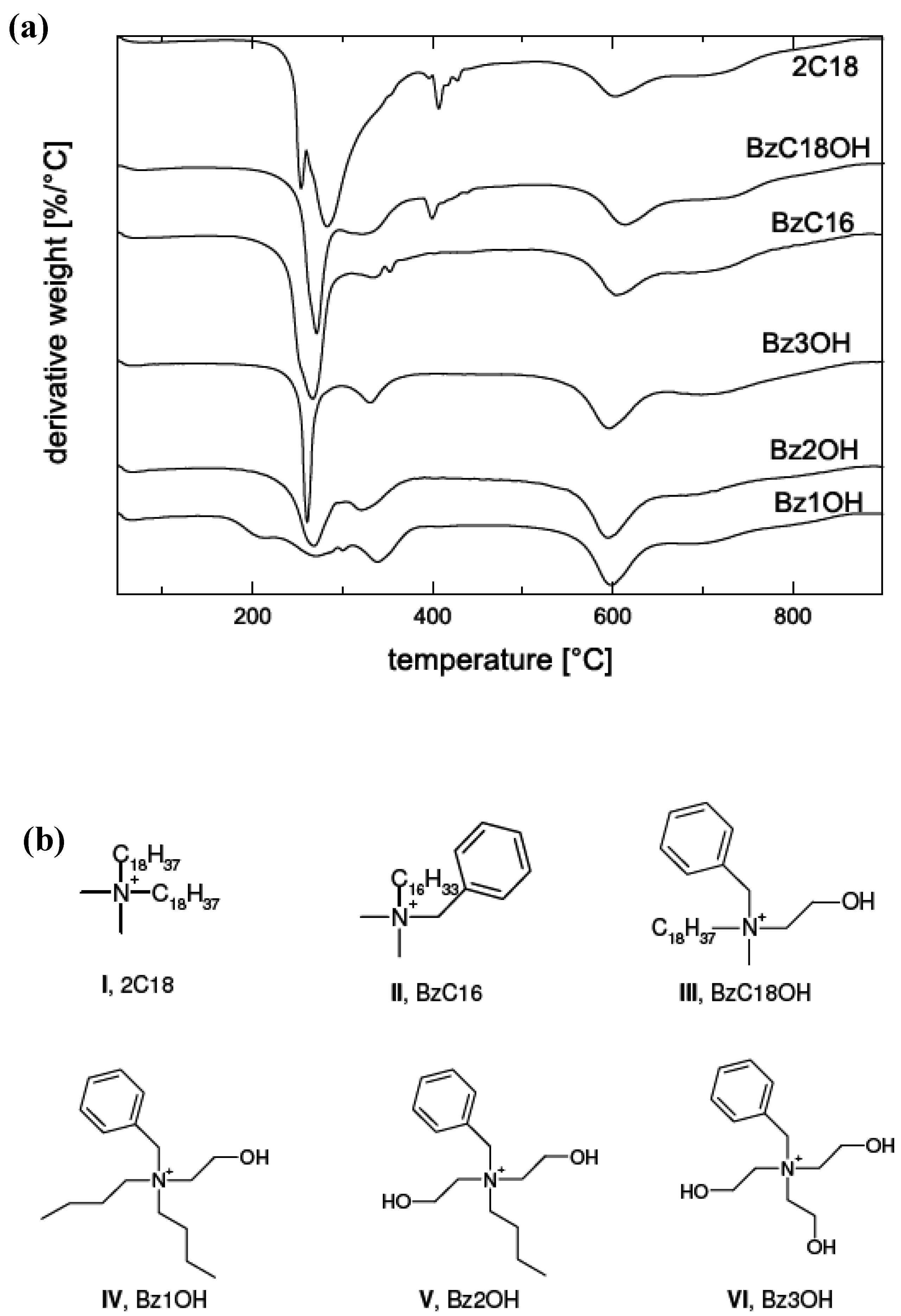
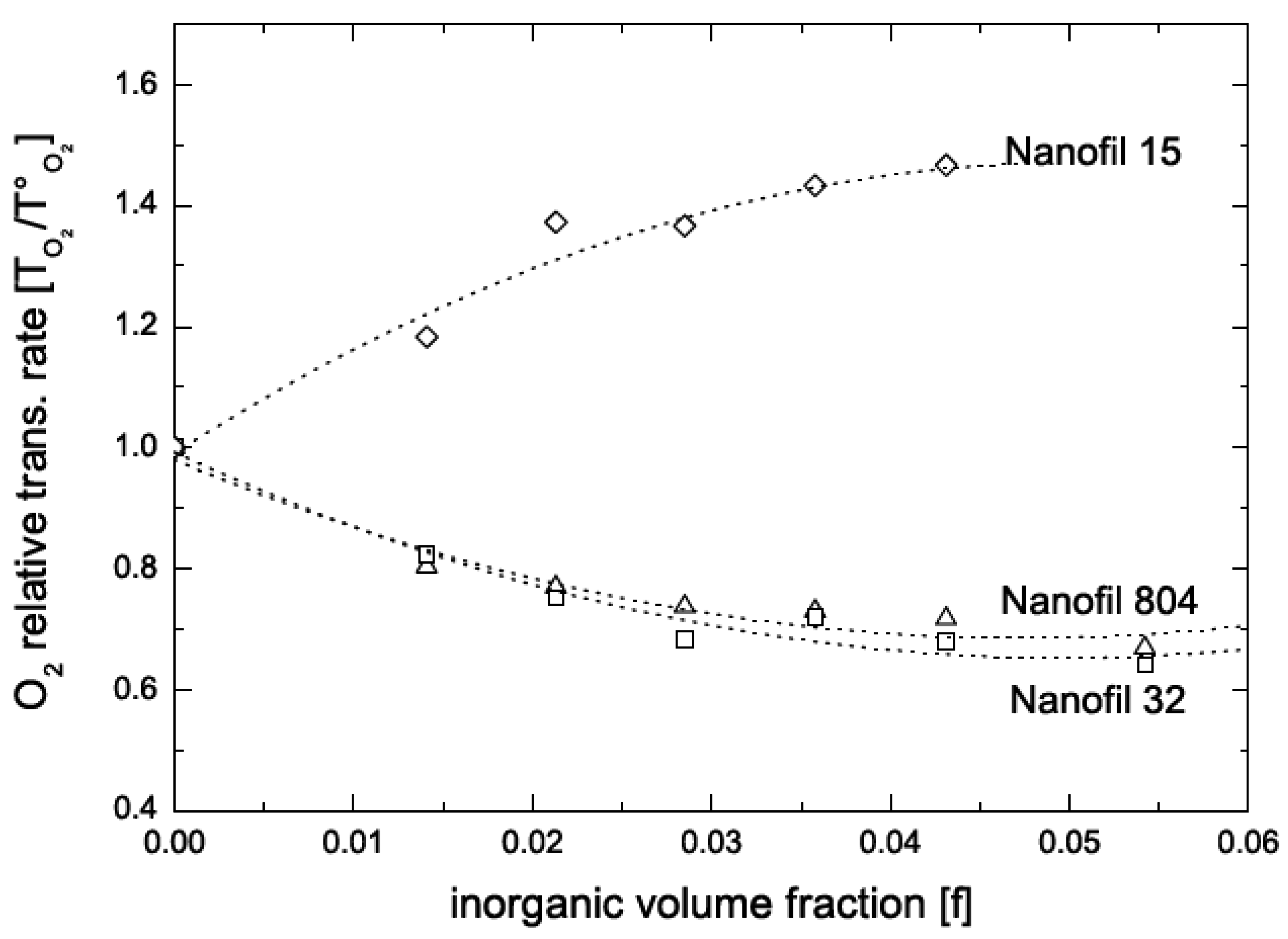
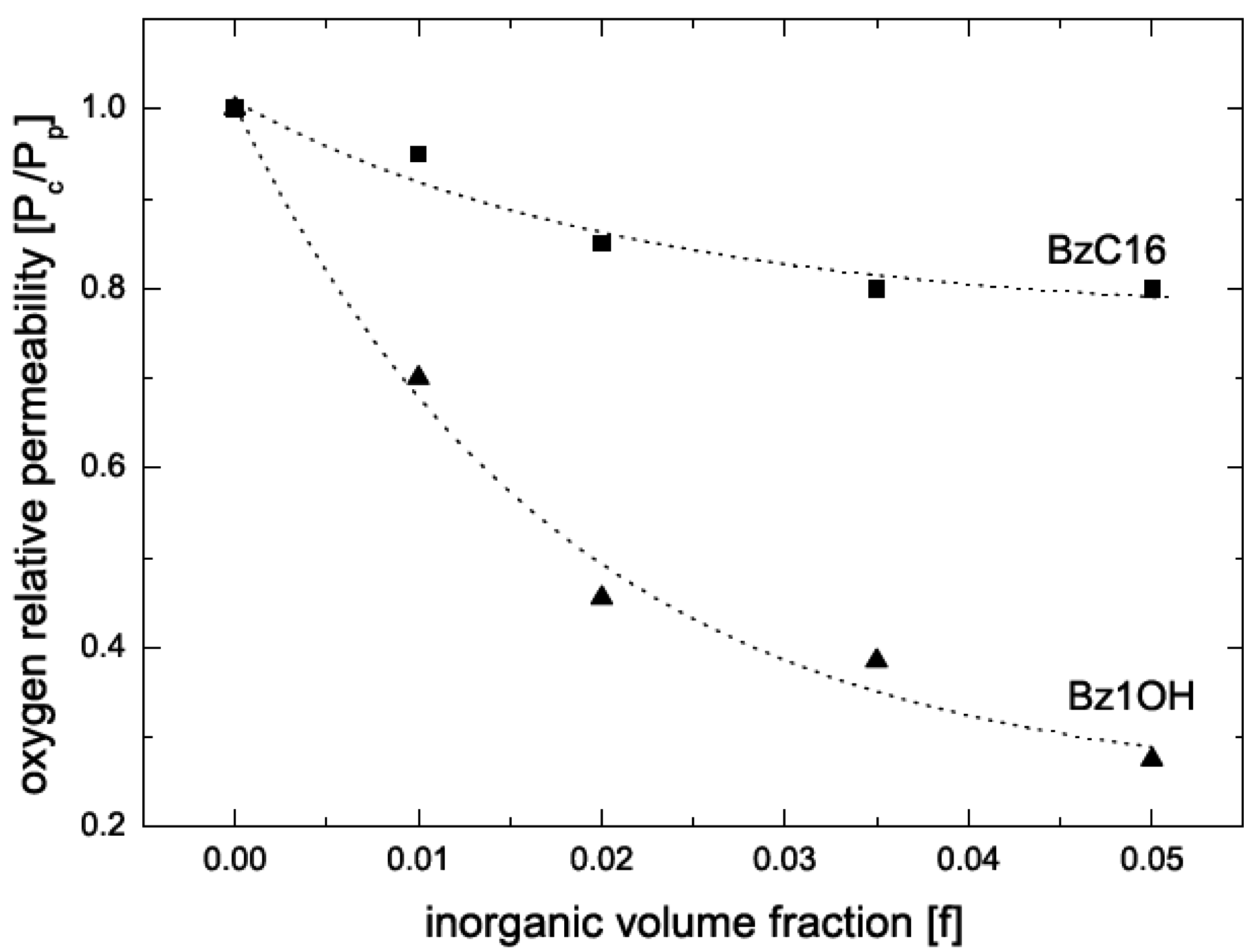
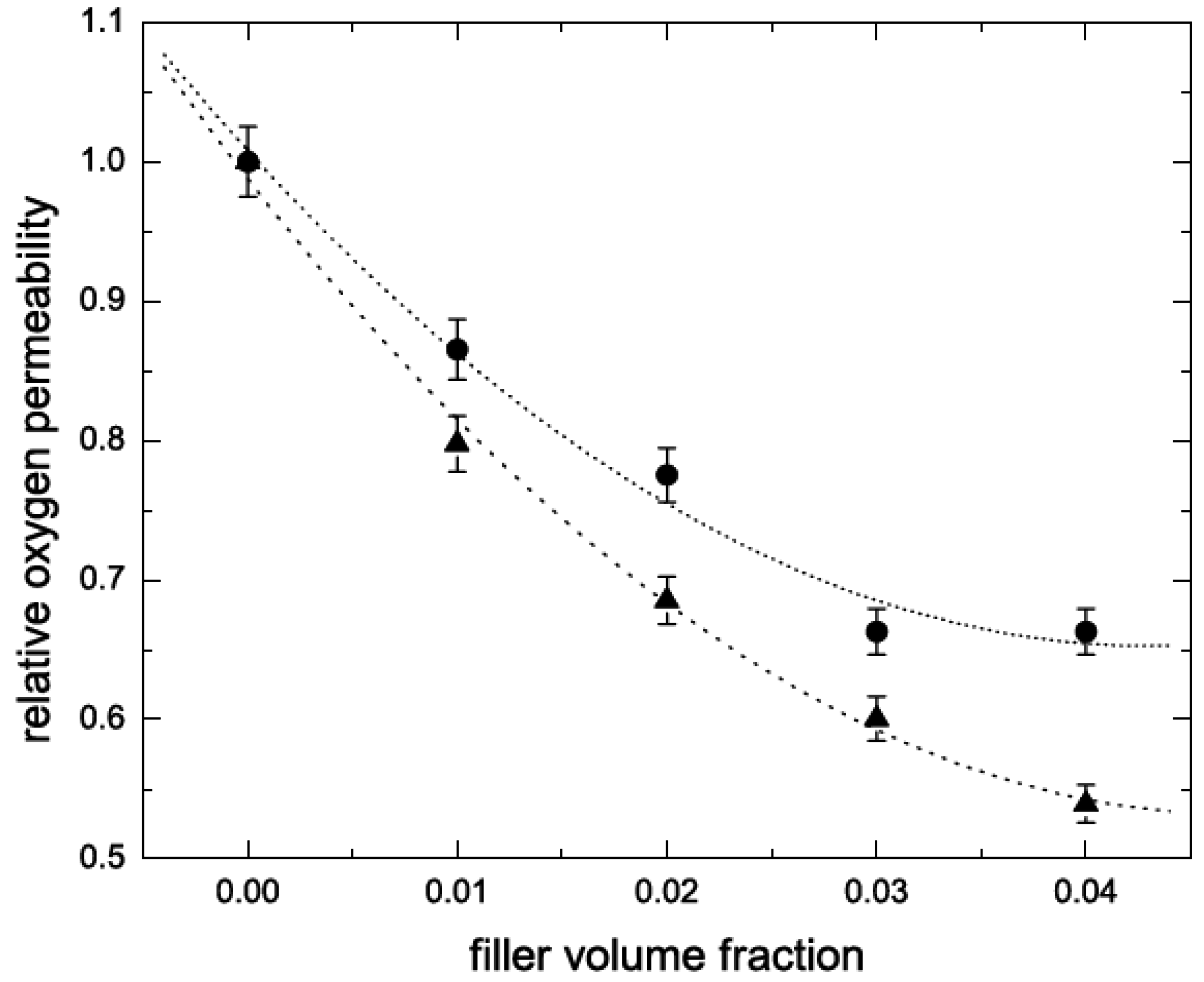
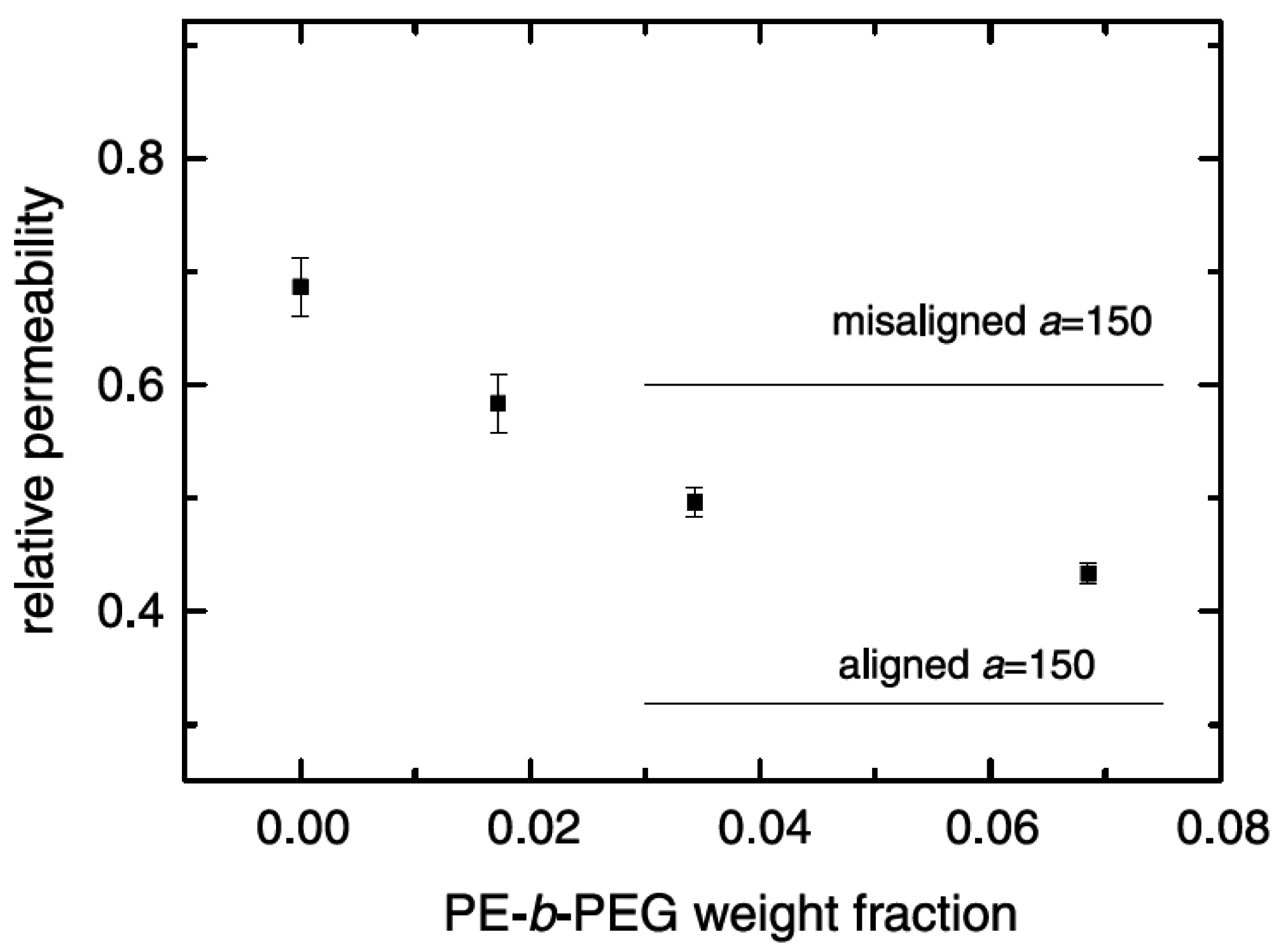
5.2. Barrier Properties


| Composite | Oxygen permeability coefficient [cm3.μm/(m2.d.mmHg)] | Water vapor trans. Rate [g.μm/(m2.d.mmHg)] |
|---|---|---|
| Neat Epoxy | 2.0 | 10 |
| Cloisite Na+ | 1.6 | 23 |
| Bz1OH | 0.77 | 6.7 |
| Bz2OH | 0.78 | 5.8 |
| Bz3OH | 1.0 | 7.1 |
| BzC18OH | 2.2 | 5.7 |
| BzC16 | 1.6 | 5.3 |
| 2C18 | 3.7 | 6.8 |


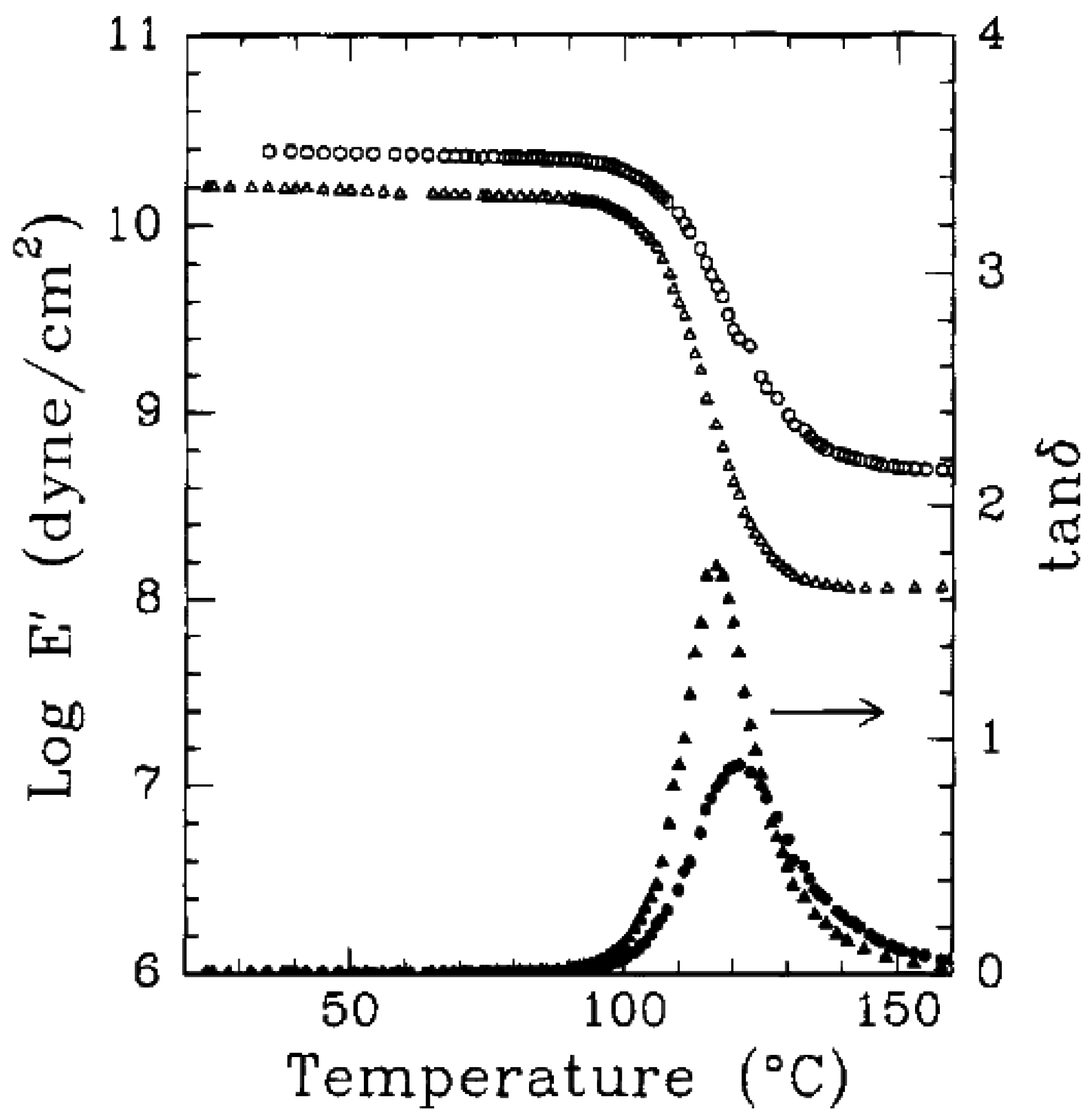
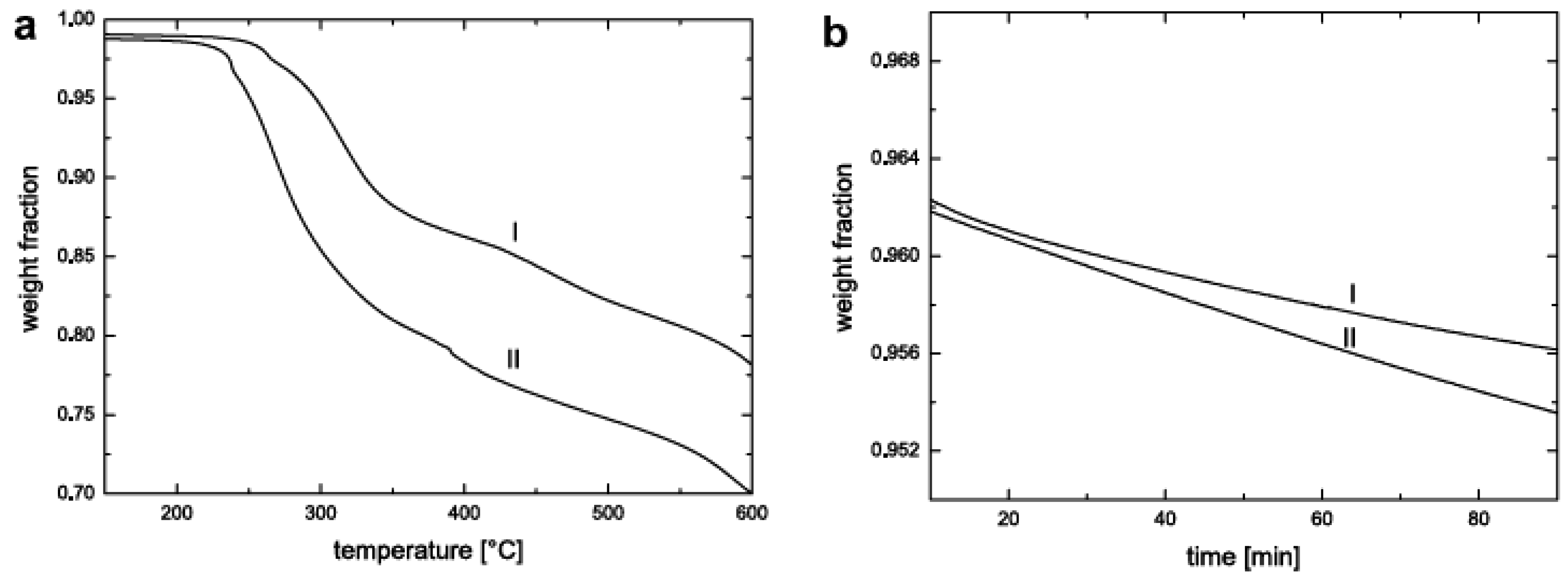
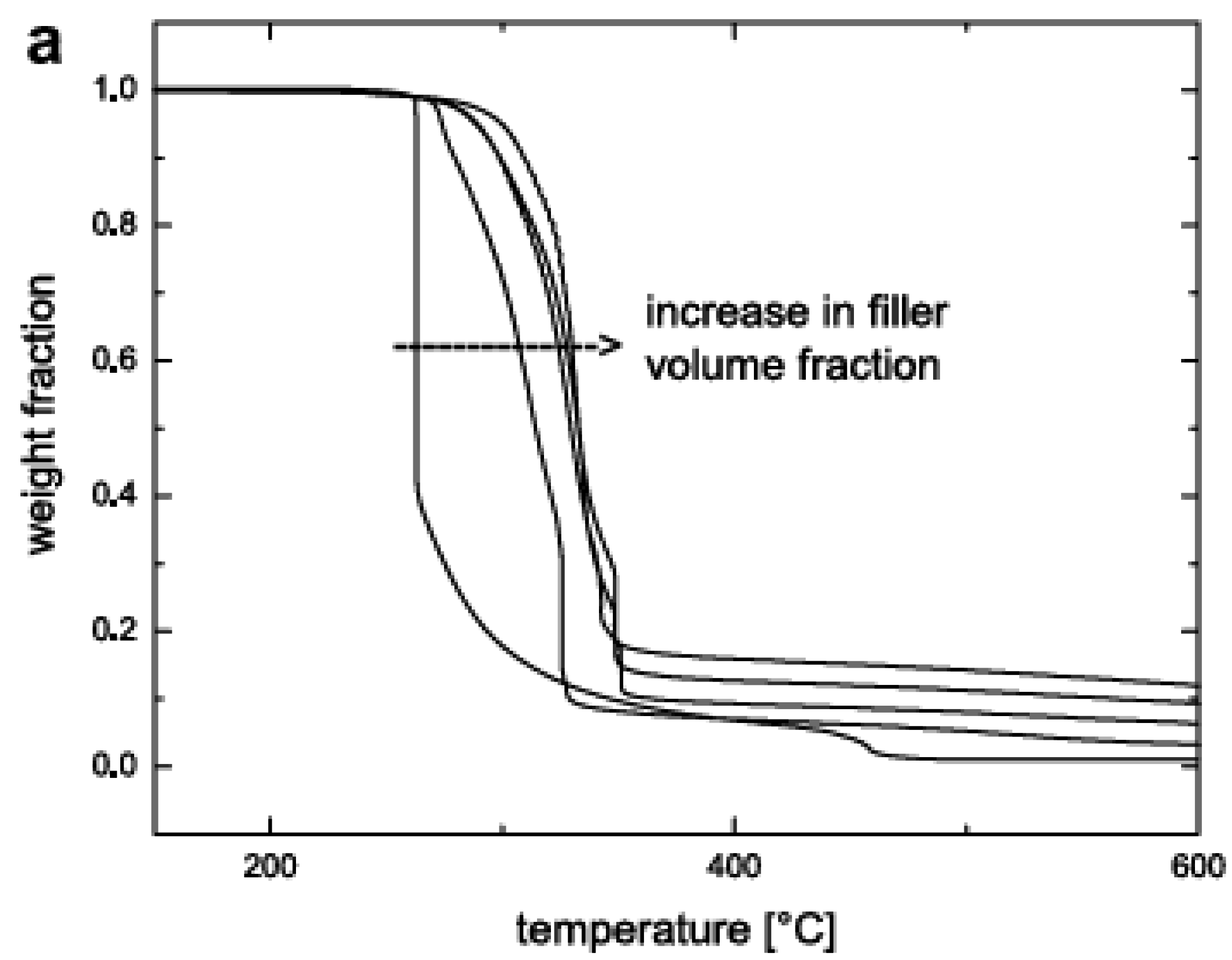
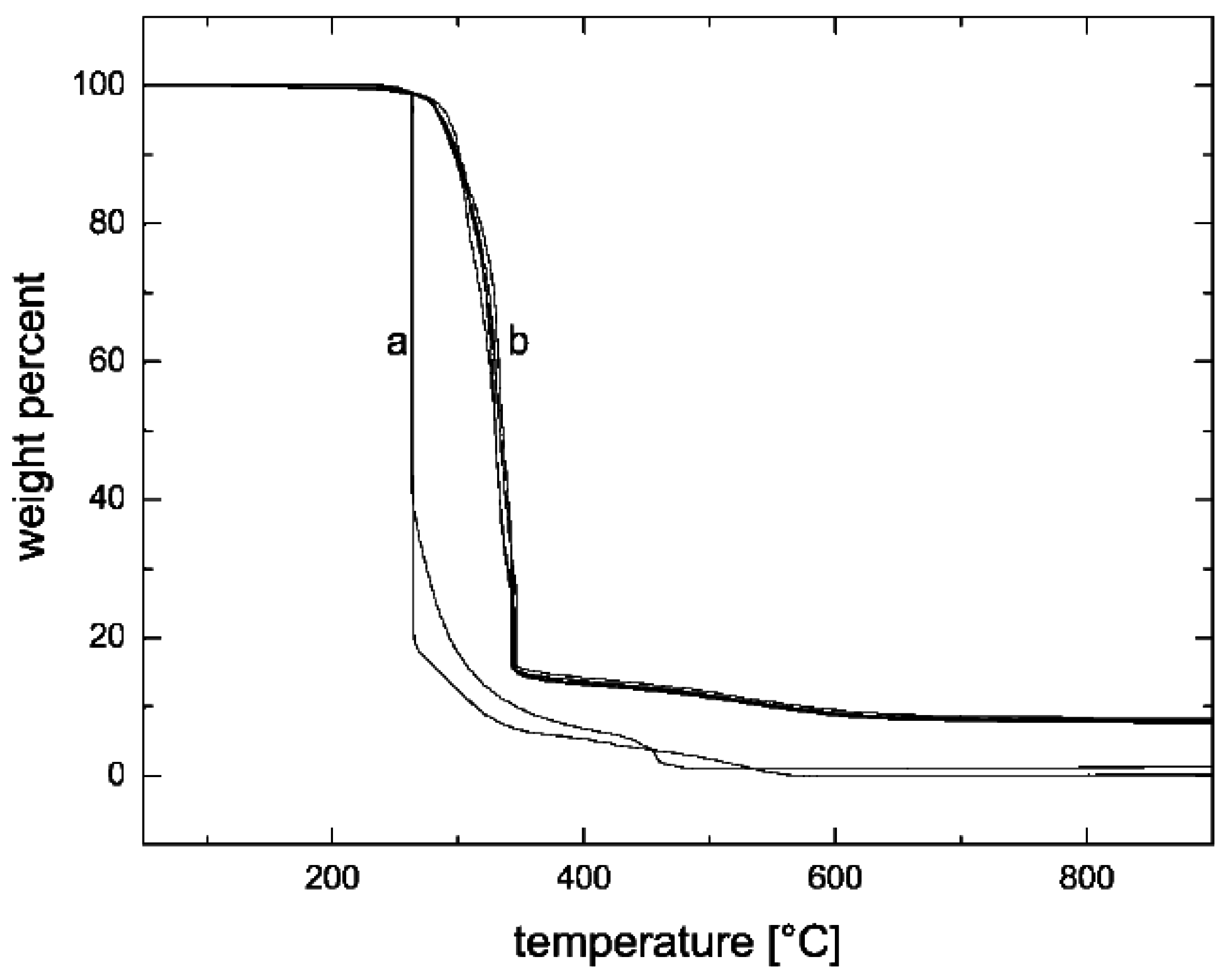
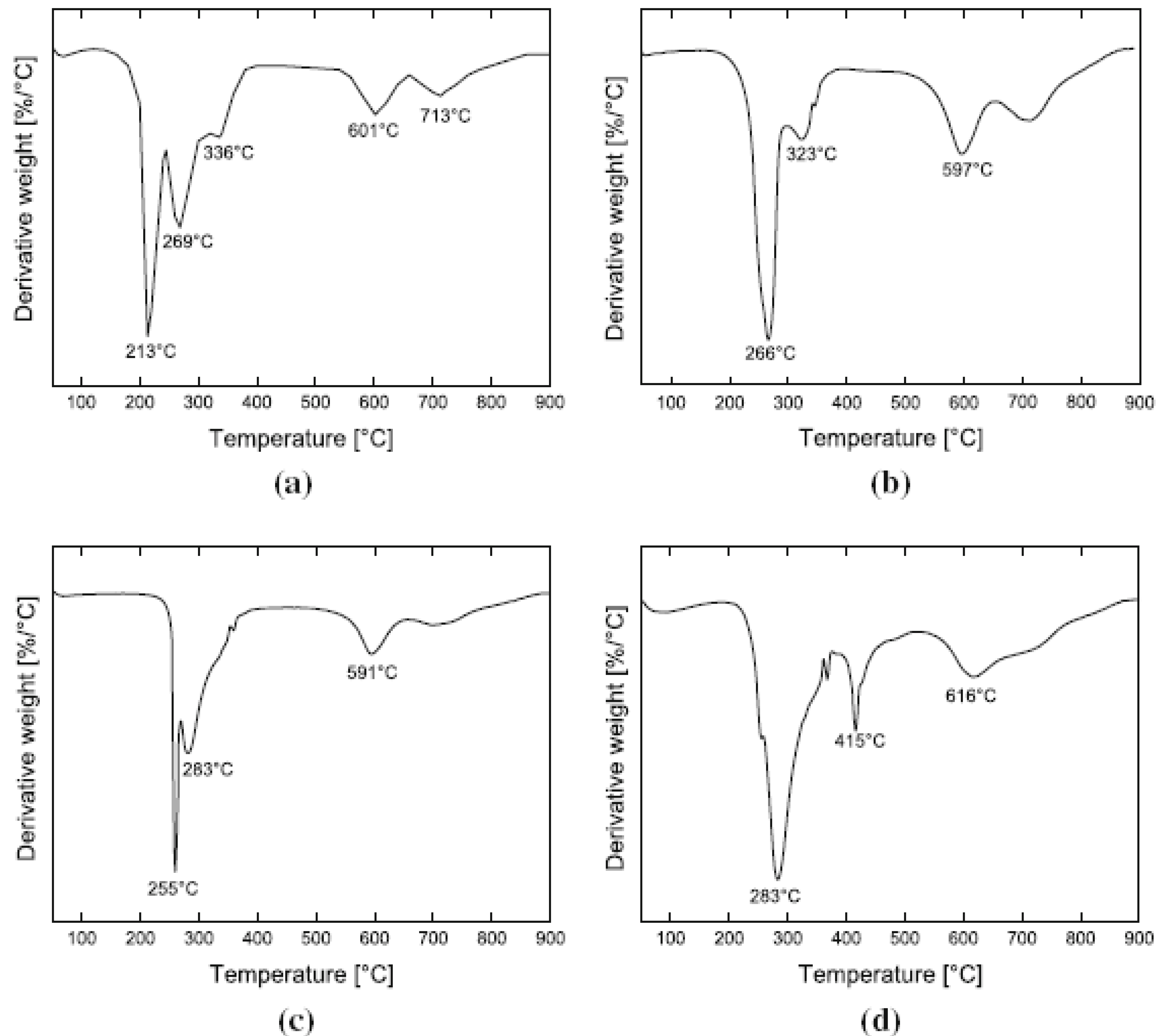
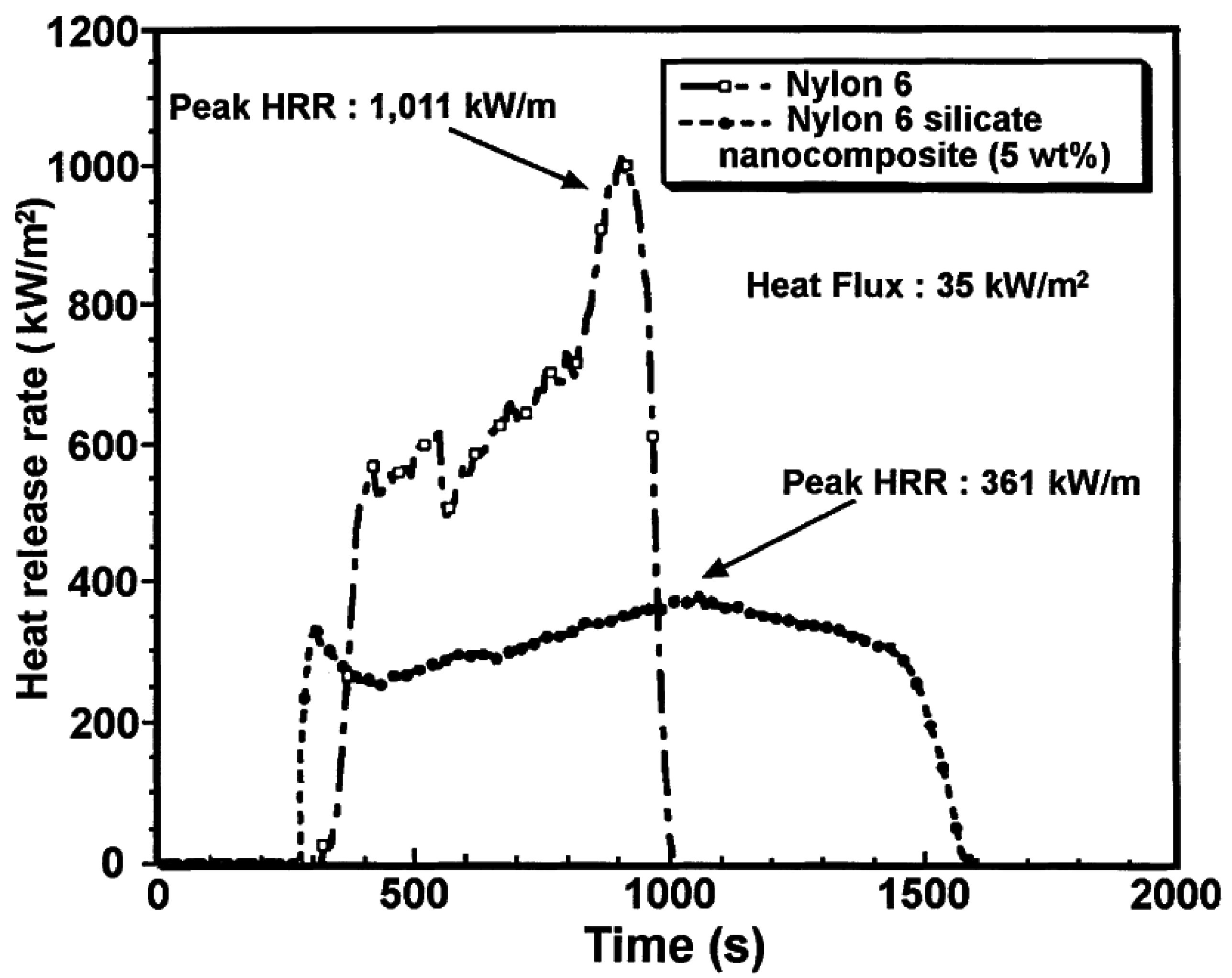
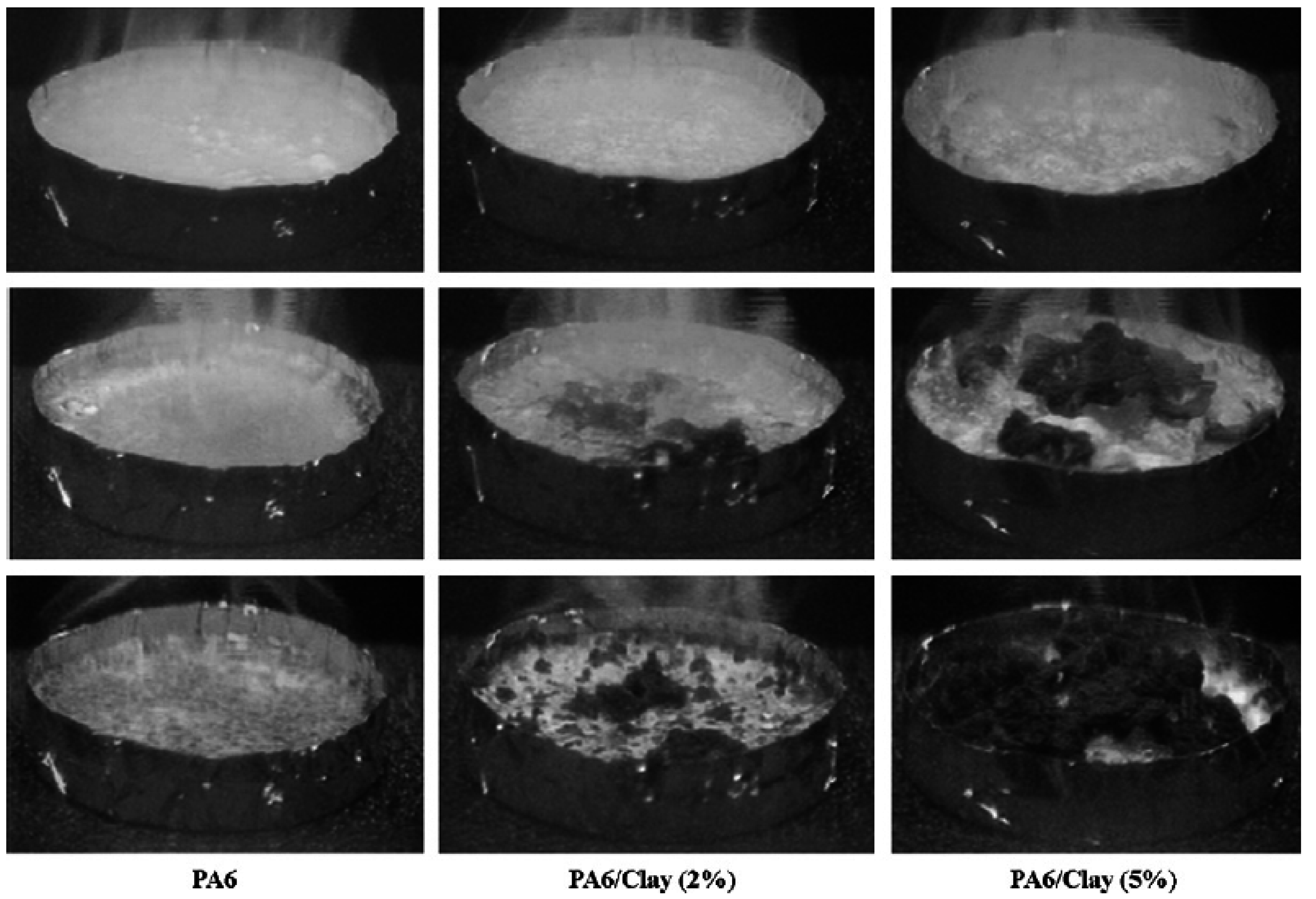
5.3. Thermal Stability and Flame Retardancy




| Sample | PHRR(kW/m2) | HRRave (kW/m2) | SEAave (m2/Kg) |
|---|---|---|---|
| PA6 | 1010 | 603 | 197 |
| PA6/2% OMLS | 686 | 390 | 271 |
| PA6/5% OMLS | 378 | 304 | 296 |
| PS | 1120 | 703 | 1460 |
| PS silicate mix3% | 1080 | 715 | 1840 |
| PS Nanocomposites 3% | 567 | 444 | 1730 |
| PSw/DBDPO/Sb2O3 30% | 491 | 318 | 2580 |
| PS | 1230 | 1315 | |
| PS/MAST-Hect 1% | 1011 | 1336 | |
| PS/MAST-Hect 3% | 894 | 1332 | |
| PS/MAST-Hect 5% | 728 | 1327 | |
| PPgMA | 1525 | 536 | 704 |
| PPgMA nanocomposites 2% | 450 | 322 | 1028 |
| PPgMA nanocomposites 4% | 381 | 275 | 968 |
| EVA/Na+ 5% | 1200 | ||
| EVA/Cloisite 30B 3% | 860 | ||
| EVA/Cloisite 30B 5% | 780 | ||
| EVA/Cloisite 30B 10% | 630 | ||
| EVA | 2303 | 430 | |
| EVA/30B | 1174 | 670 | |
| EVA/30BHect | 1289 | 593 | |
| EVA/30BMag | 2010 | 476 | |
| EVA/MMT | 1959 | 517 | |
| PU (I) | 2561 | 741 | 176 |
| PU (I)/o-MMT | 918 | 344 | 305 |
| PU (II) | 2254 | 637 | 235 |
| PU (II)/o-MMT | 641 | 363 | 412 |
| PU (III) | 2647 | 768 | 165 |
| PU (III)/o-MMT | 848 | 444 | 172 |
| PU (IV) | 2664 | 775 | 235 |
| PU (IV)/o-MMT | 797 | 435 | 412 |
| PE | 1470 | 510 | |
| PE/JS 2% | 670 | 440 | |
| PE/JS 5% | 320 | 450 | |
| PE/JS 10% | 540 | 380 | |
| PE/JS 15% | 390 | 280 |


5.4. Crystallinity
6. Summary
References and Notes
- Katz, H.S.; Milewski, J.V. Handbook of Fillers for Plastics, 2nd ed.; Van Nostrand: New York, NY, USA, 1987. [Google Scholar]
- Rothon, R. Particulate-Filled Polymer Composites; Longman: Harlow, UK, 1995. [Google Scholar]
- Brechet, Y.; Cavaille, J.Y.; Chabert, E.; Chazeau, L.; Dendievel, R.; Flandin, L.; Gauthier, C. Polymer based nanocomposites: Effect of filler-filler and filler-matrix interactions. Adv. Eng. Mater. 2001, 3, 571–577. [Google Scholar] [CrossRef]
- Pavlidoua, S.; Papaspyrides, C.D. A review on polymer–layered silicate nanocomposites. Prog. Polym. Sci. 2008, 33, 1119–1198. [Google Scholar] [CrossRef]
- Mark, J.E. Ceramic reinforced polymers and polymer-modified ceramics. Polym. Eng. Sci. 1996, 36, 2905–2920. [Google Scholar] [CrossRef]
- Reynaud, E.; Gauthier, C.; Perez, J. Nanophases in polymers. Rev. Metall. Cah. Inf. Tech. 1999, 96, 169–176. [Google Scholar]
- Calvert, P. Carbon Nanotubes; Ebbesen, T.W., Ed.; CRC Press: Boca Raton, FL, USA, 1997; pp. 277–292. [Google Scholar]
- Favier, V.; Canova, G.R.; Shrivastava, S.C.; Cavaille, J.Y. Mechanical percolation in cellulose whiskers nanocomposites. Polym. Eng. Sci. 1997, 37, 1732–1739. [Google Scholar] [CrossRef]
- Mittal, V. Organic Modifications of Clay and Polymer Surfaces for Specialty Applications. PhD Thesis, ETH Zurich, Zurich, Switzerland, 2006. [Google Scholar]
- Carter, L.W.; Hendricks, J.G.; Bolley, D.S. Elastomer reinforced with modified clay (assigned to National Lead Co.). U. S. Pat. No. 2,531,396, 1950. [Google Scholar]
- Greenland, D.J. Adsorption of poly(vinyl alcohols) by montmorillonite. J. Colloid Sci. 1963, 18, 647–664. [Google Scholar] [CrossRef]
- Yano, K.; Usuki, A.; Okada, A.; Kurauchi, T.; Kamigaito, O. Synthesis and properties of polyimide-clay hybrid. J. Polym. Sci. A Polym. Chem. 1993, 31, 2493–2498. [Google Scholar] [CrossRef]
- Kojima, Y.; Fukumori, K.; Usuki, A.; Okada, A.; Kurauchi, T. Gas permeabilities in rubber-clay hybrid. J. Mater. Sci. Lett. 1993, 12, 889–890. [Google Scholar] [CrossRef]
- Vaia, R.A.; Ishii, H.; Giannelis, E.P. Synthesis and properties of two-dimensional nanostructures by direct intercalation of polymer melts in layered silicates. Chem. Mater. 1993, 5, 1694–1696. [Google Scholar] [CrossRef]
- Mehrotra, V.; Giannelis, E.P. Conducting molecular multilayers: intercalation of conjugated polymers in layered media. Mater. Res. Soc. Symp. Proc. 1990, 171, 39–44. [Google Scholar] [CrossRef]
- Lan, T.; Kaviratna, P.D.; Pinnavaia, T.J. On the nature of polyimidie-clay hybrid composites. Chem. Mater. 1994, 6, 573–575. [Google Scholar] [CrossRef]
- Chin, I.-J.; Thurn-Albrecht, T.; Kim, H.-C.; Russell, T.P.; Wang, J. On exfoliation of montmorillonite in epoxy. Polymer 2001, 42, 5947–5952. [Google Scholar] [CrossRef]
- Lim, S.K.; Kim, J.W.; Chin, I.-J.; Kwon, Y.K.; Choi, H.J. Preparation and interaction characteristics of organically modified montmorillonite nanocomposite with miscible polymer blend of poly(ethylene oxide) and poly(methyl methacrylate). Chem. Mater. 2002, 14, 1989–1994. [Google Scholar] [CrossRef]
- Wang, Z.; Pinnavaia, T.J. Nanolayer reinforcement of elastomeric polyurethane. Chem. Mater. 1998, 10, 3769–3771. [Google Scholar] [CrossRef]
- Messersmith, P.B.; Giannelis, E.P. Synthesis and barrier properties of poly(ε-caprolactone)-layered silicate nanocomposites. J. Polym. Sci. A–Polym. Chem. 1995, 33, 1047–1057. [Google Scholar] [CrossRef]
- Yano, K.; Usuki, A.; Okada, A. Synthesis and properties of polyimide-clay hybrid films. J. Polym. Sci. A–Polym. Chem. 1997, 35, 2289–2294. [Google Scholar] [CrossRef]
- Shi, H.; Lan, T.; Pinnavaia, T.J. Interfacial effects on the reinforcement properties of polymer-organoclay nanocomposites. Chem. Mater. 1996, 8, 1584–1587. [Google Scholar] [CrossRef]
- Giannelis, E.P. Polymer layered silicate nanocomposites. Adv. Mater. 1996, 8, 29–35. [Google Scholar] [CrossRef]
- LeBaron, P.C.; Wang, Z.; Pinnavaia, T.J. Polymer layered silicate nanocomposites: An overview. Appl. Clay Sci. 1999, 15, 11–29. [Google Scholar] [CrossRef]
- Bailey, S.W. Reviews in Mineralogy; Virginia Polytechnic Institute and State University: Blacksburg, VA, USA, 1984. [Google Scholar]
- Brindley, G.W.; Brown, G. Crystal Structures of Clay Minerals and Their X-ray Identification; Mineralogical Society: London, UK, 1980. [Google Scholar]
- Beyer, G. Nanocomposites: A new class of flame retardants for polymers. Plast. Addit. Compound. 2002, 4, 22–27. [Google Scholar] [CrossRef]
- Theng, B.K.G. The Chemistry of Clay-Organic Reactions; Wiley: New York, NY, USA, 1974. [Google Scholar]
- Jasmund, K.; Lagaly, G. Tonminerale und Tone Struktur; Steinkopff: Darmstadt, Germany, 1993. [Google Scholar]
- Osman, M.A.; Ploetze, M.; Suter, U.W. Surface treatment of clay minerals thermal stability, basal plane spacing and surface coverage. J. Mater. Chem. 2003, 13, 2359–2366. [Google Scholar] [CrossRef]
- Pinnavaia, T.J. Intercalated clay catalysts. Science 1983, 220, 365–371. [Google Scholar] [CrossRef] [PubMed]
- Krishnamoorti, R.; Giannelis, E.P. Rheology of end tethered polymer layered silicate nanocomposites. Macromolecules 1997, 30, 4097–4102. [Google Scholar] [CrossRef]
- Tjong, S.C.; Meng, Y.Z. Preparation and characterization of melt-compounded polyethylene/vermiculite nanocomposites. J. Polym. Sci. B Polym. Phys. 2003, 41, 1476–1484. [Google Scholar] [CrossRef]
- Xu, J.; Li, R.K.Y.; Xu, Y.; Li, L.; Meng, Y.Z. Preparation of poly(propylene carbonate)/organo-vermiculite nanocomposites via direct melt intercalation. Eur. Polym. J. 2005, 41, 881–888. [Google Scholar] [CrossRef]
- Thomas, G.W. Methods of Soil Analysis; Page, A.L., Miller, R.H., Keeney, D.R., Eds.; American Society of Agronomy: Madison, WI, USA, 1982. [Google Scholar]
- Grim, R.E. Clay Mineralogy; Mc Graw-Hill: New York, NY, USA, 1968. [Google Scholar]
- Osman, M.A.; Suter, U.W. Determination of cation exchange capacity of muscovite mica. J. Colloid Interface Sci. 2000, 224, 112–115. [Google Scholar] [CrossRef] [PubMed]
- Meier, L.P.; Kahr, G. Determination of the cation exchange capacity (CEC) of clay minerals using the complexes of copper(II) Ion with triethylenetetramine and tetraethylenepentamine. Clays Clay Miner. 1999, 47, 386–388. [Google Scholar] [CrossRef]
- Alexandre, M.; Dubois, P. Polymer layered silicate nanocomposites: preparation, properties and uses of a new class of materials. Mater. Sci. Eng. R Rep. 2000, 28, 1–63. [Google Scholar] [CrossRef]
- Lagaly, G. Interaction of alkylamines with different types of layered compounds. Solid State Ionics 1986, 22, 43–51. [Google Scholar] [CrossRef]
- Vaia, R.A.; Teukolsky, R.K.; Giannelis, E.P. Interlayer structure and molecular environment of alkylammonium layered silicates. Chem. Mater. 1994, 6, 1017–1022. [Google Scholar] [CrossRef]
- Heinz, H.; Castelijns, H.J.; Suter, U.W. Structure and phase transitions of alkyl chains on mica. J. Am. Chem. Soc. 2003, 125, 9500–9510. [Google Scholar] [CrossRef] [PubMed]
- Mittal, V. Polypropylene-layered silicate nanocomposites: Filler matrix interactions and mechanical properties. J. Thermoplast. Compos. Mater. 2007, 20, 575–599. [Google Scholar] [CrossRef]
- Mittal, V. Optimization of Polymer Nanocomposite Properties; Wiley: Weinheim, Germany, 2009; in press. [Google Scholar]
- Osman, M.A.; Mittal, V.; Morbidelli, M.; Suter, U.W. Epoxy-layered silicate nanocomposites and their gas permeation properties. Macromolecules 2004, 37, 7250–7257. [Google Scholar] [CrossRef]
- Ray, S.S.; Okamoto, M. Polymer–layered silicate nanocomposite: a review from preparation to processing. Prog. Polym. Sci. 2003, 28, 1539–1641. [Google Scholar] [CrossRef]
- Mittal, V. Effect of the presence of excess ammonium ions on the clay surface on permeation properties of epoxy nanocomposites. J. Mater. Sci. 2008, 43, 4972–4978. [Google Scholar] [CrossRef]
- Mittal, V. Polymer Nanocomposites: Advances in Filler Surface Modifications; Mittal, V., Ed.; Nova Science Publishers: New York, NY, USA, 2009; Chapter 8, in press. [Google Scholar]
- Osman, M.A.; Mittal, V.; Suter, U.W. Poly(propylene)-layered silicate nanocomposites: Gas permeation properties and clay exfoliation. Macromol. Chem. Phys. 2007, 208, 68–75. [Google Scholar] [CrossRef]
- Mittal, V. Gas permeation and mechanical properties of polypropylene nanocomposites with thermally-stable imidazolium modified clay. Eur. Polym. J. 2007, 43, 3727–3736. [Google Scholar] [CrossRef]
- May, C.A. Epoxy Resins Chemistry and Technology, 2nd ed.; Dekker: New York, NY, USA, 1988. [Google Scholar]
- Lan, T.; Kaviratna, P.D.; Pinnavaia, T.J. Mechanism of clay tactoid exfoliation in epoxy-clay nanocomposites. Chem. Mater. 1995, 7, 2144–2150. [Google Scholar] [CrossRef]
- Kamon, T.; Furakawa, H. Curing mechanisms and mechanical properties of cured epoxy resins. Adv. Polym. Sci. 1986, 80, 173–202. [Google Scholar]
- Barton, J.M. The application of differential scanning calorimetry (DSC) to the study of epoxy resin curing reactions. Adv. Polym. Sci. 1985, 72, 111–154. [Google Scholar]
- Wilson, O.C., Jr.; Olorunyolemi, T.; Jaworski, A.; Borum, L.; Young, D.; Siriwat, A.; Dickens, E.; Oriakhi, E.; Lerner, M. Surface and interfacial properties of polymer-intercalated layered double hydroxide nanocomposites. Appl. Clay Sci. 1999, 15, 265–279. [Google Scholar] [CrossRef]
- Oriakhi, C.O.; Farr, I.V.; Lerner, M.M. Thermal characterization of poly(styrene sulfonate)/layered double hydroxide nanocomposites. Clays Clay Miner. 1997, 45, 194–202. [Google Scholar] [CrossRef]
- Fischer, H.R.; Gielgens, L.H.; Koster, T.P.M. Nanocomposites from polymers and layered minerals. Acta Polym. 1999, 50, 122–126. [Google Scholar] [CrossRef]
- Theng, B.K.G. Formation and Properties of Clay-Polymer Complexes; Elsevier: Amsterdam, The Netherland, 1979. [Google Scholar]
- Ruiz-Hitzky, E.; Aranda, P.; Casal, B.; Galvan, J.C. Nanocomposite materials with controlled ion mobility. Adv. Mater. 1995, 7, 180–184. [Google Scholar] [CrossRef]
- Ogata, N.; Kawakage, S.; Ogihara, T. Poly(vinyl alcohol)-clay and poly(ethylene oxide)-clay blend prepared using water as solvent. J. Appl. Polym. Sci. 1997, 66, 573–581. [Google Scholar] [CrossRef]
- Parfitt, R.L.; Greenland, D.J. Adsorption of poly(ethylene glycols) on montmorillonites. Clay Miner. 1970, 8, 305–323. [Google Scholar] [CrossRef]
- Billingham, J.; Breen, C.; Yarwood, J. Adsorption of polyamine, polyacrylic acid and polyethylene glycol on montmorillonite: An in situ study using ATR-FTIR. Vibr. Spectrosc. 1997, 14, 19–34. [Google Scholar] [CrossRef]
- Levy, R.; Francis, C.W. Interlayer adsorption of polyvinylpyrrolidone on montmorillonite. J. Colloid Interface Sci. 1975, 50, 442–450. [Google Scholar] [CrossRef]
- Jeon, H.G.; Jung, H.-T.; Lee, S.W.; Hudson, S.D. Morphology of polymer silicate nanocomposites: High density polyethylene and a nitrile. Polym. Bull. 1998, 41, 107–113. [Google Scholar] [CrossRef]
- Krikorian, V.; Pochan, D. Poly(l-lactide acid)/layered silicate nanocomposite: fabrication, characterization, and properties. Chem. Mater. 2003, 15, 4317–4324. [Google Scholar] [CrossRef]
- Tyan, H.-L.; Liu, Y.-C.; Wei, K.-H. Enhancement of imidization of poly(amic acid) through forming poly(amic acid)/organoclay nanocomposites. Polymer 1999, 40, 4877–4886. [Google Scholar] [CrossRef]
- Oriakhi, C.O.; Zhang, X.; Lerner, M.M. Synthesis and luminescence properties of a poly(p-phenylenevinylene)/montmorillonite layered nanocomposite. Appl. Clay Sci. 1999, 15, 109–118. [Google Scholar] [CrossRef]
- Lee, D.C.; Jang, L.W. Preparation and characterization of PMMA-clay hybrid composite by emulsion polymerization. J. Appl. Polym. Sci. 1996, 61, 1117–1122. [Google Scholar] [CrossRef]
- Noh, M.W.; Lee, D.C. Synthesis and characterization of PS-clay nanocomposite by emulsion polymerization. Polym. Bull. 1992, 42, 619–626. [Google Scholar] [CrossRef]
- Okada, A.; Usuki, A. The chemistry of polymer-clay hybrids. Mater. Sci. Eng. 1995, C3, 109–115. [Google Scholar] [CrossRef]
- Kojima, Y.; Usuki, A.; Kawasumi, M.; Okada, A.; Kurauchi, T.; Kamigaito, O. Synthesis of nylon-6-clay hybrid by montmorillonite intercalated with ε-caprolactam. J. Polym. Sci. A–Polym. Chem. 1993, 31, 983–986. [Google Scholar] [CrossRef]
- Lee, S.-S.; Ma, Y.T.; Rhee, H.-W.; Kim, J. Exfoliation of layered silicate by ring opening reaction of cyclic oligomers in PET–clay nanocomposites. Polymer 2005, 46, 2201–2210. [Google Scholar] [CrossRef]
- Messersmith, P.B.; Giannelis, E.P. Polymer-layered silicate nanocomposites: in situ intercalative polymerization of ε-caprolactone in layered silicates. Chem. Mater. 1993, 5, 1064–1066. [Google Scholar] [CrossRef]
- Dubois, P.; Alexandre, M.; Hindryckx, F.; Jerome, R. Homogeneous polyolefin-based composites. J. Macromol. Sci. Rev. Macromol. Chem. Phys. 1998, C38, 511–565. [Google Scholar] [CrossRef]
- Bergman, J.S.; Chen, H.; Giannelis, E.P.; Thomas, M.G.; Coates, G.W. Synthesis and characterization of polyolefin–silicate nanocomposites: A catalyst intercalation and in situ polymerization approach. J. Chem. Soc. Chem. Commun. 1999, 21, 2179–2180. [Google Scholar] [CrossRef]
- Jin, Y.-H.; Park, H.-J.; Im, S.-S.; Kwak, S.-Y.; Kwak, S. Polyethylene/clay nanocomposite by in situ exfoliation of montmorillonite during Ziegler–Natta polymerization of ethylene. Macromol. Rapid Commun. 2002, 23, 135–140. [Google Scholar] [CrossRef]
- Heinemann, J.; Reichert, P.; Thomann, R.; Muelhaupt, R. Polyolefin nanocomposites formed by melt compounding and transition metal catalyzed ethene homo- and copolymerization in the presence of layered silicates. Macromol. Rapid Communn. 1999, 20, 423–430. [Google Scholar] [CrossRef]
- Akelah, A.; Moet, A. Polymer-clay nanocomposites: free radical grafting of polystyrene on to organophilic montmorillonite interlayers. J. Mater. Sci. 1996, 31, 3589–3596. [Google Scholar]
- Fu, X.; Qutubuddin, S. Polymer-clay nanocomposites: Exfoliation of organophilic montmorillonite nanolayers in polystyrene. Polymer 2001, 42, 807–813. [Google Scholar] [CrossRef]
- Meier, L.P.; Shelden, R.A.; Caseri, W.R.; Suter, U.W. Polymerization of styrene with initiator ionically bound to high surface area mica: Grafting via an unexpected mechanism. Macromolecules 1994, 27, 1637–1642. [Google Scholar] [CrossRef]
- Su, S.; Wilkie, C.A. Exfoliated poly(methyl methacrylate) and polystyrene nanocomposites occur when the clay cation contains a vinyl monomer. J. Polym. Sci. A–Polym. Chem. 2003, 41, 1124–1135. [Google Scholar] [CrossRef]
- Velten, U.; Shelden, R.A.; Caseri, W.R.; Suter, U.W.; Li, Y. Polymerization of styrene with peroxide initiator ionically bound to high surface area mica. Macromolecules 1999, 32, 3590–3597. [Google Scholar] [CrossRef]
- Velten, U.; Tossati, S.; Shelden, R.A.; Caseri, W.R.; Suter, U.W.; Hermann, R.; Mueller, M. Graft polymerization of styrene on mica: Formation and behavior of molecular droplets and thin films. Langmuir 1999, 15, 6940–6945. [Google Scholar] [CrossRef]
- Zhao, H.; Farrell, B.P.; Shipp, D.A. Nanopatterns of ply(styrene-block-butyl acrylate) block copolymer brushes on the surfaces of exfoliated and intercalated clay layers. Polymer 2004, 45, 4473–4481. [Google Scholar] [CrossRef]
- Zhao, H.; Argoti, S.D.; Farrell, B.P.; Shipp, D.A. Polymer-silicate nanocomposites produced by in-situ atom transfer radical polymerization. J. Polym. Sci. A–Polym. Chem. 2004, 42, 916–924. [Google Scholar] [CrossRef]
- Messersmith, P.B.; Giannelis, E.P. Synthesis and characterization of layered silicate-epoxy nanocomposites. Chem. Mater. 1994, 6, 1719–1725. [Google Scholar] [CrossRef]
- Osman, M.A.; Mittal, V.; Morbidelli, M.; Suter, U.W. Polyurethane adhesive nanocomposites as gas permeation barrier. Macromolecules 2003, 36, 9851–9858. [Google Scholar] [CrossRef]
- Gorrasi, G.; Tortora, M.; Vittoria, V.; Galli, G.; Chiellini, E. Transport and mechanical properties of blends of poly(ε-caprolactone) and a modified montmorillonite-poly(ε-caprolactone) nanocomposite. J. Polym. Sci. B Polym. Phys. 2002, 40, 1118–1124. [Google Scholar] [CrossRef]
- Lan, T.; Pinnavaia, T.J. Clay-reinforced epoxy nanocomposites. Chem. Mater. 1994, 6, 2216–2219. [Google Scholar] [CrossRef]
- Becker, O.; Varley, R.J.; Simon, G.P. Thermal stability and water uptake of high performance epoxy layered silicate nanocomposites. Eur. Polym. J. 2004, 40, 187–195. [Google Scholar] [CrossRef]
- Koerner, H.; Misra, D.; Tan, A.; Drummy, L.; Mirau, P.; Vaia, R. Montmorillonite-thermoset nanocomposites via cryo-compounding. Polymer 2006, 47, 3426–3435. [Google Scholar] [CrossRef]
- Suh, D.; Lim, Y.; Park, O. The property and formation mechanism of unsaturated polyester–layered silicate nanocomposite depending on the fabrication methods. Polymer 2000, 41, 8557–8563. [Google Scholar] [CrossRef]
- Fornes, T.D.; Yoon, P.J.; Keskkula, H.; Paul, D.R. Nylon 6 nanocomposites: the effect of matrix molecular weight. Polymer 2001, 42, 9929–9940. [Google Scholar] [CrossRef]
- McNally, T.; Murphy, W.R.; Lew, C.Y.; Turner, R.J.; Brennan, G.P. Polyamide-12 layered silicate nanocomposites by melt compounding. Polymer 2003, 44, 2761–2772. [Google Scholar] [CrossRef]
- Davis, C.H.; Mathias, L.J.; Gilman, J.W.; Schiraldi, D.A.; Shields, J.R.; Trulove, P.; Sutto, T.E.; Delong, H.C. Effects of melt-processing conditions on the quality of poly(ethylene terephthalate) montmorillonite clay nanocomposites. J. Polym. Sci. B Polym. Phys. 2002, 40, 2661–2666. [Google Scholar] [CrossRef]
- Srivastava, S.K.; Pramanik, M.; Acharya, H. Ethylene/vinyl acetate copolymer/clay nanocomposites. J. Polym. Sci. B Polym. Phys. 2006, 44, 471–480. [Google Scholar] [CrossRef]
- Shin, S.-Y.A.; Simon, L.C.; Soares, J.B.P.; Scholz, G. Polyethylene–clay hybrid nanocomposites: In-situ polymerization using bifunctional organic modifiers. Polymer 2003, 44, 5317–5321. [Google Scholar] [CrossRef]
- Gopakumar, T.G.; Lee, J.A.; Kontopoulou, M.; Parent, J.S. Influence of clay exfoliation on the physical properties of montmorillonite/polyethylene composites. Polymer 2002, 43, 5483–5491. [Google Scholar] [CrossRef]
- Thellen, C.; Orroth, C.; Froio, D.; Ziegler, D.; Lucciarini, J.; Farrell, R.; D’Souza, N.A.; Ratto, J.A. Influence of montmorillonite layered silicate on plasticized poly(l-lactide) blown films. Polymer 2005, 46, 11716–11727. [Google Scholar] [CrossRef]
- Di, Y.; Iannace, S.; Maio, E.D.; Nicolais, L. Nanocomposites by melt intercalation based on polycaprolactone and organoclay. J. Polym. Sci.B Polym. Phys. 2003, 41, 670–678. [Google Scholar] [CrossRef]
- Vaia, R.A.; Giannelis, E.P. Lattice of polymer melt intercalation in organically-modified layered silicates. Macromolecules 1997, 30, 7990–7999. [Google Scholar] [CrossRef]
- Balazs, A.C.; Singh, C.; Zhulina, E.; Lyatskaya, Y. Modeling the phase behavior of polymer/clay nanocomposites. Acc. Chem. Res. 1999, 32, 651–657. [Google Scholar] [CrossRef]
- Ginzburg, V.V.; Singh, C.; Balazs, A.C. Theoretical phase diagrams of polymer/clay composites: The role of grafted organic modifiers. Macromolecules 2000, 33, 1089–1099. [Google Scholar] [CrossRef]
- Heinz, H.; Suter, U.W. Surface structure of organoclays. Angew. Chem. Int. Ed. 2004, 43, 2239–2243. [Google Scholar] [CrossRef]
- Hasegawa, R.; Aoki, Y.; Doi, M. Optimum graft density for dispersing particles in polymer melts. Macromolecules 1996, 29, 6656–6662. [Google Scholar] [CrossRef]
- Edgecombe, S.R.; Gardiner, J.M.; Matsen, M.W. Suppressing autophobic dewetting by using a bimodal brush. Macromolecules 2002, 35, 6475–6477. [Google Scholar] [CrossRef]
- Kato, M.; Usuki, A.; Okada, A. Synthesis of polypropylene oligomer-clay intercalation compounds. J. Appl. Polym. Sci. 1997, 66, 1781–1785. [Google Scholar] [CrossRef]
- Kawasumi, M.; Hasegawa, N.; Kato, M.; Usuki, A.; Okada, A. Preparation and mechanical properties of polypropylene-clay hybrids. Macromolecules 1997, 30, 6333–6338. [Google Scholar] [CrossRef]
- Usuki, A.; Kato, M.; Okada, A.; Kurauchi, T. Synthesis of polypropylene-clay hybrid. J. Appl. Polym. Sci. 1997, 63, 137–138. [Google Scholar] [CrossRef]
- Hasegawa, N.; Kawasumi, M.; Kato, M.; Usuki, A.; Okada, A. Preparation and mechanical properties of polypropylene-clay hybrids using a maleic anhydride-modified polypropylene oligomer. J. Appl. Polym. Sci. 1998, 67, 87–92. [Google Scholar] [CrossRef]
- Reichert, P.; Nitz, H.; Klinke, S.; Brandsch, R.; Thomann, R.; Muelhaupt, R. Poly(propylene)/organoclay nanocomposite formation: Influence of compatibilizer functionality and organoclay modification. Macromol. Mater. Eng. 2000, 275, 8–17. [Google Scholar] [CrossRef]
- Manias, E.; Touny, A.; Wu, L.; Strawhecker, K.; Lu, B.; Chung, T.C. Polypropylene/montmorillonite nanocomposites. Review of synthetic routes and materials properties. Chem. Mater. 2001, 13, 3516–3523. [Google Scholar] [CrossRef]
- Zhang, Q.; Fu, Q.; Jiang, L.; Lei, Y. Preparation and properties of polypropylene/mont-morillonite layered nanocomposites. Polym. Int. 2000, 49, 1561–1564. [Google Scholar] [CrossRef]
- Oya, A.; Kurokawa, Y.; Yasuda, H. Factors controlling mechanical properties of clay mineral/polypropylene nanocomposites. J. Mater. Sci. 2000, 35, 1045–1050. [Google Scholar] [CrossRef]
- Xu, W.; Liang, G.; Wang, W.; Tang, S.; He, P.; Pan, W.P. PP-PP-g-MAH-Org-MMT nanocomposites. I. Intercalation behavior and microstructure. J. Appl. Polym. Sci. 2003, 88, 3225–3231. [Google Scholar] [CrossRef]
- Ellis, T.S.; D’Angelo, J.S. Thermal and mechanical properties of a polypropylene nanocomposite. J. Appl. Polym. Sci. 2003, 90, 1639–1647. [Google Scholar] [CrossRef]
- Su, S.; Jiang, D.D.; Wilkie, C.A. Poly(methyl methacrylate), polypropylene and polyethylene nanocomposite formation by melt blending using novel polymerically-modified clays. Polym. Degrad. Stab. 2004, 83, 321–331. [Google Scholar] [CrossRef]
- Hasegawa, N.; Okamoto, H.; Kawasumi, M.; Kato, M.; Tsukigase, A.; Usuki, A. Polyolefin-clay hybrids based on modified polyolefins and organophilic clay. Macromol. Mater. Eng. 2000, 280–281, 76–79. [Google Scholar] [CrossRef]
- Pinnavaia, T.J. Polymer-Clay Nanocomposites; John Wiley: Chichester, UK, 2000. [Google Scholar]
- Sathe, S.N.; Rao, G.S.S.; Devi, S. Grafting of maleic anhydride onto polypropylene: Synthesis and characterization. J. Appl. Polym. Sci. 1994, 53, 239–245. [Google Scholar] [CrossRef]
- Sun, T.; Garces, J.M. High performance polypropylene-clay nanocomposites by in-situ polymerization with metallocene/clay catalysts. Adv. Mater. 2002, 14, 128–130. [Google Scholar] [CrossRef]
- Zheng, X.; Jiang, D.D.; Wilkie, C.A. Polystyrene nanocomposites based on an oligomerically-modified clay containing maleic anhydride. Polym. Degrad. Stab. 2006, 91, 108–113. [Google Scholar] [CrossRef]
- Hasegawa, N.; Okamoto, H.; Kato, M.; Usuki, A.; Sato, N. Nylon 6-montmorillonite nanocomposites prepared by compounding nylon 6 with Na-montmorillonite slurry. Polymer 2003, 44, 2933–2937. [Google Scholar] [CrossRef]
- Mittal, V. Polymer Nanocomposites: Advances in Filler Surface Modification Techniques; Mittal, V., Ed.; Nova Science Publishers: New York, NY, USA, 2009; Chapter 1, in press. [Google Scholar]
- Osman, M.A.; Rupp, J.E.P.; Suter, U.W. Gas permeation properties of polyethylene-layered silicate nanocomposites. J. Mater. Chem. 2005, 15, 1298–1304. [Google Scholar]
- Mittal, V. Novel Polymers and Nanoscience; Adeli, M., Ed.; Research Signpost Publishers: Trivandrum, Kerala, India, 2008; pp. 73–92. [Google Scholar]
- Mittal, V. Advances in Polymer Nanocomposites Technology; Mittal, V., Ed.; Nova Science Publishers: New York, NY, USA, 2009; Chapter 1, in press. [Google Scholar]
- Mittal, V.; Herle, V. Physical adsorption of organic molecules on the surface of layered silicate clay platelets: A thermogravimetric study. J. Colloid Interface Sci. 2008, 327, 295–301. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Frost, R.L.; He, H.; Xi, Y.; Liu, H. Adsorbed para-nitrophenol on HDTMAB organoclay-A TEM and infrared spectroscopic study. J. Colloid Interface Sci. 2007, 307, 357–363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Q.; He, H.; Frost, R.L.; Xi, Y. Adsorption of p-nitrophenol on mono-, bi-, and trialkyl surfactant-intercalated organoclays: A comparative study. J. Phys. Chem. C 2007, 111, 7487–7493. [Google Scholar] [CrossRef]
- Zhou, Q.; Frost, R.L.; He, H.; Xi, Y. Changes in the surfaces of adsorbed para-nitrophenol on HDTMA organoclay—The XRD and TG study. J. Colloid Interface Sci. 2007, 307, 50–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Q.; Frost, R.L.; He, H.; Xi, Y.; Zbik, M. TEM, XRD, and thermal stability of adsorbed paranitrophenol on DDOAB organoclay. J. Colloid Interface Sci. 2007, 311, 24–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mittal, V. Esterification reactions on the surface of layered silicate clay platelets. J. Colloid Interface Sci. 2007, 315, 135–141. [Google Scholar] [CrossRef] [PubMed]
- Fornes, T.D.; Paul, D.R. Modelling properties of nylon 6/clay nanocomposites using composite theories. Polymer 2003, 44, 4993–5013. [Google Scholar] [CrossRef]
- Osman, M.A.; Rupp, J.E.P.; Suter, U.W. Tensile properties of polyethylene-layered silicate nanocomposites. Polymer 2005, 46, 1653–1660. [Google Scholar] [CrossRef]
- Osman, M.A.; Rupp, J.E.P.; Suter, U.W. Effect of non-ionic surfactants on the exfoliation and properties of polyethylene-layered silicate nanocomposites. Polymer 2005, 46, 8202–8209. [Google Scholar] [CrossRef]
- Mittal, V. Mechanical and gas permeation properties of compatibilized polypropylene–layered silicate nanocomposites. J. Appl. Polym. Sci. 2008, 107, 1350–1361. [Google Scholar] [CrossRef]
- Liu, L.M.; Qi, Z.N.; Zhu, X.G. Studies on nylon-6 nanocomposites by melt-intercalation process. J. Appl. Polym. Sci. 1999, 71, 1133–1138. [Google Scholar] [CrossRef]
- Wang, Z.; Pinnavaia, T.J. Hybrid organic-inorganic nanocomposites: Exfoliation of magadiite nanolayers in an elastomeric epoxy polymer. Chem. Mater. 1998, 10, 1820–1826. [Google Scholar] [CrossRef]
- Chang, J.-H.; Kim, S.J.; Joo, Y.L.; Im, S. Poly(ethylene terephthalate) nanocomposites by in situ interlayer polymerization: The thermomechanical properties and morphology of the hybrid fiber. Polymer 2004, 45, 919–926. [Google Scholar] [CrossRef]
- Hauser, E.A.; Kollman, R.C. Clay complexes with conjugated unsaturated aliphatic compounds. US Patent No. 2,951,087, 1960. [Google Scholar]
- Wang, S.J.; Long, C.F.; Wang, X.Y.; Li, Q.; Qi, Z.N. Synthesis and properties of silicon rubber organo-montmorillonite hybrid nanocomposites. J. Appl. Polym. Sci. 1998, 69, 1557–1561. [Google Scholar] [CrossRef]
- Finnigan, B.; Martin, D.; Halley, P.; Truss, R.; Campell, K. Morphology and properties of thermoplastic polyurethane nanocomposites incorporating hydrophilic layered silicates. Polymer 2004, 45, 2249–2260. [Google Scholar] [CrossRef]
- Yao, K.J.; Song, M.; Hourston, D.J.; Luo, D.Z. Polymer/layered clay nanocomposites: 2 polyurethane nanocomposites. Polymer 2002, 43, 1017–1020. [Google Scholar] [CrossRef]
- Goettler, L.A. Overview of property development in layered silicate polymer nanocomposites. Ann. Tech. Confr. Soc. Plast. Eng. 2005, 1980–1982. [Google Scholar]
- Phang, I.Y.; Liu, T.; Mohamed, A.; Pramoda, K.P.; Chen, L.; Shen, L.; Chow, S.Y.; He, C.; Lu, X.; Hu, X. Morphology, thermal and mechanical properties of nylon 12/organoclay nanocomposites prepared by melt compounding. Polym. Int. 2005, 54, 456–464. [Google Scholar] [CrossRef]
- Liu, X.; Wu, Q. PP/clay nanocomposites prepared by grafting melt intercalation. Polymer 2001, 42, 10013–10019. [Google Scholar] [CrossRef]
- Nam, P.H.; Maiti, P.; Okamoto, M.; Kotaka, T.; Hasegawa, N.; Usuki, A. A hierarchical structure and properties of intercalated polypropylene/clay nanocomposites. Polymer 2001, 42, 9633–9640. [Google Scholar] [CrossRef]
- Laus, M.; Francesangeli, O.; Sandrolini, F. New hybrid nanocomposites based on an organophilic clay and poly(styrene-b-butadiene) copolymers. J. Mater. Res. 1997, 12, 3134–3139. [Google Scholar] [CrossRef]
- Krikorian, V.; Pochan, D. Poly(l-lactide acid)/layered silicate nanocomposite: fabrication, characterization, and properties. Chem. Mater. 2003, 15, 4317–4324. [Google Scholar] [CrossRef]
- Ray, S.S.; Maiti, P.; Okamoto, M.; Yamada, K.; Ueda, K. New polylactide/ layered silicate nanocomposites. 1. Preparation, characterization and properties. Macromolecules 2002, 35, 3104–3110. [Google Scholar] [CrossRef]
- Xu, R.; Manias, E.; Snyder, A.J.; Runt, J. New biomedical poly(urethane urea)-layered silicate nanocomposites. Macromolecules 2001, 34, 337–339. [Google Scholar] [CrossRef]
- Chang, J.H.; An, Y.U. Nanocomposites of polyurethane with various organoclays: thermomechanical properties, morphology and gas permeability. J. Polym. Sci. B Polym. Phys. 2002, 40, 670–677. [Google Scholar] [CrossRef]
- Tortora, M.; Gorrasi, G.; Vittoria, V.; Galli, G.; Ritrovati, S.; Chiellini, E. Structural characterization and transport properties of organically modified montmorillonite/polyurethane nanocomposites. Polymer 2002, 43, 6147–6157. [Google Scholar] [CrossRef]
- Gorrasi, G.; Tortora, M.; Vittoria, V.; Kaempfer, D.; Muelhaupt, R. Transport properties of organic vapors in nanocomposites of organophilic layered silicate and syndiotactic polypropylene. Polymer 2003, 44, 3679–3685. [Google Scholar] [CrossRef]
- Manias, E. A direct blending approach for polypropylene/clay nanocomposites enhances properties. MRS Bull. 2001, 26, 862–863. [Google Scholar]
- Ke, Z.; Yongping, B. Improve gas barrier property of PET film with montmorillonite by in situ interlayer polymerization. Mater. Lett. 2005, 59, 3348–3351. [Google Scholar] [CrossRef]
- Ogasawara, T.; Ishida, Y.; Ishikawa, T.; Aoki, T.; Ogura, T. Helium gas permeability of montmorillonite/epoxy nanocomposites. Compos. Part A 2006, 37, 2236–2240. [Google Scholar] [CrossRef]
- Ray, S.S.; Yamada, K.; Okamoto, M.; Ueda, K. New polylactide/layered silicate nanocomposites. 2. Concurrent improvements of material properties, biodegradability and melt rheology. Polymer 2003, 44, 857–866. [Google Scholar] [CrossRef]
- Ray, S.S.; Boushmina, M. Biodegradable polymers and their layered silicate nanocomposites: in greening the 21st century materials world. Prog. Mater. Sci. 2005, 50, 962–1079. [Google Scholar] [CrossRef]
- Ray, S.S.; Okamoto, M. Polymer-layered silicate nanocomposite: A review from preparation to processing. Prog. Polym. Sci. 2003, 28, 1539–1641. [Google Scholar] [CrossRef]
- Vyazovkin, S.; Dranka, I.; Fan, X.; Advincula, R. Kinetics of the thermal and thermo-oxidative degradation of a polystyrene–clay nanocomposite. Macromol. Rapid Commun. 2004, 25, 498–503. [Google Scholar] [CrossRef]
- Zanetti, M.; Bracco, P.; Costa, L. Thermal degradation behavior of PE/clay nanocomposites. Polym. Degrad. Stab. 2004, 85, 657–665. [Google Scholar] [CrossRef]
- Bandyopadhyay, S.; Chen, R.; Giannelis, E.P. Biodegradable organic-inorganic hybrids based on poly(l-lactide). J. Macromol. Sci. Rev. 1999, 81, 159–160. [Google Scholar]
- Paul, M.-A.; Alexandre, M.; Degee, P.; Henrist, C.; Rulmont, A.; Dubois, P. New nanocomposite materials based on plasticized poly(l-lactide) and organo-modified montmorillonites: Thermal and morphological study. Polymer 2003, 44, 443–450. [Google Scholar] [CrossRef]
- Berta, M.; Lindsay, C.; Pans, G.; Camino, G. Effect of chemical structure on combustion and thermal behaviour of polyurethane elastomer layered silicate nanocomposites. Polym. Degrad. Stab. 2006, 91, 1179–1191. [Google Scholar] [CrossRef]
- Zhao, C.; Qin, H.; Gong, F.; Feng, M.; Zhang, S.; Yang, M. Mechanical, thermal and flammability properties of polyethylene/clay nanocomposites. Polym. Degrad. Stab. 2005, 87, 183–189. [Google Scholar] [CrossRef]
- Beyer, G. Flame retardant properties of EVA-nanocomposites and improvements by combination of nanofillers with aluminium trihydrate. Fire Mater. 2002, 25, 193–197. [Google Scholar] [CrossRef]
- Mittal, V. New Research on Nanocomposites; Krause, L.M., Walter, J.T., Eds.; Nova Science Publishers: New York, NY, USA, 2008; pp. 281–306. [Google Scholar]
- Gilman, J.W. Flammability and thermal stability studies of polymer layered-silicate (clay) nanocomposites. Appl. Clay Sci. 1999, 15, 31–49. [Google Scholar] [CrossRef]
- Porter, D.; Metcalfe, E.; Thomas, M.J.K. Nanocomposite fire retardants-a review. Fire Mater. 2000, 24, 45–52. [Google Scholar] [CrossRef]
- Preston, C.M.L.; Amarasinghe, G.; Hopewell, J.L.; Shanks, R.A.; Mathys, Z. Evaluation of polar ethylene copolymers as fire retardant nanocomposite matrices. Polym. Degrad. Stab. 2004, 84, 533–544. [Google Scholar] [CrossRef]
- Kashiwagi, T.; Harris, R.H., Jr.; Zhang, X.; Briber, R.M.; Cipriano, B.H.; Raghavan, S.R.; Awad, W.H.; Shields, J.R. Flame retardant mechanism of polyamide 6–clay nanocomposites. Polymer 2004, 45, 881–891. [Google Scholar] [CrossRef]
- Rybnikar, F. Interactions in the system isotactic polypropylene-calcite. J. Appl. Polym. Sci. 1991, 42, 2727–2737. [Google Scholar] [CrossRef]
- Supaphol, P.; Harnsiri, W.; Junkasem, J. Effects of calcium carbonate and its purity on crystallization and melting behavior, mechanical properties, and processability of syndiotactic polypropylene. J. Appl. Polym. Sci. 2004, 92, 201–212. [Google Scholar] [CrossRef]
- Svoboda, P.; Zeng, C.; Wang, H.; Lee, L.J.; Tomasko, D.L. Morphology and mechanical properties of polypropylene/organoclay nanocomposites. J. Appl. Polym. Sci. 2002, 85, 1562–1570. [Google Scholar] [CrossRef]
- Maiti, P.; Nam, P.H.; Okamoto, M.; Hasegawa, N.; Usuki, A. Influence of crystallization on intercalation, morphology, and mechanical properties of polypropylene/clay nanocomposites. Macromolecules 2002, 35, 2042–2052. [Google Scholar] [CrossRef]
- Maiti, P.; Nam, P.H.; Okamoto, M.; Kotaka, T.; Hasegawa, N.; Usuki, A. The effect of crystallization on the structure and morphology of polypropylene/clay nanocomposites. Polym. Eng. Sci. 2002, 42, 1864–1871. [Google Scholar] [CrossRef]
- Kodgire, P.; Kalgaonkar, R.; Hambir, S.; Bulakh, N.; Jog, J.P. PP/clay nanocomposites: effect of clay treatment on morphology and dynamic mechanical properties. J. Appl. Polym. Sci. 2001, 81, 1786–1792. [Google Scholar] [CrossRef]
- Ma, J.; Zhang, S.; Qi, Z.; Li, G.; Hu, Y. Crystallization behaviors of polypropylene/mont-morillonite nanocomposites. J. Appl. Polym. Sci. 2002, 83, 1978–1985. [Google Scholar] [CrossRef]
© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Mittal, V. Polymer Layered Silicate Nanocomposites: A Review. Materials 2009, 2, 992-1057. https://doi.org/10.3390/ma2030992
Mittal V. Polymer Layered Silicate Nanocomposites: A Review. Materials. 2009; 2(3):992-1057. https://doi.org/10.3390/ma2030992
Chicago/Turabian StyleMittal, Vikas. 2009. "Polymer Layered Silicate Nanocomposites: A Review" Materials 2, no. 3: 992-1057. https://doi.org/10.3390/ma2030992
APA StyleMittal, V. (2009). Polymer Layered Silicate Nanocomposites: A Review. Materials, 2(3), 992-1057. https://doi.org/10.3390/ma2030992