
The First 20 Hours of Geopolymerization: An in Situ WAXS Study of Flyash-Based Geopolymers

Abstract
:1. Introduction
Geopolymer Kinetics
2. Materials and Methods
2.1. Geopolymer Synthesis
2.2. WAXS Data Collection
2.3. WAXS Data Processing
2.4. WAXS Phase Identification
2.5. WAXS Data Analysis
2.6. SAXS Data Collection
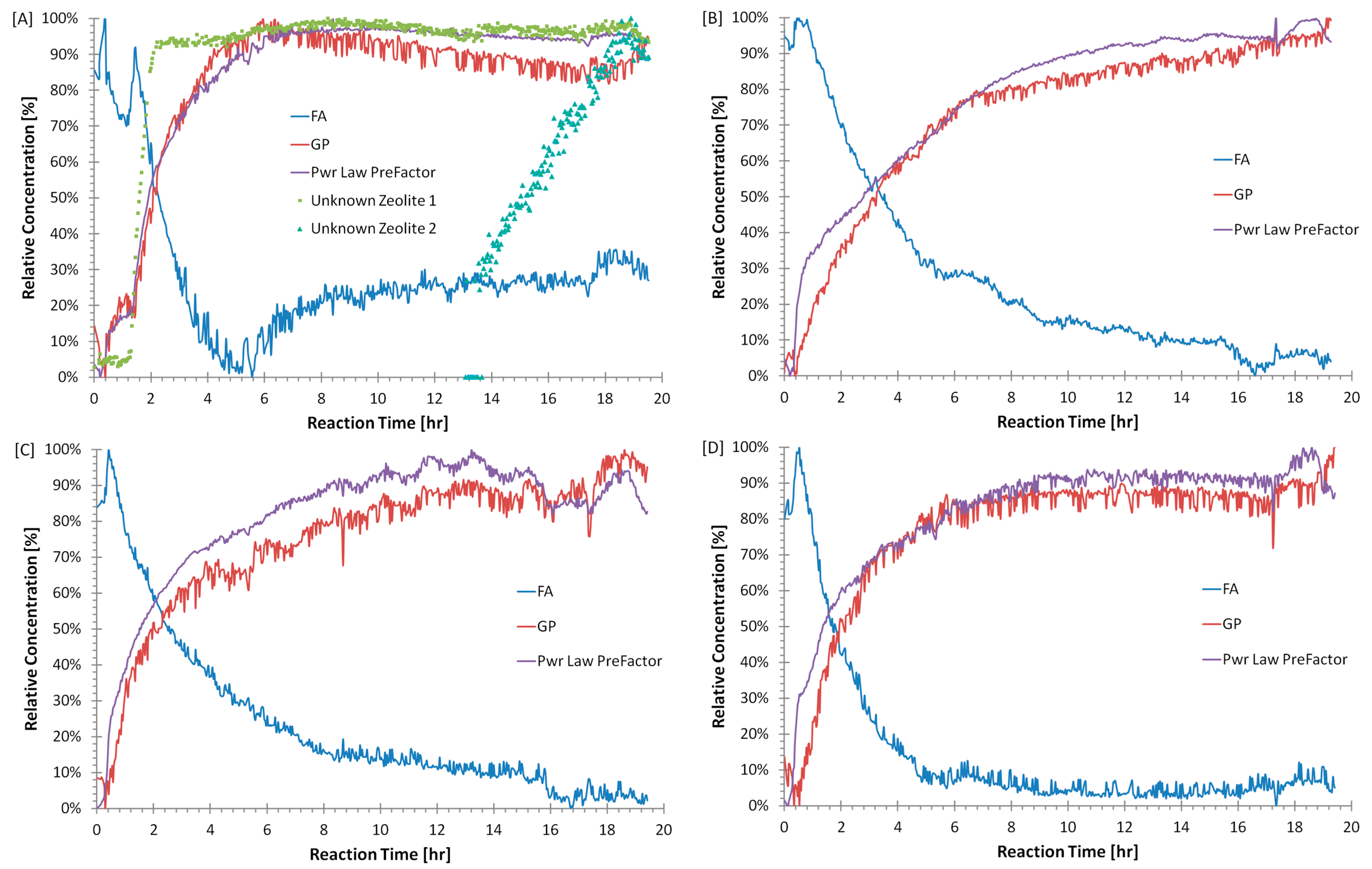
3. Results
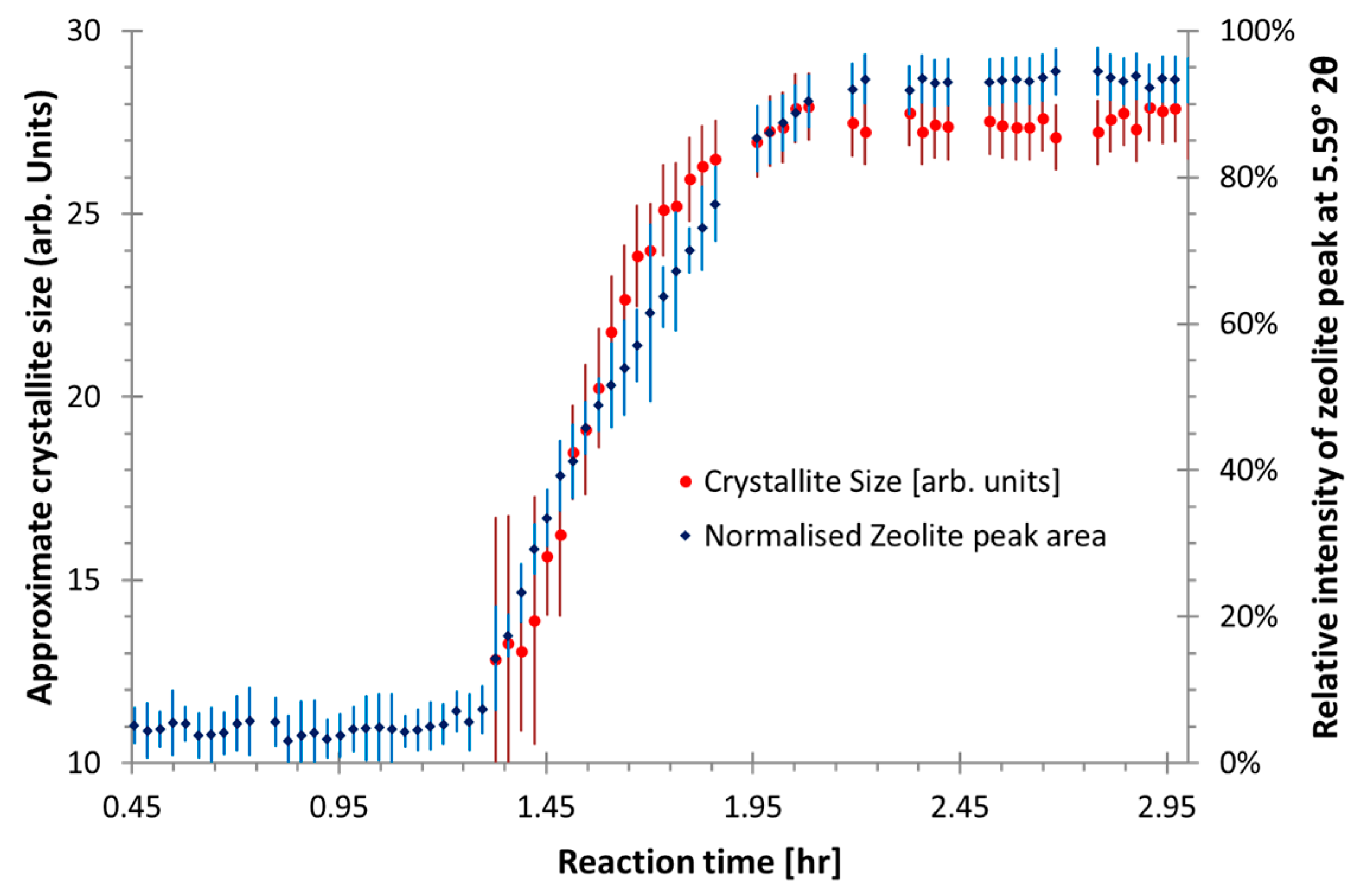
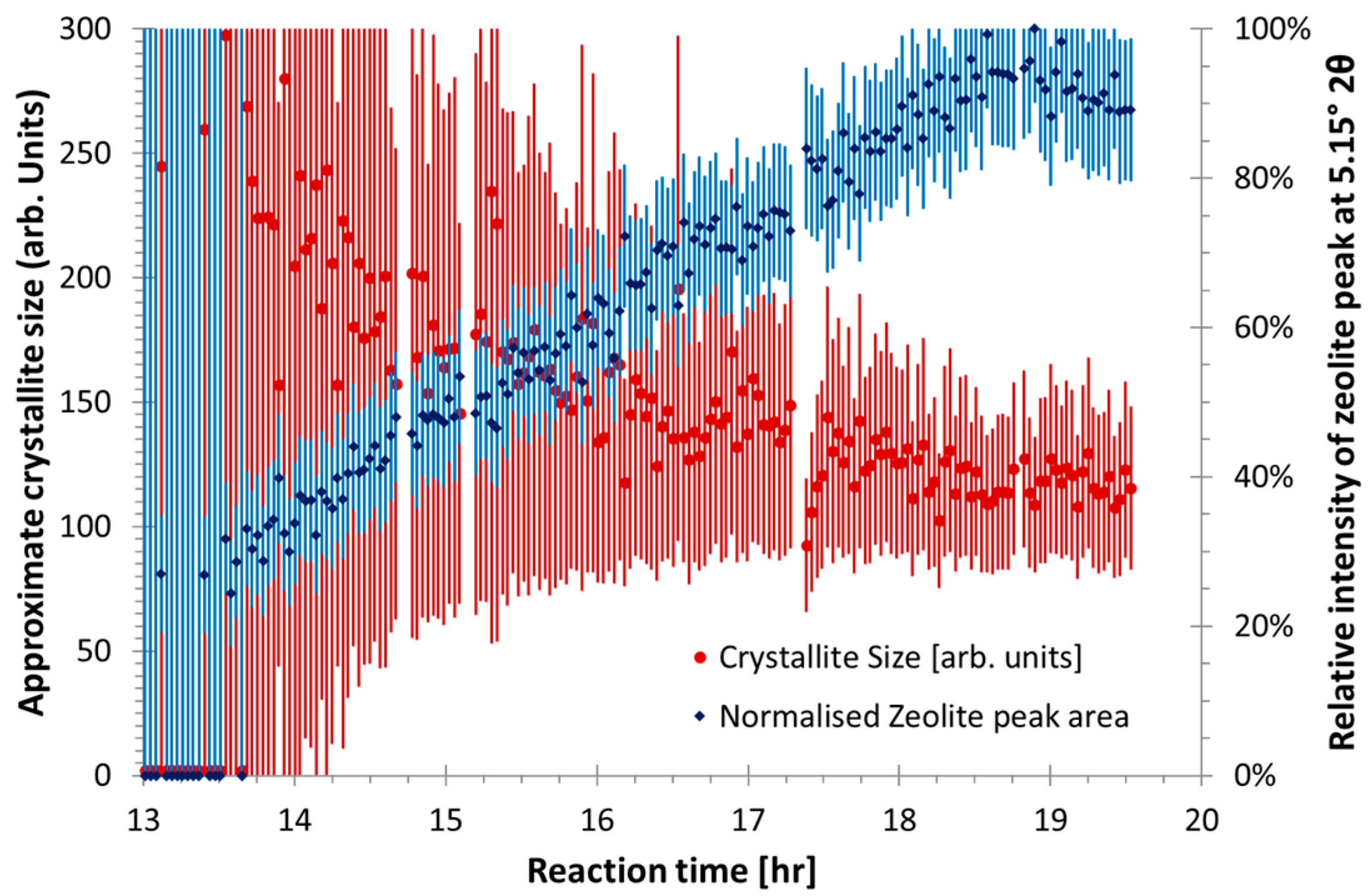
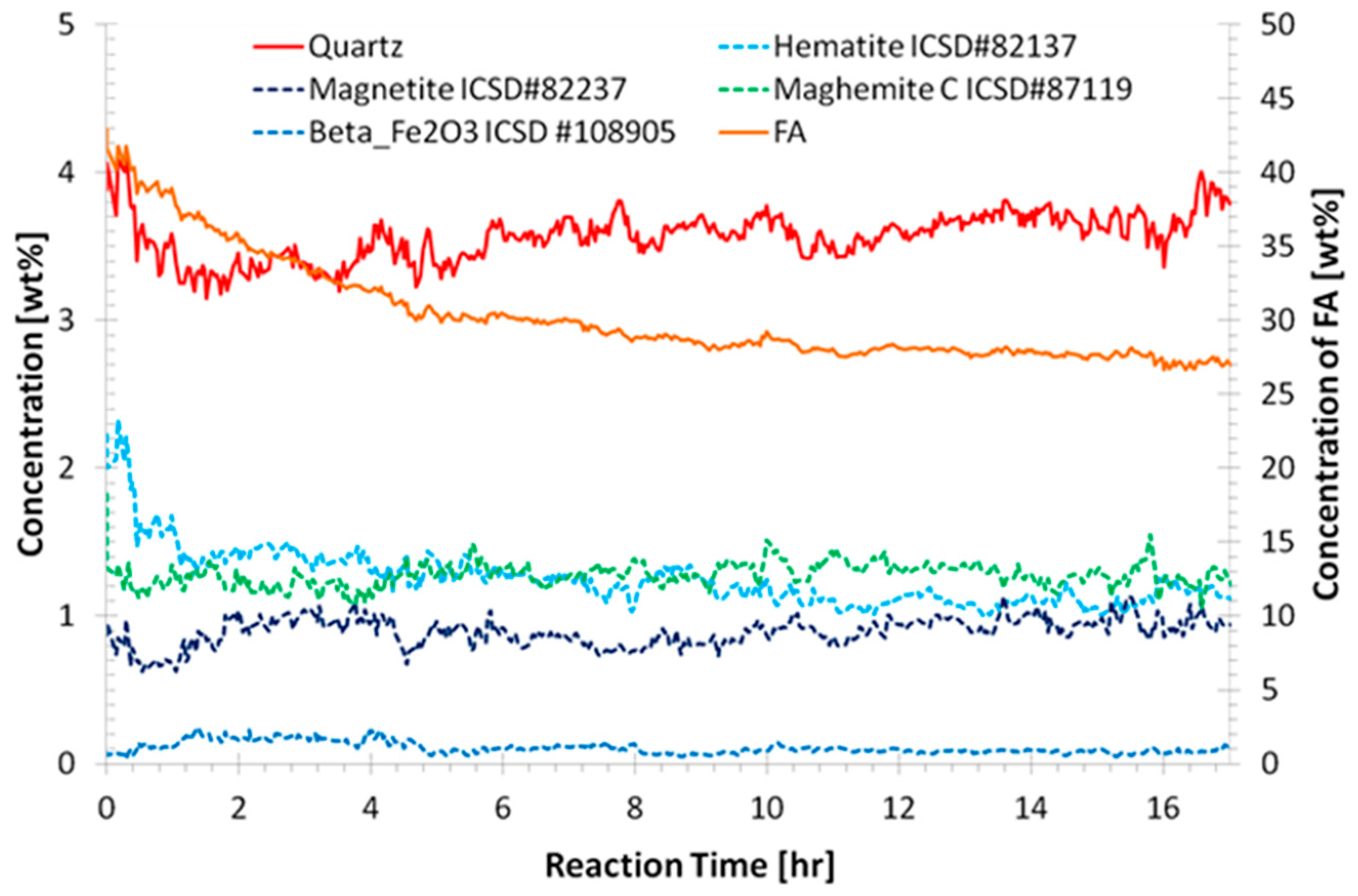
3.1. Preliminary Analysis of WAXS Data
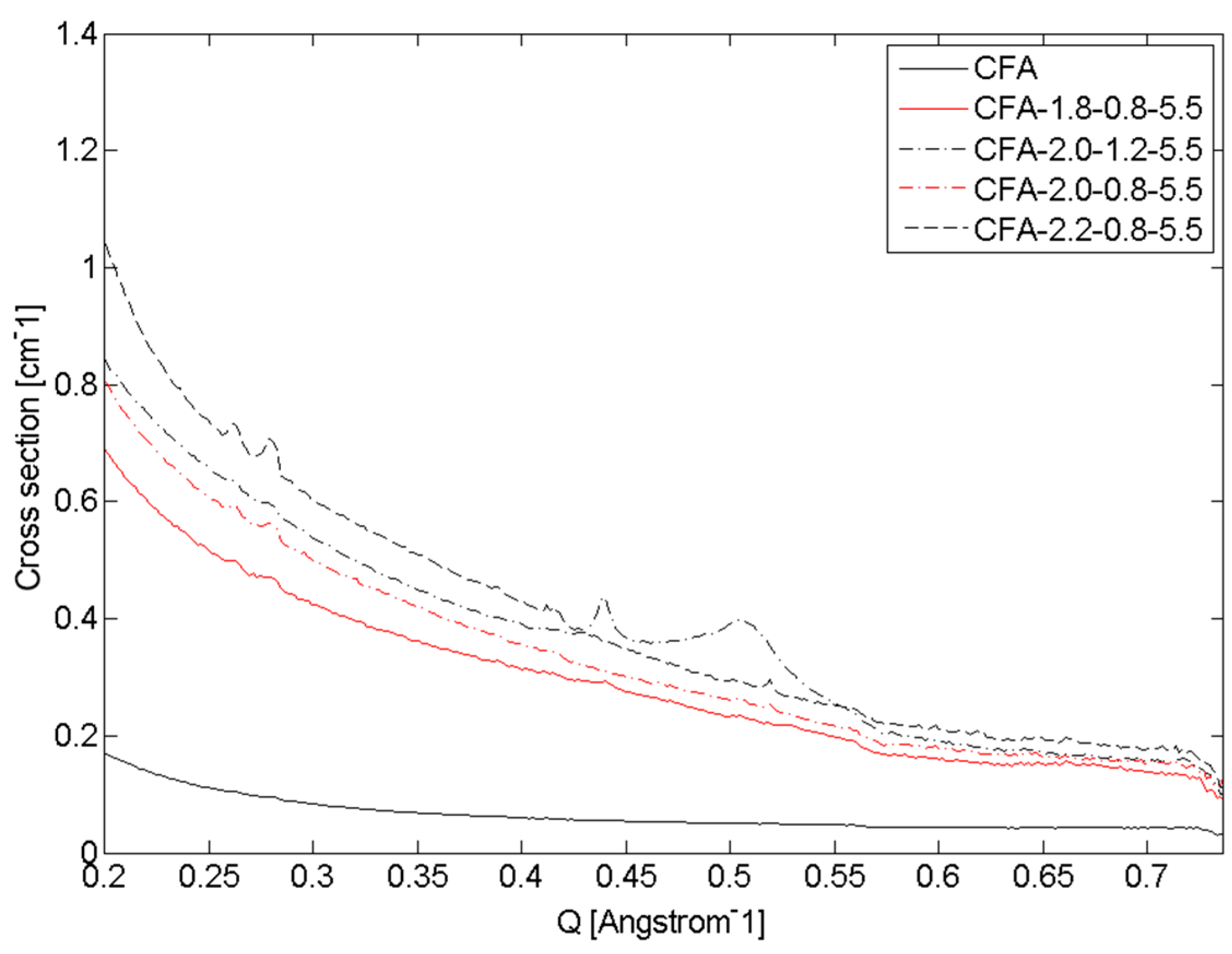
3.2. Quantitative Analysis of WAXS Data
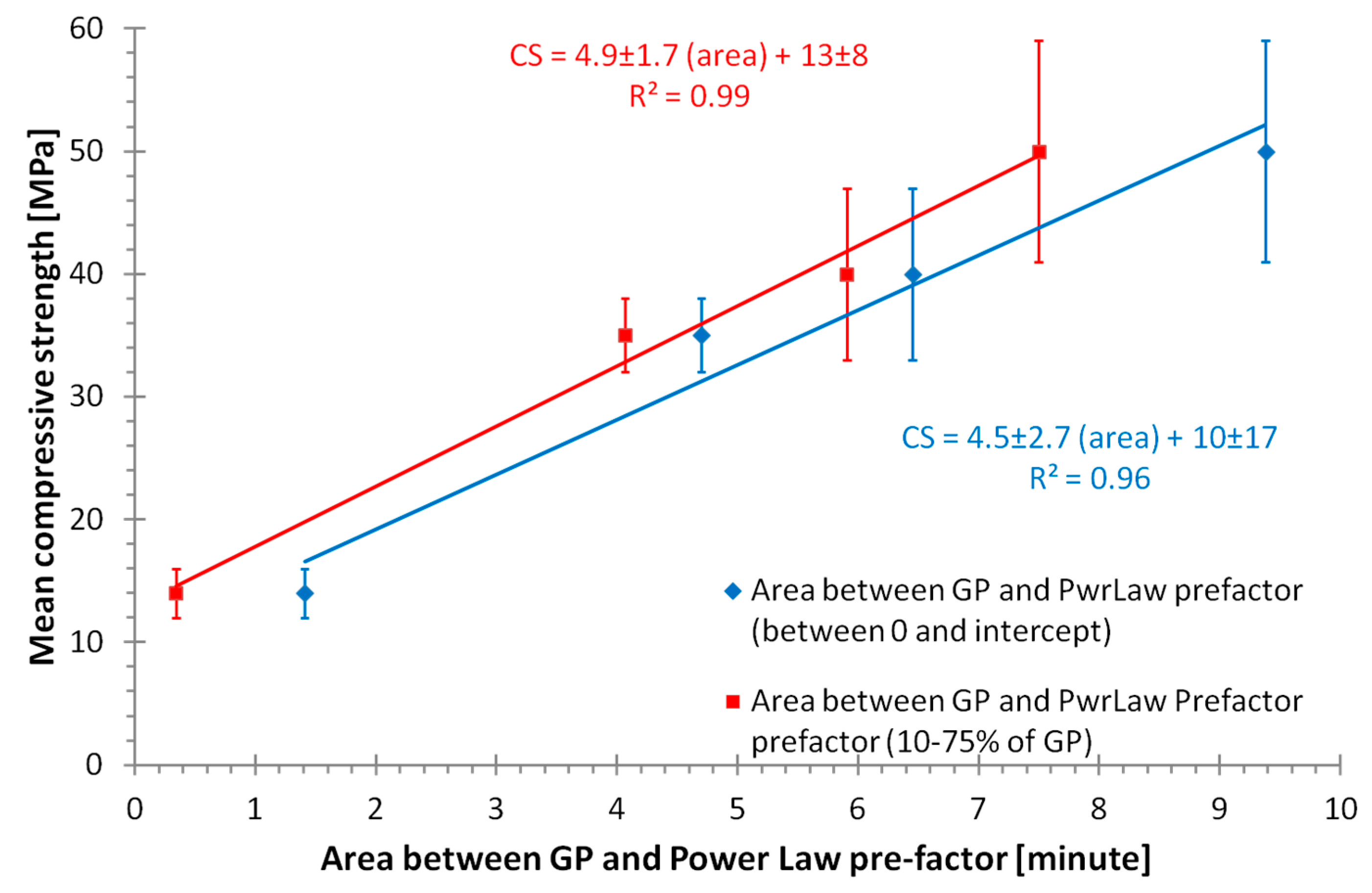
4. Discussion
5. Conclusions
- Confirmation that there are multiple mechanisms of zeolite-like phase formation even within the same geopolymer sample.
- A new method has been identified to separately measure the progress of feedstock dissolution and geopolymer formation in situ, which will allow a large number of optimization experiments to be completed in the future.
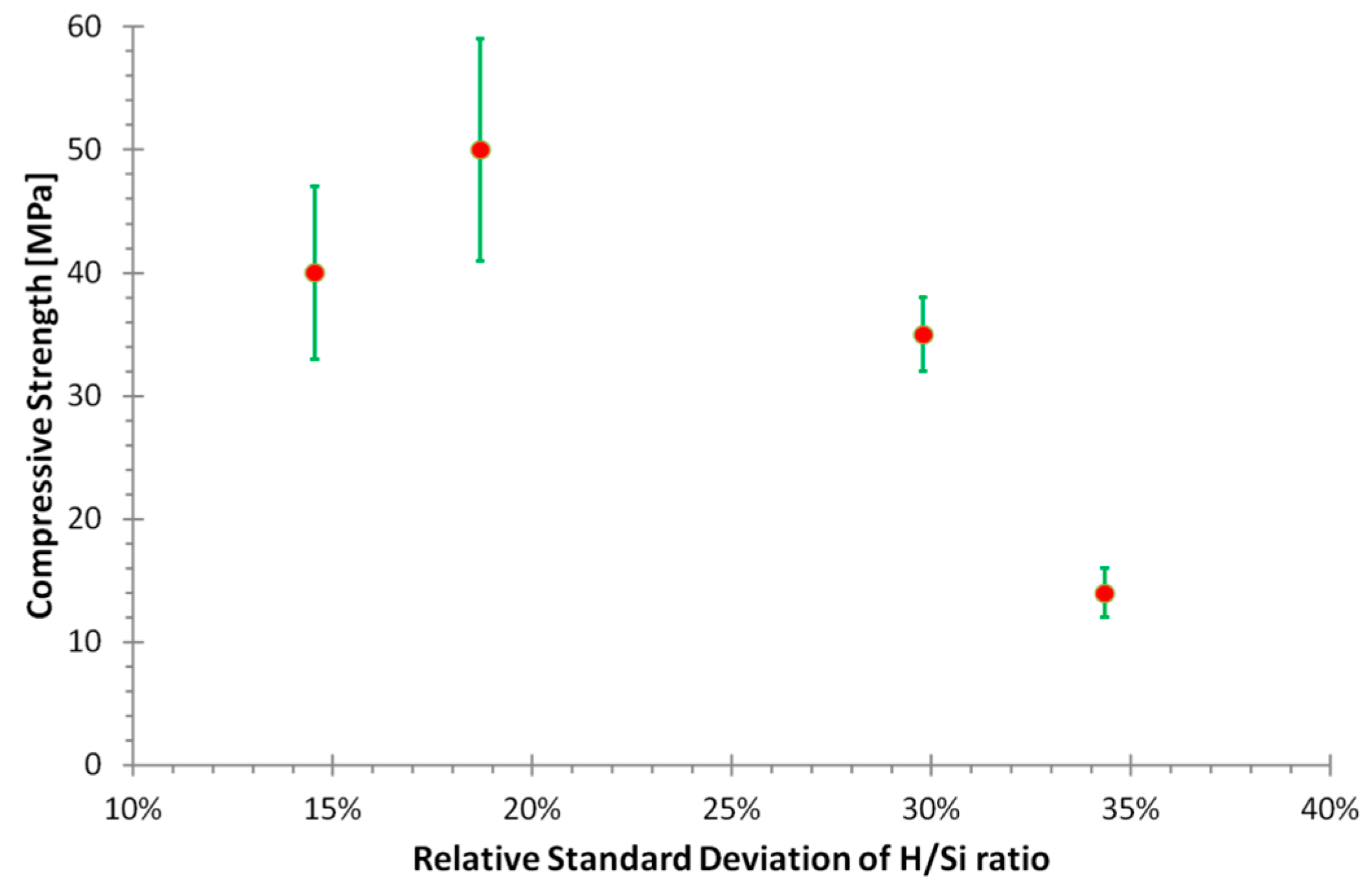
- Identification of significant temporal variation in the composition of the solution from which the geopolymer forms; the relative standard deviation of the H/Si ratio during formation was correlated to the physical properties.
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Provis, J.L.; van Deventer, J.S.J. Geopolymerization kinetics. 1. In situ energy-dispersive X-ray diffractometry. Chem. Eng. Sci. 2007, 62, 2309–2317. [Google Scholar] [CrossRef]
- Madsen, I.C.; Scarlett, N.V.Y.; Riley, D.P.; Raven, M.D. Quantitative Phase Analysis Using the Rietveld Method. In Modern Diffraction Methods; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2012; pp. 283–320. [Google Scholar]
- Williams, R.P.; Hart, R.D.; van Riessen, A. Quantification of the Extent of Reaction of Metakaolin-Based Geopolymers Using X-Ray Diffraction, Scanning Electron Microscopy, and Energy-Dispersive Spectroscopy. J. Am. Ceram. Soc. 2011, 94, 2663–2670. [Google Scholar] [CrossRef]
- Kern, A.; Madsen, I.; Scarlett, N.Y. Quantifying Amorphous Phases. In Uniting Electron Crystallography and Powder Diffraction; Kolb, U., Shankland, K., Meshi, L., Avilov, A., David, W.I.F., Eds.; Springer: Dordrecht, The Netherlands, 2012; pp. 219–231. [Google Scholar]
- Madsen, I.C.; Scarlett, N.V.Y.; Kern, A. Description and survey of methodologies for the determination of amorphous content via X-ray powder diffraction. Z. Kristallogr. Cryst. Mater. 2011, 226, 944–955. [Google Scholar] [CrossRef]
- Rees, C.A.; Provis, J.L.; Lukey, G.C.; van Deventer, J.S.J. In Situ ATR-FTIR Study of the Early Stages of Fly Ash Geopolymer Gel Formation. Langmuir 2007, 23, 9076–9082. [Google Scholar] [CrossRef] [PubMed]
- Rahier, H.; Denayer, J.F.; van Mele, B. Low-temperature synthesized aluminosilicate glasses Part IV Modulated DSC study on the effect of particle size of metakaolinite on the production of inorganic polymer glasses. J. Mater. Sci. 2003, 38, 3131–3136. [Google Scholar] [CrossRef]
- Provis, J.L.; Walls, P.A.; van Deventer, J.S.J. Geopolymerisation kinetics. 3. Effects of Cs and Sr salts. Chem. Eng. Sci. 2008, 63, 4480–4489. [Google Scholar] [CrossRef]
- Panzarella, B.; Tompsett, G.; Conner, W.C.; Jones, K. In situ SAXS/WAXS of Zeolite Microwave Synthesis: NaY, NaA, and Beta Zeolites. Chem. Phys. Chem. 2007, 8, 357–369. [Google Scholar] [CrossRef] [PubMed]
- De Moor, P.-P.E.A.; Beelen, T.P.M.; Komanschek, B.U.; van Santen, R.A. Nanometer scale precursors in the crystallization of Si-TPA-MFI. Microporous Mesoporous Mater. 1998, 21, 263–269. [Google Scholar] [CrossRef]
- Dokter, W.H.; Beelen, T.P.M.; van Garderen, H.F.; van Santen, R.A.; Bras, W.; Derbyshire, G.E.; Mant, G.R. Simultaneous monitoring of amorphous and crystalline phases in silicalite precursor gels. An in situ hydrothermal and time-resolved small- and wide-angle X-ray scattering study. J. Appl. Crystallogr. 1994, 27, 901–906. [Google Scholar] [CrossRef]
- Walton, R.I.; Millange, F.; O’Hare, D.; Davies, A.T.; Sankar, G.; Catlow, C.R.A. An in Situ Energy-Dispersive X-ray Diffraction Study of the Hydrothermal Crystallization of Zeolite A. 1. Influence of Reaction Conditions and Transformation into Sodalite. J. Phys. Chem. B 2000, 105, 83–90. [Google Scholar] [CrossRef]
- Norby, P.; Hanson, J.C. Hydrothermal synthesis of the microporous aluminophosphate CoAPO-5; in situ time-resolved synchrotron X-ray powder diffraction studies. Catal. Today 1998, 39, 301–309. [Google Scholar] [CrossRef]
- Jozić, D.; Zorica, S.; Tibljaš, D.; Bernstorff, S. In situ SAXS/WAXS study of the developing process of geopolymer structures. In Proceedings of the 15th European Conference on Composite Materials, Venice, Italy, 24–28 June 2012.
- De Silva, P.; Sagoe-Crenstil, K.; Sirivivatnanon, V. Kinetics of geopolymerization: Role of Al2O3 and SiO2. Cem. Concr. Res. 2007, 37, 512–518. [Google Scholar] [CrossRef]
- Zhang, Z.; Wang, H.; Provis, J.L.; Bullen, F.; Reid, A.; Zhu, Y. Quantitative kinetic and structural analysis of geopolymers. Part 1. The activation of metakaolin with sodium hydroxide. Thermochim. Acta 2012, 539, 23–33. [Google Scholar] [CrossRef]
- Zhang, Z.; Provis, J.L.; Wang, H.; Bullen, F.; Reid, A. Quantitative kinetic and structural analysis of geopolymers. Part 2. Thermodynamics of sodium silicate activation of metakaolin. Thermochim. Acta 2013, 565, 163–171. [Google Scholar] [CrossRef]
- Steins, P.; Poulesquen, A.; Frizon, F.; Diat, O.; Jestin, J.; Causse, J.; Lambertin, D.; Rossignol, S. Effect of aging and alkali activator on the porous structure of a geopolymer. J. Appl. Crystallogr. 2014, 47, 316–324. [Google Scholar] [CrossRef]
- Maitland, C.F.; Buckley, C.E.; O’Connor, B.H.; Butler, P.D.; Hart, R.D. Characterization of the pore structure of metakaolin-derived geopolymers by neutron scattering and electron microscopy. J. Appl. Crystallogr. 2011, 44, 697–707. [Google Scholar] [CrossRef]
- Kriven, W.M.; Bell, J.L.; Gordon, M. Geopolymer refractories for the glass manufacturing industry. Ceram. Eng. Sci. Proc. 2004, 25, 57–79. [Google Scholar]
- Bell, J.L.; Kriven, W.M. Nanoporosity in Aluminosilicate, Geopolymeric Cements. Microsc. Microanal. 2004, 10 (Suppl. S2), 590–591. [Google Scholar] [CrossRef]
- Duxson, P.; Provis, J.L.; Lukey, G.C.; Mallicoat, S.W.; Kriven, W.M.; van Deventer, J.S.J. Understanding the relationship between geopolymer composition, microstructure and mechanical properties. Colloids Surf. A Physicochem. Eng. Asp. 2005, 269, 47–58. [Google Scholar] [CrossRef]
- Phair, J.W.; Schulz, J.C.; Bertram, W.K.; Aldridge, L.P. Investigation of the microstructure of alkali-activated cements by neutron scattering. Cem. Concr. Res. 2003, 33, 1811–1824. [Google Scholar] [CrossRef]
- Williams, R.P.; van Riessen, A. Determination of the reactive component of fly ashes for geopolymer production using XRF and XRD. Fuel 2010, 89, 3683–3692. [Google Scholar] [CrossRef]
- Williams, R.P. Optimising Geopolymer Formation. Ph.D. Thesis, Curtin University, Bentley, WA, Australia, 14 December 2015. [Google Scholar]
- Powder Diffraction File Database; PDF 4+ (2009); The International Centre for Diffraction Data (ICDD): Newtown Square, PA, USA, 2009.
- Inorganic Crystal Structure Database (ICSD); 2010/1; FIZ: Karlsruhe, Germany, 2010.
- Downs, R.T.; Hall-Wallace, M. The American Mineralogist Crystal Structure Database. Am. Mineral. 2003, 88, 247–250. [Google Scholar]
- Stinton, G.W.; Evans, J.S.O. Parametric Rietveld refinement. J. Appl. Crystallogr. 2007, 40, 87–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Müller, M.; Dinnebier, R.E.; Jansen, M.; Wiedemann, S.; Plüg, C. Kinetic analysis of the phase transformation from α- to β-copper phthalocyanine: A case study for sequential and parametric Rietveld refinements. Powder Diffr. 2009, 24, 191–199. [Google Scholar] [CrossRef]
- Treacy, M.M.; Higgins, J.B. Collection of Simulated XRD Powder Patterns for Zeolites, 5th Revised ed.; Elsevier: Amsterdam, The Netherlands, 2007. [Google Scholar]
- Balzar, D. Voigt-Function Model in Diffraction Line-Broadening Analysis. In Defect and Microstructure Analysis by Diffraction; Snyder, R., Fiala, J., Bunge, H.J., Eds.; Oxford University Press: Oxford, UK, 2000; p. 808. [Google Scholar]
- Williams, R.P.; van Riessen, A. Development of alkali activated borosilicate inorganic polymers (AABSIP). J. Eur. Ceram. Soc. 2011, 31, 1513–1516. [Google Scholar] [CrossRef]
- Rowles, M.R.; O’Connor, B.H. Chemical and Structural Microanalysis of Aluminosilicate Geopolymers Synthesized by Sodium Silicate Activation of Metakaolinite. J. Am. Ceram. Soc. 2009, 92, 2354. [Google Scholar] [CrossRef]
- Provis, J.L.; van Deventer, J.S.J. Geopolymerisation kinetics. 2. Reaction kinetic modelling. Chem. Eng. Sci. 2007, 62, 2318–2329. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample Name | Si/Al | Na/Al | H/Si | CS (MPa) |
|---|---|---|---|---|
| CFA-1.8-0.8-5.5 | 1.8 | 0.8 | 5.5 | 14(2) |
| CFA-2.0-0.8-5.5 | 2.0 | 0.8 | 5.5 | 35(3) |
| CFA-2.0-1.2-5.5 | 2.0 | 1.2 | 5.5 | 50(9) |
| CFA-2.2-0.8-5.5 | 2.2 | 0.8 | 5.5 | 40(7) |
| Sample Name | Initial (wt %) | Final (wt %) | Extent of Dissolution (%) | CS (MPa) |
|---|---|---|---|---|
| CFA-1.8-0.8-5.5 | 46 | 33 | 29 | 14(2) |
| CFA-2.0-0.8-5.5 | 44 | 20 | 54 | 35(3) |
| CFA-2.0-1.2-5.5 | 43 | 27 | 38 | 50(9) |
| CFA-2.2-0.8-5.5 | 42 | 24 | 42 | 40(7) |
| Sample Name | Flyash Amorphous Phase (Å) | Geopolymer Amorphous Phase (Å) |
|---|---|---|
| CFA-1.8-0.8-5.5 | 3.96 | 2.92 |
| CFA-2.0-0.8-5.5 | 4.00 | 3.02 |
| CFA-2.0-1.2-5.5 | 4.08 | 2.99 |
| CFA-2.2-0.8-5.5 | 4.03 | 3.06 |
| Sample Name | Solution during Reaction | Final Extent | ||||
|---|---|---|---|---|---|---|
| Si/Al | Na/Al | H/Si | Si/Al | Na/Al | H/Si | |
| CFA-1.8-0.8-5.5 | 2.3(4) | 4.4(28) | 22.5(77) | 2.23(2) | 3.9(2) | 21.6(6) |
| CFA-2.0-0.8-5.5 | 2.5(6) | 2.2(16) | 11.2(33) | 2.24(1) | 1.46(4) | 8.9(2) |
| CFA-2.0-1.2-5.5 | 2.8(4) | 4.3(17) | 13.8(26) | 2.51(3) | 3.2(1) | 11.7(3) |
| CFA-2.2-0.8-5.5 | 3.2(8) | 2.3(12) | 10.8(16) | 3.0(1) | 2.1(2) | 10.4(5) |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Williams, R.P.; Van Riessen, A. The First 20 Hours of Geopolymerization: An in Situ WAXS Study of Flyash-Based Geopolymers. Materials 2016, 9, 552. https://doi.org/10.3390/ma9070552
Williams RP, Van Riessen A. The First 20 Hours of Geopolymerization: An in Situ WAXS Study of Flyash-Based Geopolymers. Materials. 2016; 9(7):552. https://doi.org/10.3390/ma9070552
Chicago/Turabian StyleWilliams, Ross P., and Arie Van Riessen. 2016. "The First 20 Hours of Geopolymerization: An in Situ WAXS Study of Flyash-Based Geopolymers" Materials 9, no. 7: 552. https://doi.org/10.3390/ma9070552
APA StyleWilliams, R. P., & Van Riessen, A. (2016). The First 20 Hours of Geopolymerization: An in Situ WAXS Study of Flyash-Based Geopolymers. Materials, 9(7), 552. https://doi.org/10.3390/ma9070552