Synthesis, Structure and Thermal Behavior of Oxalato-Bridged Rb+ and H3O+ Extended Frameworks with Different Dimensionalities

Abstract
:
1. Introduction
2. Experimental
2.1. Syntheses and Characterizations
2.1.1. {Rb(HC2O4)(H2C2O4)(H2O) 2}∞1
2.1.2. {H3O(HC2O4)(H2C2O4).2H2O}∞1
2.1.3. {Rb(HC2O4)}∞3
2.2. X-Ray Crystallography

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | 1 | 2 | 3 |
|---|---|---|---|
| Crystal data | |||
| Empirical formula | C4 H7 O10 Rb | C4 H10 O11 | C2 H1 O4 Rb |
| Formula mass | 300.57 | 234.12 | 174.50 |
| Space group | P-1 | P-1 | P21/c |
| a (Å) | 6.2969(10) | 6.3310(1) | 4.2940(3) |
| b (Å) | 7.206(3) | 7.22649(10) | 13.6229(2) |
| c (Å) | 10.641(2) | 10.5541(2) | 7.6689(1) |
| α (°) | 94.549(2) | 94.253(2) | 90.00 |
| β (°) | 100.3990(10) | 100.2630(5) | 101.50(5) |
| γ (°) | 97.676(2) | 97.719(2) | 90.00 |
| V (Å3) | 468.0(2) | 468.440(14) | 439.60(9) |
| Z | 2 | 2 | 4 |
| T (K) | 298(2) | 298(2) | 298(2) |
| ρcal (g.m-3) | 2.133 | 1.660 | 2.637 |
| λ(Mo Kα1) (Å) | 0.71070 | 0.71070 | 0.71070 |
| μ (mm-1) | 5.332 | 0.175 | 11.150 |
| Data collection | |||
| θ max (°) | 30 | 27.49 | 30 |
| h | -8 ≤ h ≤ 8 | -8 ≤ h ≤ 8 | -6 ≤ h ≤ 6 |
| k | -10 ≤ k ≤ 10 | -9 ≤ k ≤ 9 | -18 ≤ k ≤19 |
| l | -14 ≤ l ≤ 14 | -13 ≤ l ≤ 13 | -10 ≤ l ≤ 10 |
| Reflections collected/unique | 17,670/2,712 | 15,199/2,143 | 9,372/1,278 |
| Rint | 0.1234 | 0.0466 | 0.1126 |
| Tmax; Tmin | 0.527; 0.289 | 0.968; 0.958 | 0.315; 0.111 |
| Refinement | |||
| Refinement method | Full-matrix | Full-matrix | Full-matrix |
| least-squares on F2 | least-squares on F2 | least-squares on F2 | |
| Parameters | 165 | 177 | 69 |
| Goodness-of-fit on F2 | 1.160 | 1.056 | 1.123 |
| Final R indices [I>2σ(I)] | R1 = 0.0351 | R1 = 0.0458 | R1 = 0.0317 |
| wR1 = 0.0834 | wR1 = 0.1158 | wR1 = 0.0690 | |
| R indices (all data) | R2 = 0.0396 | R2 = 0.0670 | R2 = 0.0433 |
| wR2 = 0.0854 | wR2 = 0.1256 | wR2 = 0.0744 | |
| ∆ρ max;∆ρ min (eÅ-3) | 1.626, -1.125 | 0.556, -0.468 | 0.803, -0.670 |
| Compound 1 a | |||
| Rb-O41i | 2.9554(18) | O2w-Rb-O42 | 54.09(6) |
| Rb-O21ii | 2.9744(19) | O1w-Rb-O22iv | 148.33(5) |
| Rb-O11 | 2.9845(19) | O11-C1-C2-O21 | 178.5(2) |
| Rb-O1w | 3.025(2) | O11-C1-C2-O22 | -1.6(3) |
| Rb-O2w | 3.109(3) | O12-C1-C2-O21 | -0.3(3) |
| Rb-O22iv | 3.173(2) | O12-C1-C2-O22 | 179.5(2) |
| Rb-O42 | 3.238(2) | O31-C3-C3v-O32v | 1.2(3) |
| Rb-O31 | 2.9719(19) | O41-C4-C4iii-O42iii | 0.5(3) |
| Rb-O12ii | 2.9700(2) | ||
| Compound 2 | |||
| O3w-O41 | 2.939(2) | O11-C1-C2-O21 | -178.9(2) |
| O3w-O31 | 2.950(2) | O11-C1-C2-O22 | 0.7(3) |
| O3w-O11 | 2.972(2) | O12-C1-C2-O21 | 0.3(3) |
| O3w-H5w | 0.923(10) | O12-C1-C2-O22 | 179.95(19) |
| O3w-H6w | 0.922(10) | H5w-O3w-H6w | 107(3) |
| O3w-H7w | 0.93(3) | H5w-O3w-H7w | 121(3) |
| H6w-O3w-H7w | 104(3) | ||
| Compound 3 b | |||
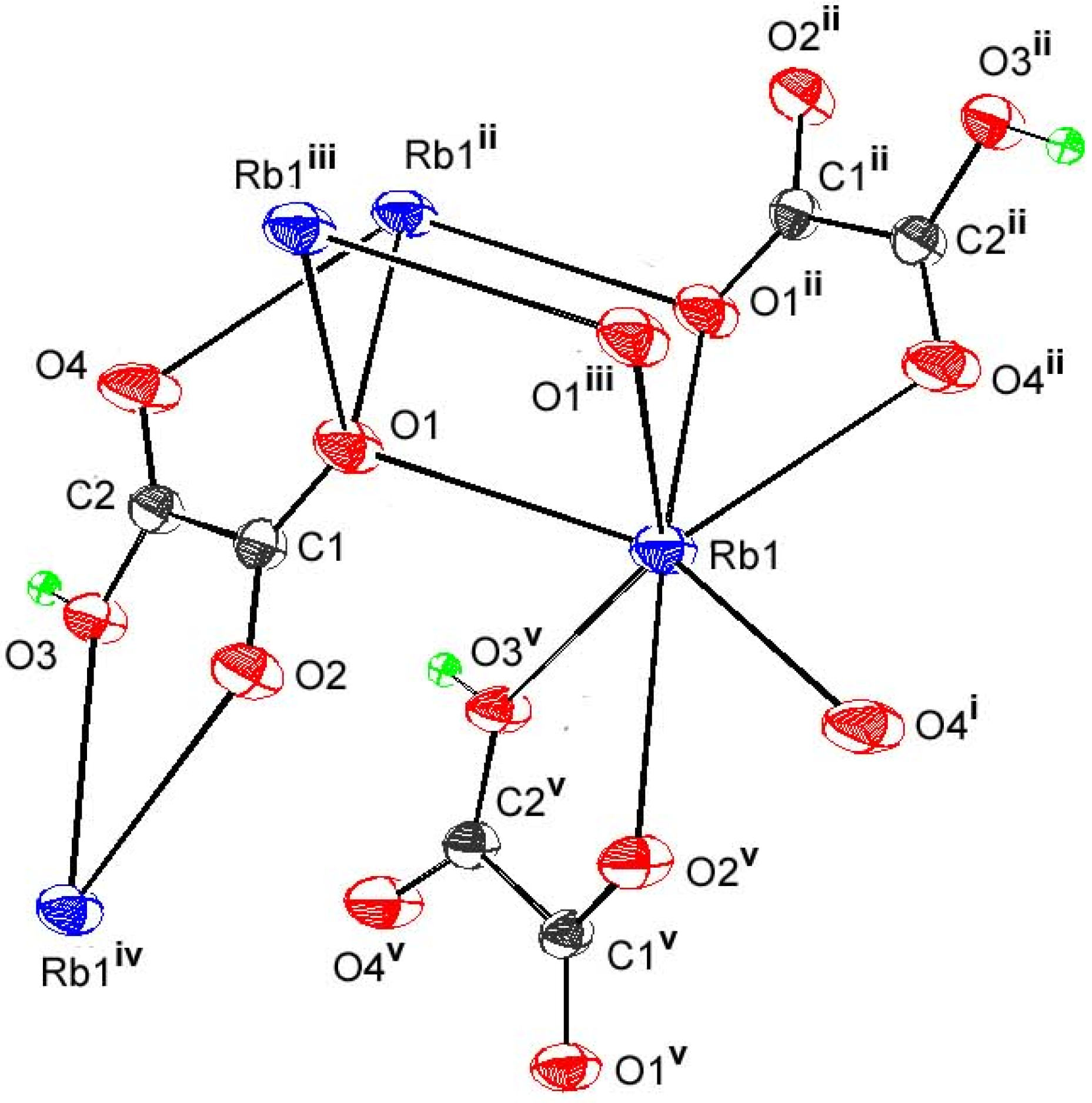
| Rb-O2v | 2.839(2) | Rbiv-O3-H | 127(3) |
| Rb-O4i | 2.908(3) | Rbvi-O3-H | 73(3) |
| Rb-O1ii | 2.940(2) | O1-C1-C2-O3 | 164.8(2) |
| Rb-O1 | 2.945(3) | O1-C1-C2-O4 | -14.8(4) |
| Rb-O4ii | 2.957(3) | O2-C1-C2-O3 | -15.8(4) |
| Rb-O1iii | 2.980(3) | O2-C1-C2-O4 | 164.5(2) |
| Rb-O3v | 3.027(2) | ||
3. Results and Discussions
3.1. Syntheses and Characterizations
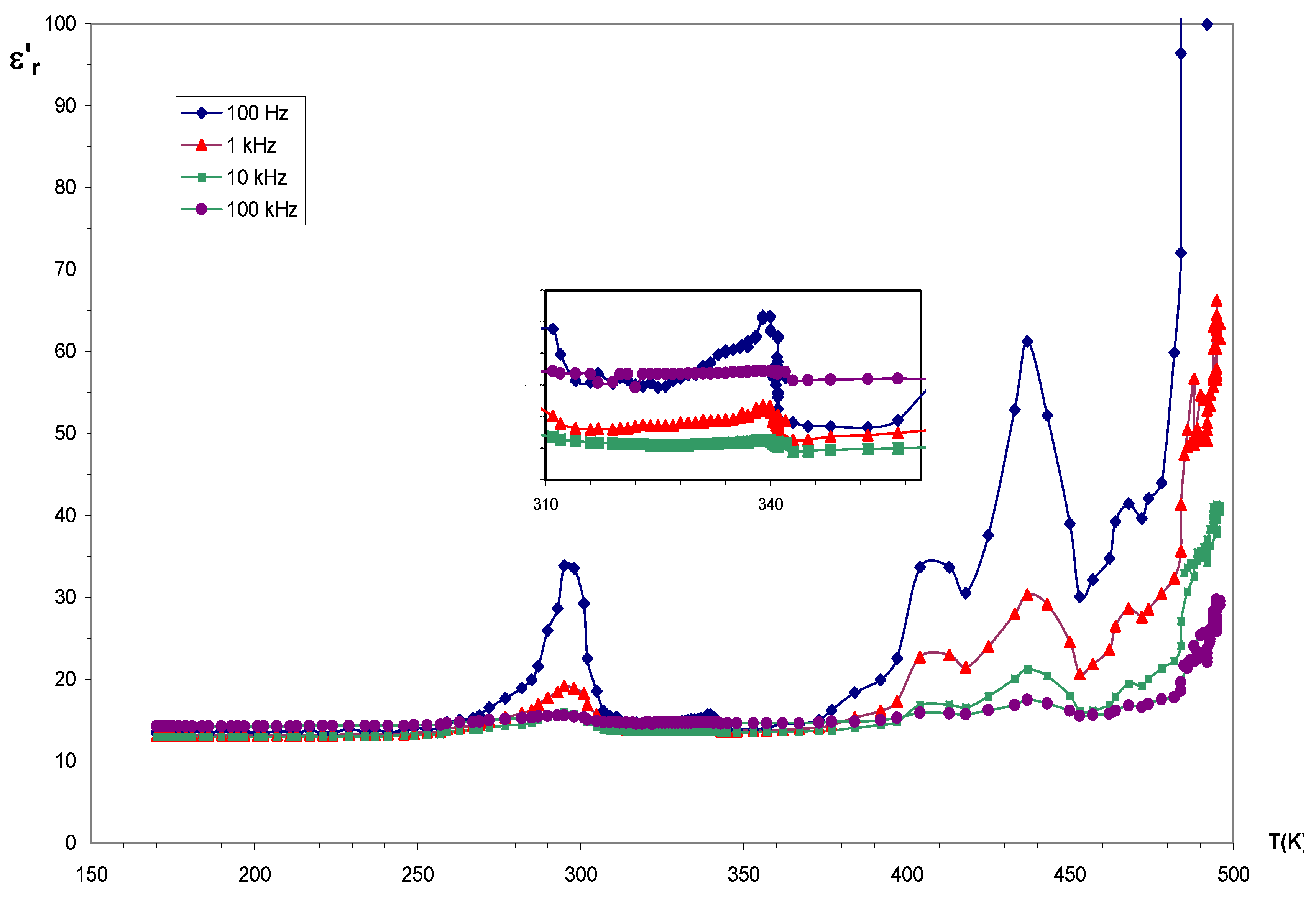
3.2. Thermal and Electric Behaviors
3.2.1. {Rb (HC2O4) (H2C2O4) (H2O)2}∞1
3.2.2. {H3O(HC2O4)(H2C2O4).2H2O}∞1
3.2.3. {Rb(HC2O4)}∞3

3.3. Crystal Structures
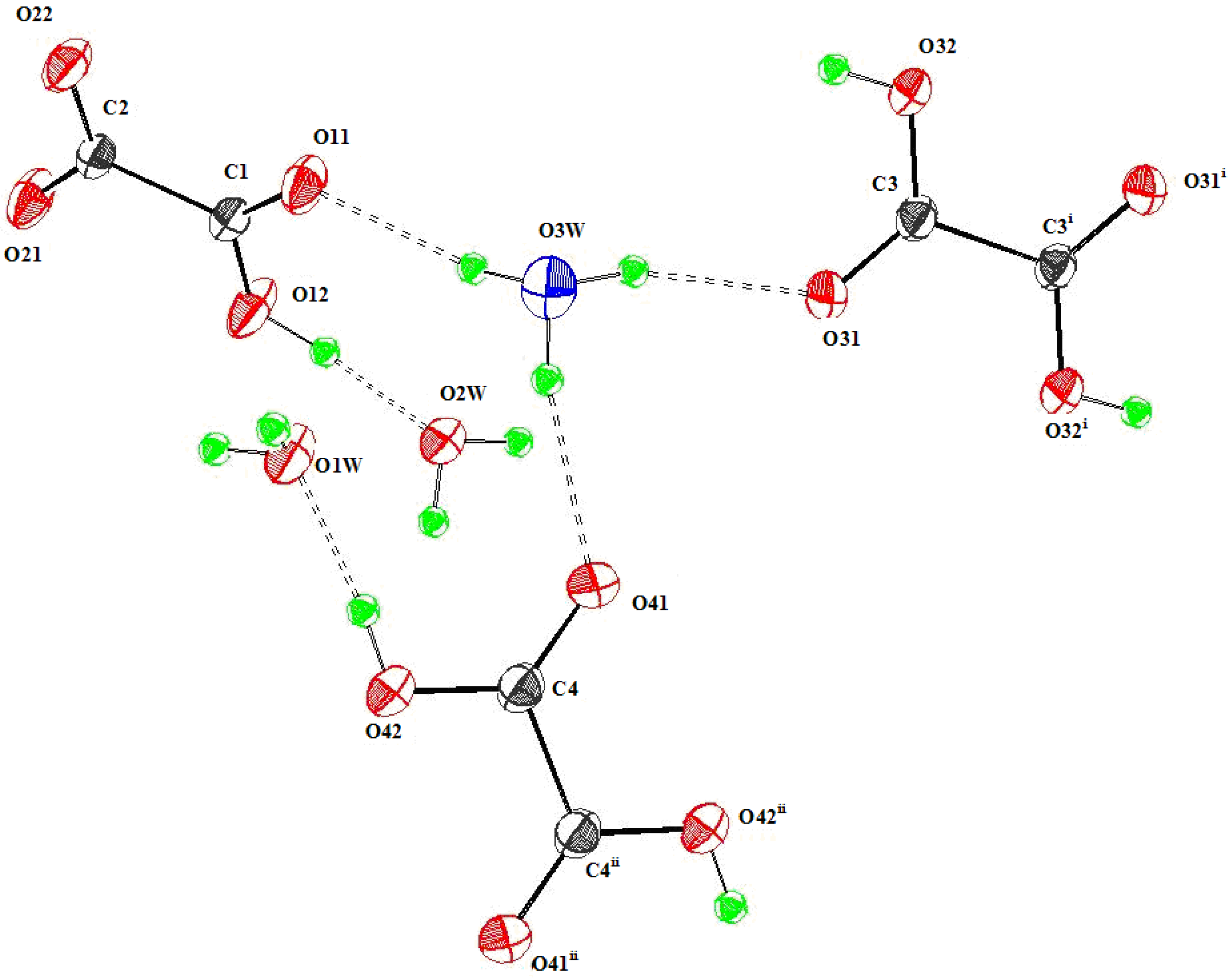
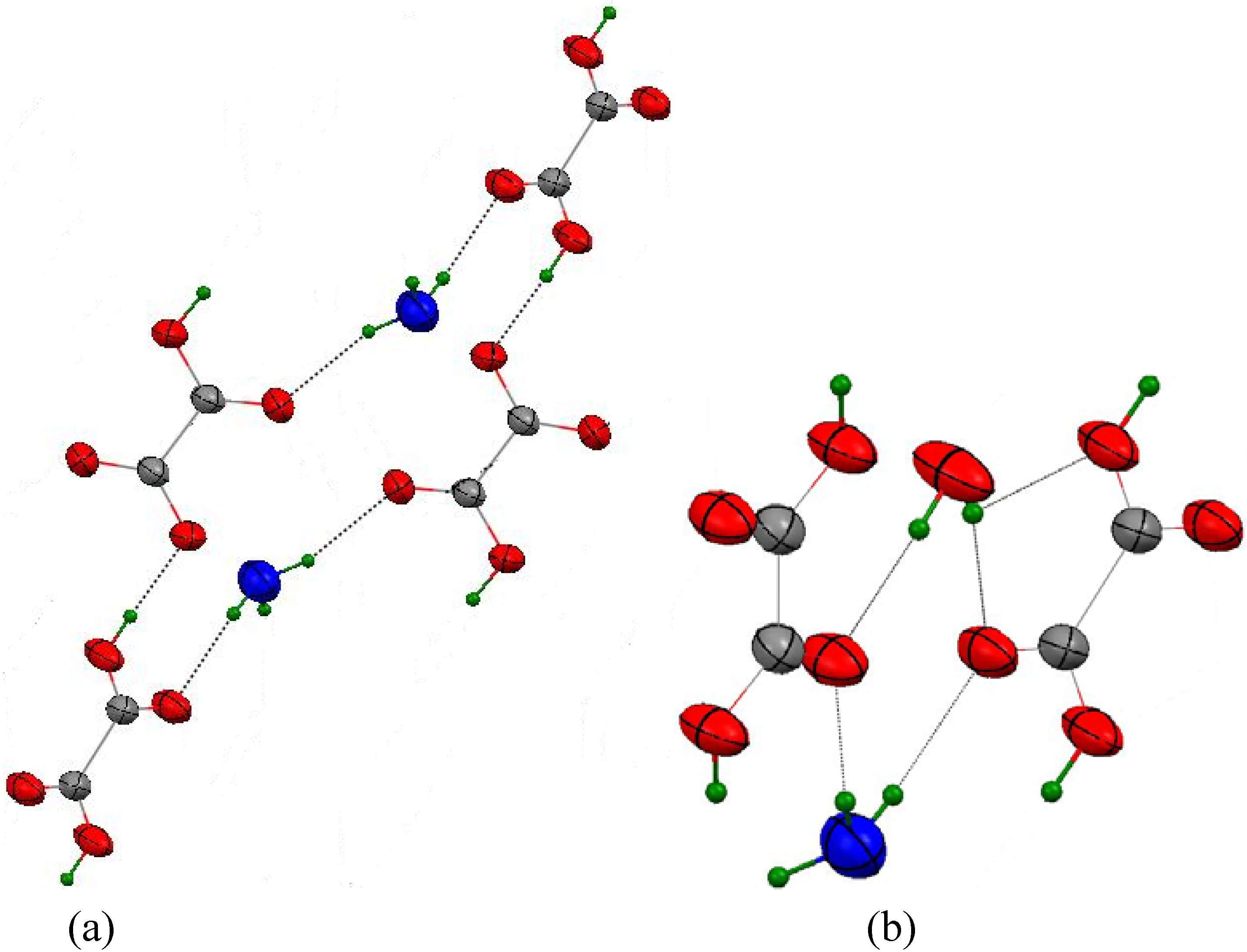
3.3.1. {Rb(HC2O4)(H2C2O4)(H2O)2}∞1

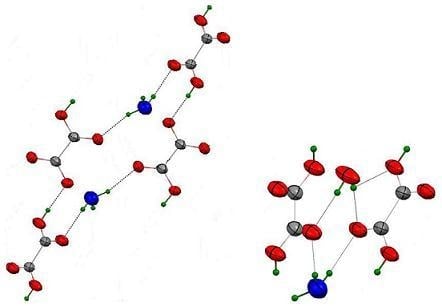
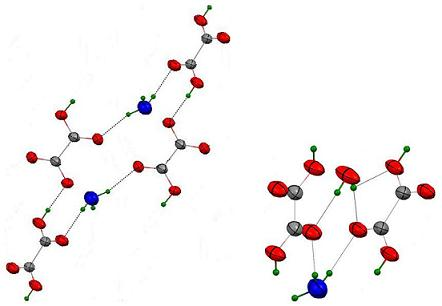
3.3.2. {H3O(HC2O4)(H2C2O4).2H2O}∞1


3.3.3. {Rb(HC2O4)}∞3

4. Conclusion
Supplementary Materials
Acknowledgement
References
- Li, H.; Eddaoudi, M.; Groy, T.L.; Yaghi, O.M. Establishing microporosity in open metal-organic frameworks: Gas sorption isotherms for Zn(BDC)(BDC = 1,4-Benzenedicarboxylate. J. Am. Chem. Soc. 1998, 120, 8571–8572. [Google Scholar] [CrossRef]
- Li, H.; Eddaoudi, M.; O’Keeffe, M.; Yaghi, O.M. Design and synthesis of an exceptionally stable and highly porous metal-organic framework. Nature 1999, 402, 276–279. [Google Scholar] [CrossRef]
- Eddaoudi, M.; Moler, D.B.; Li, H.; Chen, B.; Reineke, T.M.; O’Keeffe, M.; Yaghi, O.M. Modular chemistry: Secondary building units as a basis for the design of highly porous and robust metal-organic carboxylate frameworks. Acc. Chem. Res. 2001, 34, 319–330. [Google Scholar] [CrossRef] [PubMed]
- Eddaoudi, M.; Kim, J.; Rosi, N.; Vodak, D.; Wachter, J.; O’Keeffe, M.; Yaghi, O.M. Systematic design of pore size and functionality in isoreticular MOFs and their application in methane storage. Science 2002, 295, 469–472. [Google Scholar] [CrossRef] [PubMed]
- Chae, H.K.; Siberio-Pérez, D.Y.; Kim, J.; Go, Y.; Eddaoudi, M.; Matzger, A.J.; O’Keefe, M.; Yaghi, O.M. A route to high surface area, porosity and inclusion of large molecules in crystals. Nature 2004, 427, 523–527. [Google Scholar] [CrossRef] [PubMed]
- Ockwig, N.W.; Delgado-Friedrchs, O.; O’Keeffe, M.; Yaghi, O.M. Reticular chemistry: Occurrence and taxonomy of nets and grammar for the design of frameworks. Acc. Chem. Res. 2005, 38, 176–182. [Google Scholar] [CrossRef] [PubMed]
- Cheetham, A.K.; Férey, G.; Loiseau, T. Open-framework inorganic materials. Angew. Chem. Int. Ed. 1999, 38, 3268–3292. [Google Scholar] [CrossRef]
- Rao, C.N.R.; Natarajan, S.; Vaidhyanathan, R. Metal carboxylates with open architectures. Angew. Chem. Int. Ed. 2004, 43, 1466–1496. [Google Scholar] [CrossRef]
- Kobel, W.; Hanack, M. Bis axially coordinated (phthalocyaninato)ruthenium(II) compounds. Inorg. Chem. 1986, 25, 103–107. [Google Scholar] [CrossRef]
- Mosaad, M.M.; El-Shawarby, A.; El-Tanahy, Z.H.; Abdel-Kader, M.M. Electronical and thermal properties of ammonium and potassium oxalates. J. Mat. Sci. Mater. Electronics. 1995, 6, 235–239. [Google Scholar] [CrossRef]
- Rodrigues, V.H.; Paixâo, J.A.; Costa, M.R.; Matos-Beja, A. Betaine betainium hydrogen oxalate. Acta Cryst. 2001, C57, 213–215. [Google Scholar]
- Schaack, G. Experimental results on phase transitions in betaine compounds. Ferroelectrics 1990, 104, 147–158. [Google Scholar] [CrossRef]
- Fujita, M.; Kwon, Y.J.; Washizu, S.; Ogura, K. Preparation, clathration ability, and catalysis of a two-dimensional square network material composed of cadmium(II) and 4,4’-bipyridine. J. Am. Chem. Soc. 1994, 116, 1151–1152. [Google Scholar] [CrossRef]
- Carlucci, L.; Ciani, G.; Proserpio, D.M.; Sironi, A. 1-,2-, and 3-dimensional polymeric frames in the coordination chemistry of AgBF4 with pyrazine. The first example of three interpenetrating 3-dimensional triconnected nets. J. Am. Chem. Soc. 1995, 117, 4562–4569. [Google Scholar] [CrossRef]
- Audebrand, N.; Raite, S.; Louër, D. The layer crystal structure of [In2(C2O4)3(H2O)3].7H2O and microstructure of nanocrystalline In2O3 obtained from thermal decomposition. Solid State Sciences 2003, 5, 783–794. [Google Scholar] [CrossRef]
- Benmerad, B.; Guehria-Laïdoudi, A.; Bernardinelli, G.; Balegroune, F. Polymeric tetraaquatris(malonato)-dilanthanum(III) monohydrate. Acta Cryst. 2000, C56, 321–323. [Google Scholar]
- Benmerad, B.; Guehria-Laïdoudi, A.; Dahaoui, S.; Lecomte, C. A polynuclear coordination glutarate of lanthanum(III) with an uncommon cage feature. Acta Cryst. 2004, C60, m119–m122. [Google Scholar]
- Benmerad, B.; Guehria-Laïdoudi, A.; Dahaoui, S.; Lecomte, C. The first suberate lanthanum(III) complex without uncoordinated water. Acta Cryst. 2004, C60, m407–m409. [Google Scholar]
- Djeghri, A.; Balegroune, F.; Guehria-Laïdoudi, A.; Toupet, L. Synthesis and crystal of bis (malonato) tetra (aqua) barium(II) cobalt(II): [BaCo(C3H2O4)2(H2O)4]. J. Chem. Cryst. 2005, 35, 603–607. [Google Scholar] [CrossRef]
- Aliouane, K.; Taïbi, K.; Guehria-Laïdoudi, A.; Dahaoui, S.; Lecomte, C. Poly[diaqua-µ3-hydrogenglutarato-µ2-hydrogenglutarato-strontium(II)]. Acta Cryst. 2006, 62, m2316–m2318. [Google Scholar]
- Djeghri, A.; Balegroune, F.; Guehria-Laïdoudi, A.; Toupet, L. Poly[tetraaquadi-µ3-malonato-cobalt(II)calcium(II)]. Acta Cryst. 2006, C62, m126–m128. [Google Scholar]
- Djeghri, A.; Balegroune, F.; Guehria-Laïdoudi, A.; Toupet, L. Synthesis and structural characterization of the dinuclear bis(glutarato)bis(urea) copper(II)complex, dihydrate. J. Chem. Cryst. 2006, 36, 239–242. [Google Scholar] [CrossRef]
- Rahahlia, N.; Benmerad, B.; Guehria-Laïdoudi, A.; Dahaoui, S.; Lecomte, C. Poly[tetraaquadi-µ4-glutarato-cerium(III) chloride dihydrate. Acta Cryst. 2006, E62, m2145–m2147. [Google Scholar]
- Rahahlia, N.; Aliouane, K.; Guehria-Laïdoudi, A.; Dahaoui, S.; Lecomte, C. Poly[[tetraaquadi-µ4-succinato-µ5-succinato-diytterbium(III)] hexahydrate]. Acta Cryst. 2006, E62, m2543–m2545. [Google Scholar]
- Aliouane, K.; Djeghri, A.; Guehria-Laïdoudi, A.; Dahaoui, S.; Lecomte, C. Synthesis, structure and thermal behaviour of a barium-glutarate framework. J. Mol. Struct. 2007, 832, 150–155. [Google Scholar] [CrossRef]
- Rahahlia, N.; Benmerad, B.; Guehria-Laïdoudi, A.; Dahaoui, S.; Lecomte, C. Three-dimensional ionic frameworks built up from La(III) and Ce(III) succinates. J. Mol. Struct. 2007, 833, 42–48. [Google Scholar] [CrossRef]
- Djehni, S.; Balegroune, F.; Guehria-Laïdoudi, A.; Dahaoui, S.; Lecomte, C. Catena-poly[[aquabarium(II)]-µ-aqua-bis(µ-2’-carboxybiphenyl-2-carboxylato]. Acta Cryst. 2007, C63, m91–m93. [Google Scholar]
- Tkachev, V.V.; Davidovitch, R.L.; Atovvmyan, L.O. Crystal Structure of RbH3(C2O4)2.2H2O. Russ. J. Coord. Chem. 1995, 21, 436–438. [Google Scholar]
- Wenger, M.; Bernstein, J. Cocrystal design gone awry? A new dimorphic hydrate of oxalic acid. Mol. Pharmaceutics. 2007, 4, 355–359. [Google Scholar] [CrossRef]
- Aakeröy, C.B.; Fasulo, M.E.; Desper, J. Cocrystal or salt: Does it really matter? Mol. Pharmaceutics. 2007, 4, 317–322. [Google Scholar] [CrossRef]
- Childs, S.L.; Stahly, G.P.; Park, A. The salt-cocrystal continuum: the influence of crystal structure on ionization state. Mol. Pharmaceutics. 2007, 4, 323–338. [Google Scholar] [CrossRef]
- Alcock, N.W. The analytical method for absorption correction. In Crystallographic Computing; Ahmed, F.R., Hall, S.R., Huber, C.P., Eds.; Munksgaard: Copenhagen, Denmark, 1970; pp. 271–278. [Google Scholar]
- Duisenberg, A.J.M.; Kroon-Batenburg, L.M.J.; Schreurs, A.M.M. An intensity evaluation method: EVAL-14. J. Appl. Cryst. 2003, 36, 220–229. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Shelxs-97. Program for the solution of crystal structures. University of Göttingen, Germany, 1997. [Google Scholar]
- Sheldrick, G.M. Shelxl-97. Program for crystal structure determination. University of Göttingen, Germany, 1997. [Google Scholar]
- Farrugia, L.J. WinGX suite for small-molecule single-crystal crystallography. J. Appl. Cryst. 1999, 32, 837–838. [Google Scholar] [CrossRef]
- Benmerad, B.; Gueria-Laïdoudi, A.; Balegroune, F.; Birkedal, H.; Chapuis, G. Polymeric aqua(glutarato)(hydrogenglutarato) lanthanum(III) monohydrate. Acta Cryst. 2000, C56, 789–792. [Google Scholar]
- Curie, M.; Speakman, J.C.; Curry, N.A. The crystal structures oft he acid salts of some dibasic acids. Part I. A neutron-diffraction study of ammonium (and potassium) tetroxalate. J. Chem. Soc 1967, A1967, 1862–1869. [Google Scholar]
- Küppers, H.; Siegert, H. The elastic constants oft he triclinic crystals, ammonium and potassium tetroxalate dihydrate. Acta Cryst. 1970, A26, 401–405. [Google Scholar] [CrossRef]
- Küppers, H. Elastic properties of ammonium oxalate hydrate, ammonium hydrogen oxalate hemihydrate, and ammonium tetroxalate dihydrate. Acta Cryst. 1972, A28, 522–527. [Google Scholar] [CrossRef]
- Emsley, J.; Jones, D.J.; Kuroda, R. The hydrogen bonds of potassium tetroxalate: An X-ray redetermination of the crystal structure of KHC2O4.H2C2O4.2H2O). J. Inorg. Nucl. Chem. 1981, 43, 2243–2246. [Google Scholar] [CrossRef]
- Gilmore, C.J.; Speakman, J.C. The structure of potassium tetroxalate: A redetermination and a cautionary tale. Acta Cryst. 1982, B38, 2809–2813. [Google Scholar] [CrossRef]
- Clegg, W. Crystal structure of caesium tetroxalate dihydrate. Z. Kristallogr. 1984, 169, 211–217. [Google Scholar] [CrossRef]
- Haussühl, H. Elastic and thermoelastic constants of triclinic thallium trihydrogen oxalate dihydrate, TlH3(C2O4)2. 2H2O. Z. Kristallogr. 1993, 204, 115–119. [Google Scholar] [CrossRef]
- Fujita, J.; Nakamoto, K.; Kosbayashi, M. Infrared spectra of metallic complexes. II. The absorption bands of coordinated water in aquo complexes. J. Am. Chem. Soc. 1956, 78, 3963–3965. [Google Scholar] [CrossRef]
- Sengupta, A. K.; Sinha, K. Oxalatofluoro-indium compounds. J. Fluor. Chem. 1990, 47, 345–351. [Google Scholar] [CrossRef]
- Deb, N.; Baruah, S.D.; Dass, N.N. Thermal dehydration and decomposition of M[M(C2O4)2].xH2O (x = 3 for M = Sr(II) and x = 6 for M = Hg(II)). J. Therm. Anal. Cal. 1995, 45, 457–469. [Google Scholar] [CrossRef]
- Nakamoto, K. Infrared and Raman Inorganic and Coordination Compound, part B, 5th ed.; Wiley Interscience Publication, J. Wiley & sons, Inc: New York, NY, USA, 1997. [Google Scholar]
- Deb, N.; Baruah, S.D.; Sen-Sarma, N.; Dass, N.N. Synthesis, characterization and thermal investigation of M[M(C2O4)3].xH2O (x = 4 for M = Cr(III) and x = 2 for M = Sb(III) and x = 9 for M = La(III)). Thermochimica Acta 1998, 320, 53–67. [Google Scholar] [CrossRef]
- Albu, T.V.; Mindru, I.; Patron, L.; Segal, E.; Brezeanu, M. Thermal behaviour of some solid coordination compounds with malic acid as ligand. Thermochimica Acta 1999, 340–341, 235–240. [Google Scholar]
- Singh, B.P.; Singh, B. Synthesis and magnetic properties of one-dimensional metal oxalate networks as molecular-based magnets. Bull. Mater. Sci. 2000, 23, 11–16. [Google Scholar] [CrossRef]
- Chen, X.F.; Cheng, P.; Liu, X.; Zhao, B.; Liao, D.Z.; Yan, S.P.; Jiang, Z.H. Two-dimensional coordination polymers of copper(II) with oxalate: Lattice water control of structure. Inorg. Chem. 2001, 40, 2652–2659. [Google Scholar] [CrossRef] [PubMed]
- Del Arco, M.; Gutiérrez, S.; Martin, C.; Rives, V. Intercalation of [Cr(C2O4)3]3- complex in Mg, Al layered double hydroxides. Inorg. Chem. 2003, 42, 4232–4240. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.F.; Liu, L.; Ma, J.G.; Yi, L.; Cheng, P.; Liao, D.Z.; Yan, S.P.; Jiang, Z.H. Synthesis, reaction and structure of a series of chromium (III) complexes containing oxalate ligand. J. Mol. Struc. 2005, 750, 94–100. [Google Scholar] [CrossRef]
- Vlad, M.; Labadi, L.; Siaty, L.; Tudose, R.; Linert, W.; Costisor, O. Synthesis, characterisation and thermal properties of [Cu(VO)2(C2O4)3(4,4’-bpy)2.2H2O]A 2D polymer. J. Therm. Anal. Cal. 2008, 91, 925–928. [Google Scholar] [CrossRef]
- Vlad, M.; Labadi, L.; Sasca, V.; Saity, L.; Costisor, O. Synthesis and thermal decomposition of the coordination polymer of 4,4’-bipyridine and oxalate anion with Cu(II) and VO(IV). Chem. Bull. Politechnica Univ.Timisoara 2006, 51, 17–19. [Google Scholar]
- Stoyanov, E.S.; Kim, K.C.; Reed, C.A. The nature of the H3O+ hydronium ion in benzene and chlorinated hydrocarbon solvents. Conditions of existence and reinterpretation of infrared data. J. Am. Chem. Soc. 2006, 128, 1948–1958. [Google Scholar] [CrossRef] [PubMed]
- Dusausoy, Y.; Protas, J.; Mutin, J.C.; Watelle, G. Structure cristalline de BaC2O4.H2C2O4.2H2O. Acta Cryst. 1970, B26, 1567–1574. [Google Scholar] [CrossRef]
- Thongtem, T.; Thongtem, S. Preparation and characterization of Li1-xNi1+xO2 powder used as cathode materials. Adv. Technol. Mater. Mater. Process. J. 2005, 7, 71–76. [Google Scholar]
- Ito, R.; Masuda, Y.; Ito, Y. Thermal analyses of rubidium and cesium oxalate monohydrates. Thermochim Acta. 1987, 127, 159–170. [Google Scholar] [CrossRef]
- Dinnebier, R.E.; Vensky, S.; Panthöfer, M.; Jansen, M. Crystal and molecular structures of alkali oxalates: First proof of a staggered oxalate anion in the solid state. Inorg. Chem. 2003, 42, 1499–1507. [Google Scholar] [CrossRef] [PubMed]
- Dinnebier, R.E.; Vensky, S.; Jansen, M.; Hanson, J.C. Crystal structures and topological aspects of the high-temperature phases and decomposition products of the alkali-metal oxalates M2[C2O4] (M = K, Rb, Cs). Chem. Eur. J. 2005, 11, 1119–1129. [Google Scholar] [CrossRef] [PubMed]
- Martin Britto Dahas, S.A.; Natarajan, S. Growth and characterization of Lithium hydrogen oxalate monohydrate, a new semiorganic NLO material. Mater. Lett. 2008, 62, 1136–1138. [Google Scholar]
- Portalone, G.; Colapietro, M. Redetermination of ammonium oxalate oxalic acid dihydrate. Acta Cryst. 2006, E62, o4725–o4727. [Google Scholar]
- Kolitsh, U. RbCrIII(C2O4)2.2H2O, Cs2Mg(C2O4).4H2O and Rb2CuII(C2O4)2.2H2O : Three new complex oxalate hydrates. Acta Cryst. 2004, C60, m129–m133. [Google Scholar]
- Steiner, T. The Hydrogen Bond in the Solid State. Angew. Chem. Int. Ed. 2002, 41, 48–76. [Google Scholar] [CrossRef]
- Bernal, I. The composition, charge and architecture of hydronium ion as observed in the crystalline state. C.R. Chimie. 2006, 9, 1454–1466. [Google Scholar] [CrossRef]
- Delaplane, R.G.; Ibers, J.A. An X-ray study of oxalic acid dihydrate (COOH)2.2H2O and of its deuterium analogue, (COOD)2.2D2O: Isotope effect in hydrogen bonding and anisotropic extinction effects. Acta Cryst. 1969, B25, 2423–2437. [Google Scholar]
- Zobel, D.; Luger, P.; Dreissig, W.; Koritsanszky, T. Charge density studies on small organic molecules around 20 K: Oxalic acid dihydrate at 15 K and acetamide at 23 K. Acta Cryst. 1992, B48, 837–848. [Google Scholar] [CrossRef]
- Etter, M.C.; Mc Donald, J.C.; Bernstein, J. Graph-set analysis of hydrogen-bond patterns in organic crystals. Acta Cryst. 1990, B46, 256–262. [Google Scholar] [CrossRef]
- Desiraju, G.R. Supramolecular synthons in crystal engineering—A new organic synthesis. Angew.Chem. Int. Ed. Engl. 1995, 34, 2311–2327. [Google Scholar] [CrossRef]
- Steward, J.J.P. Application of the PM6 method to modeling the solid state. J. Mol. Model. 2008, 14, 499–535. [Google Scholar] [CrossRef] [PubMed]
- Li You, X.; Weng, S. Poly[hydronium[dyprosium(III)-μ3 -(ethylenediaminetetraacetato-к8 N,N’,O,O’,O”,O”’:O”’’:O””’)] monohydrate]. Acta Cryst. 2007, E63, m1819–1819. [Google Scholar]
- Guillou, N.; Auffredic, J.P.; Louer, M.; louer, D. The crystal structure of hydronium cerium (III) nitrate hydrate, Ce(NO3)5(H2O)2.H2O. J. Solid State Chem. 1993, 106, 295–300. [Google Scholar] [CrossRef]
- Zhang, L.-J.; Xu, J.A.; Shi, Z.; Xu, W.; Wang, T.G. Hydrothermal synthesis and characterization of the first oxalate-bta mixed-ligand three-dimensional frameworks:{[M2(μ8-bta)( μ2-C2O4)].(H3O)2(H2O)}n (M= CoII, FeII ; bta=benzene-1,2,4,5-tetracarboxylate). Dalton Trans 2003, 1148–1152. [Google Scholar]
- Serezhkina, L.B.; Vologzhanina, A.V.; Maruknou, A.V.; Pushkin, D.V.; Serezhkin, V.N. Synthesis and crystal structure of Na3(H3O)[UO2(SeO3)2]2 .H2O. Crystallogr. Reports. 2009, 54, 852–857. [Google Scholar] [CrossRef]
- Stahly, G.P. Diversity in single- and multiple-component crystals. The search for and prevalence of polymorphs and cocrystals. Cryst Growth Des. 2007, 7, 1007–1026. [Google Scholar] [CrossRef]
- Desiraju, G.R. Counterpoint: What’s in a name? Cryst Growth Des. 2004, 4, 1089–1090. [Google Scholar] [CrossRef]
- Ma, Z.; Moulton, B. Supramolecular medicinal chemistry: Mixed-ligand coordination complexes. Mol. Pharmaceutics. 2007, 4, 373–385. [Google Scholar] [CrossRef]
- Pedersen, B.F. The crystal structure of potassium hydrogen oxalate, KHC2O4. Acta Chem. Scand. 1969, 22, 2953–2964. [Google Scholar] [CrossRef]
- Einpahr, H.; March, P.E.; Donobue, J. The crystal structure of potassium binoxalate. Acta Cryst. 1972, B28, 2194–2195. [Google Scholar] [CrossRef]
- Kholodkovskaya, L.N.; Trunov, V.K.; Tskhelashvili, N.B. Refinement of the crystal structures of potassium and rubidium hydrogen oxalates MHC2O4 (M = K, Rb). J. Struct. Chem. 1990, 31, 509–511. [Google Scholar] [CrossRef]
- Fujita, M.; Powell, A.; Creutz, C.A. From the molecule to nanoscale: Synthesis, structure and properties. In Comprehensive Coordination Chemistry II; Mc Cleverly, J.A., Meyer, T.J., Eds.; Elsevier Pergamon: Amsterdam, the Netherlands, 2004. [Google Scholar]
- Zhang, X.L.; Chen, X.M.; Ng, S.W. Melaminium bis(hydrogen oxalate). Acta Cryst. 2005, E61, o156–o157. [Google Scholar]
- Price, D.J.; Powell, A.K.; Wood, P.T. Hydrothermal crystallisation and X-ray structure of anhydrous strontium oxalate. Polyhedron 1999, 18, 2499–2503. [Google Scholar] [CrossRef]
- Viroveis, A.V.; Naumov, D.Y.; Boldyreva, E.V.; Podberezskaya, N.V. Structure of lead (II) oxalate dihydrate. Acta Cryst. 1993, C49, 1882–1884. [Google Scholar]
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Kherfi, H.; Hamadène, M.; Guehria-Laïdoudi, A.; Dahaoui, S.; Lecomte, C. Synthesis, Structure and Thermal Behavior of Oxalato-Bridged Rb+ and H3O+ Extended Frameworks with Different Dimensionalities. Materials 2010, 3, 1281-1301. https://doi.org/10.3390/ma3021281
Kherfi H, Hamadène M, Guehria-Laïdoudi A, Dahaoui S, Lecomte C. Synthesis, Structure and Thermal Behavior of Oxalato-Bridged Rb+ and H3O+ Extended Frameworks with Different Dimensionalities. Materials. 2010; 3(2):1281-1301. https://doi.org/10.3390/ma3021281
Chicago/Turabian StyleKherfi, Hamza, Malika Hamadène, Achoura Guehria-Laïdoudi, Slimane Dahaoui, and Claude Lecomte. 2010. "Synthesis, Structure and Thermal Behavior of Oxalato-Bridged Rb+ and H3O+ Extended Frameworks with Different Dimensionalities" Materials 3, no. 2: 1281-1301. https://doi.org/10.3390/ma3021281
APA StyleKherfi, H., Hamadène, M., Guehria-Laïdoudi, A., Dahaoui, S., & Lecomte, C. (2010). Synthesis, Structure and Thermal Behavior of Oxalato-Bridged Rb+ and H3O+ Extended Frameworks with Different Dimensionalities. Materials, 3(2), 1281-1301. https://doi.org/10.3390/ma3021281