Development and Deployment of High-Throughput Retrotransposon-Based Markers Reveal Genetic Diversity and Population Structure of Asian Bamboo


 ,
,  , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Material
2.2. Isolation of Genomic DNA
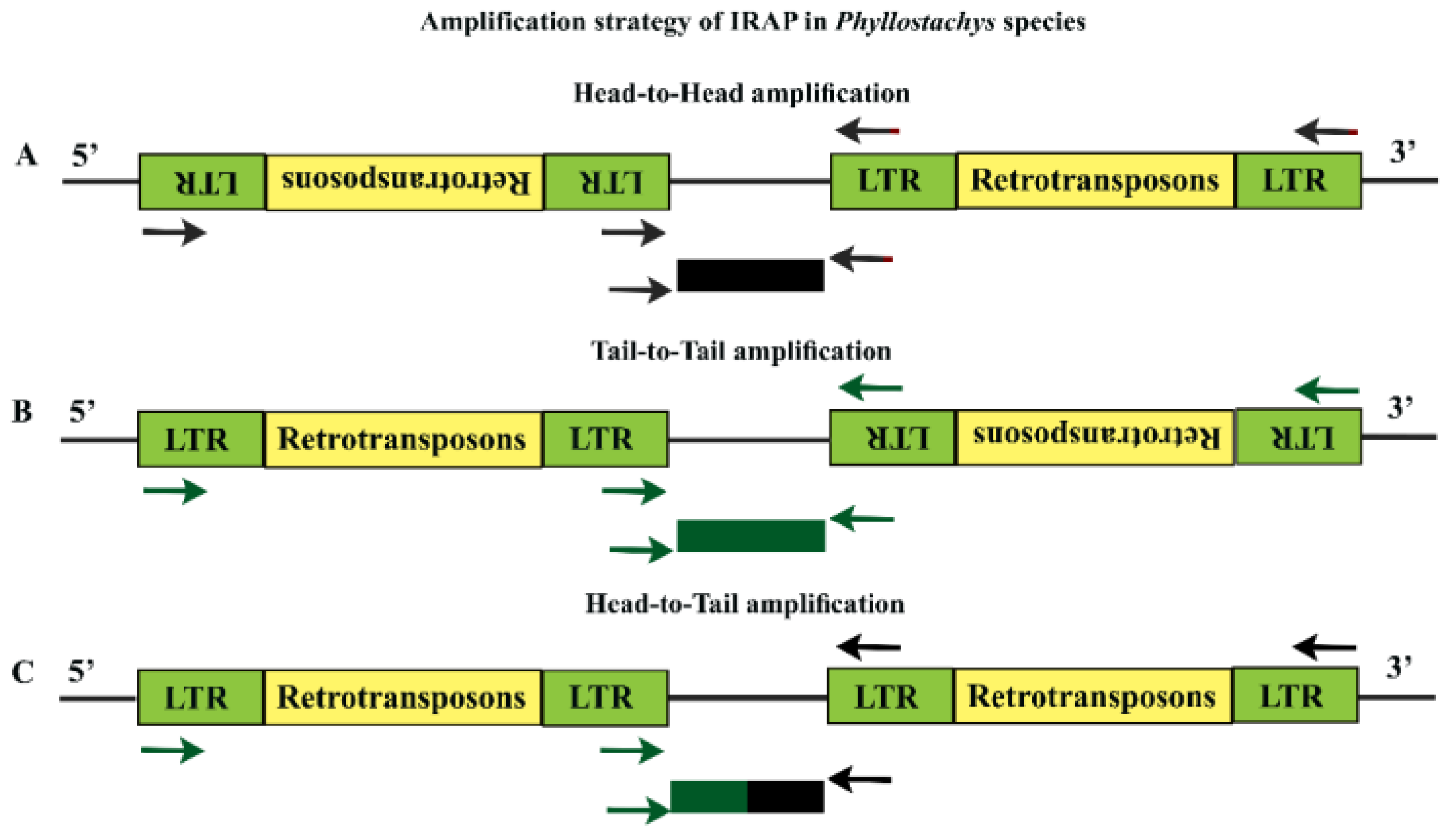
2.3. Isolation of LTR-Retrotransposons and IRAP-Primer Design
2.4. PCR Amplification of IRAP and Electrophoresis
2.5. Cloning and Sequencing of IRAP Fragments
2.6. Marker Statistics and Genetic Relations
2.7. Genetic Distance and Diversity of Bamboo Accessions
2.8. IRAP-Statistical Fitness Analysis
2.9. Analysis of Population Structure
2.10. Analysis of Molecular Variance
3. Results
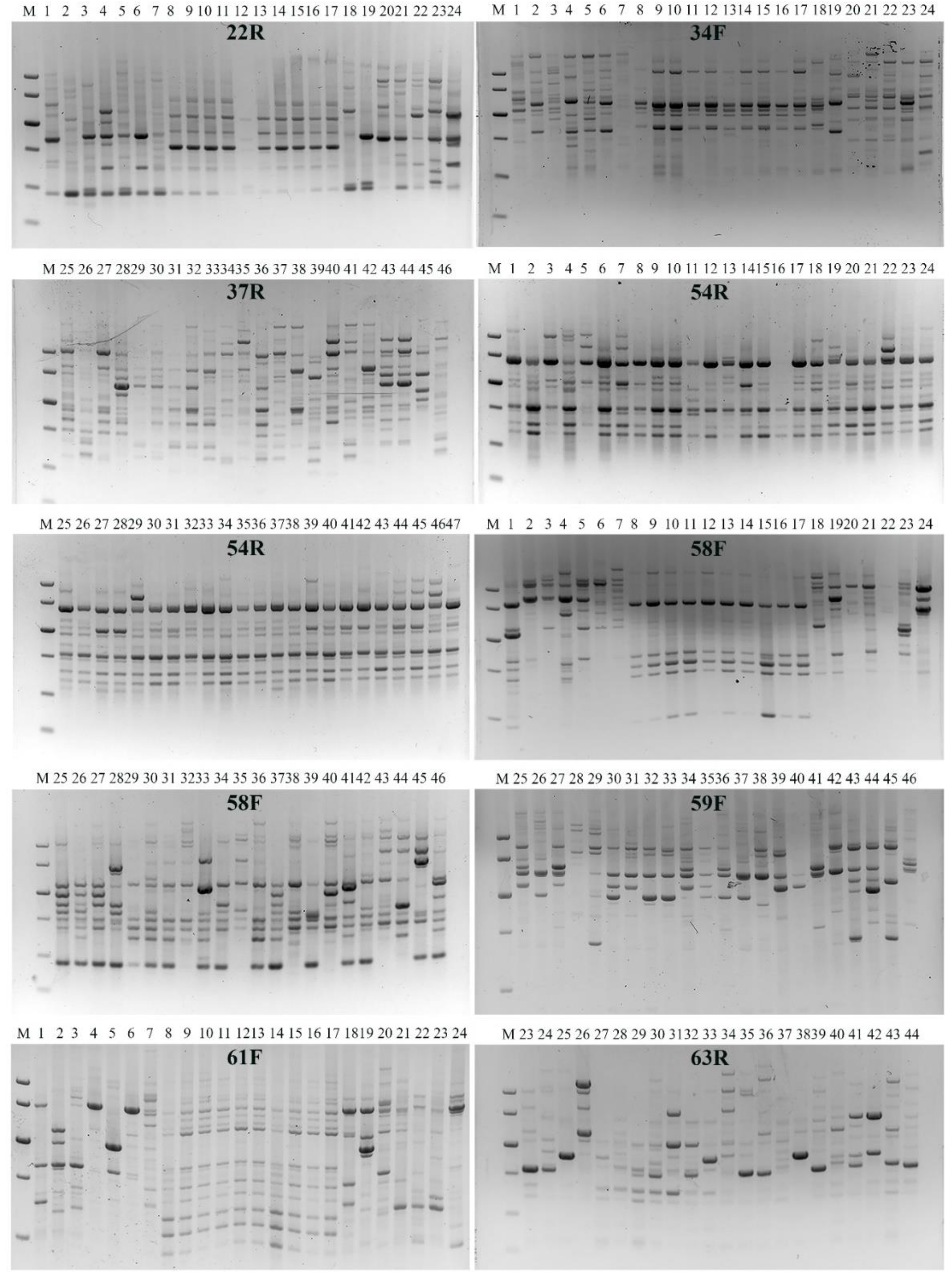
3.1. Development of IRAP Makers and Functionality Assay
3.2. IRAP Amplicon Variation within and between the Accessions
3.3. Multiple Sequence Alignment of IRAP-PCR Products
3.4. Diversity Analysis of Asian Bamboo Accessions
3.5. Statistical Fitness Analysis of Clustering Pattern
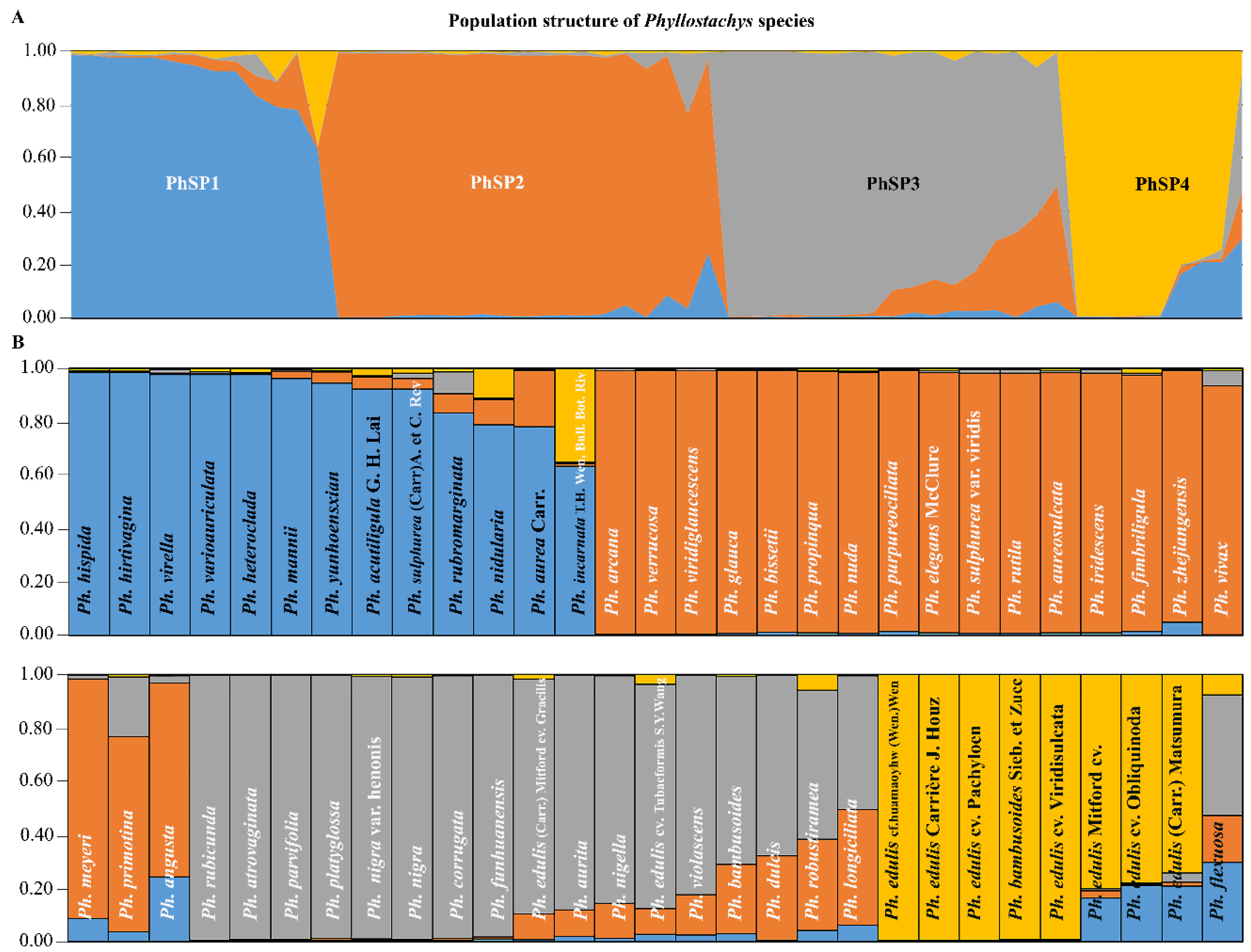
3.6. Population Structure of Asian Bamboo Accessions
3.7. Analysis of Molecular Variance
4. Discussions
4.1. Genetic Diversity of Asian Bamboo Species
4.2. Population Structure of Asian Bamboo
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AFLP | Amplified fragment length polymorphism |
| AMOVA | Analysis of the molecular variance |
| CCC | Cophenetic correlation coefficient |
| CTAB | Cetyltrimethylammonium bromide |
| df | Degree of freedom |
| EST-SSR | Expressed sequence tags—SSR |
| IPTG | Isopropyl β-D-1-thiogalactopyranoside |
| IRAP | Inter-retrotransposon amplified polymorphism |
| ISSRs | Inter-simple sequence repeats |
| LTR | Long terminal repeat |
| MCMC | Markov Chain Monte Carlo |
| MS | Mean of squared deviations |
| PCA | Principal component analysis |
| PhC | Phyllostachys cluster |
| PhSP | Phyllostachys sub-population |
| PIC | Polymorphic information content |
| RAPD | Randomly amplified polymorphic DNA |
| RFLP | Restriction fragment length polymorphism |
| SCoT | Start codon targeted |
| siRNAs | Small interfering RNAs |
| SNP | Single nucleotide polymorphism |
| SS | Sums of squared deviations |
| SSRs | Simple sequence repeats |
| TEs | Transposable elements |
| UPGMA | Unweighted pair-group method with arithmetic average |
References
- Yeasmin, L.; Ali, M.N.; Gantait, S.; Chakraborty, S. Bamboo: An overview on its genetic diversity and characterization. 3 Biotech 2015, 5, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biswas, P.; Chakraborty, S.; Dutta, S.; Pal, A.; Das, M. Bamboo flowering from the perspective of comparative genomics and transcriptomics. Front. Plant Sci. 2016, 7, 1900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, J.-L.; Yue, J.-J.; Gu, X.-P.; Lin, C.-S. Flowering of woody bamboo in tissue culture systems. Front. Plant Sci. 2017, 8, 1589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Tang, D.; Lin, X.; Ding, M.; Tong, Z. Genome-wide identification of MADS-box family genes in moso bamboo (Phyllostachys edulis) and a functional analysis of PeMADS5 in flowering. BMC Plant Biol. 2018, 18, 176. [Google Scholar] [CrossRef] [PubMed]
- Tao, G.; Fu, Y.; Zhou, M. Advances in studies on molecular mechanisms of rapid growth of bamboo species. J. Agric. Biotechnol. 2018, 26, 871–887. [Google Scholar]
- Zheng-ping, W.; Chris, S. Phyllostachys Siebold & Zuccarini, Abh. Math.-Phys. Cl. Königl. Bayer. Akad. Wiss. 3: 745. 1843, nom. cons., not Torrey (1836), nom. rej. Flora China 2006, 22, 163–180. [Google Scholar]
- Panee, J. Potential medicinal application and toxicity evaluation of extracts from bamboo plants. J. Med. Plant Res. 2015, 9, 681–692. [Google Scholar]
- Friar, E.; Kochert, G. Bamboo germplasm screening with nuclear restriction fragment length polymorphisms. Theor. Appl. Genet. 1991, 82, 697–703. [Google Scholar] [CrossRef]
- Friar, E.; Kochert, G. A study of genetic variation and evolution of Phyllostachys (Bambusoideae: Poaceae) using nuclear restriction fragment length polymorphisms. Theor. Appl. Genet. 1994, 89, 265–270. [Google Scholar] [CrossRef]
- Gielis, J.; Everaert, I.; De Loose, M. Genetic variability and relationships in Phyllostachys using random amplified polymorphic DNA. In The Bamboos; Chapman, G.P., Ed.; Linnean Society Symposium Series Academic: London, UK, 1997; Volume 19, pp. 107–124. [Google Scholar]
- Zhang, S.; Ma, Q.; Ding, Y. RAPD analysis for the genetic diversity of Phyllostachys edulis China forestry. Sci. Technol. 2007, 21. [Google Scholar]
- Hodkinson, T.R.; Renvoize, S.A.; Chonghaile, G.N.; Stapleton, C.M.; Chase, M.W. A comparison of ITS nuclear rDNA sequence data and AFLP markers for phylogenetic studies in Phyllostachys (Bambusoideae, Poaceae). J. Plant Res. 2000, 113, 259–269. [Google Scholar] [CrossRef]
- Suyama, Y.; Obayashi, K.; Hayashi, I. Clonal structure in a dwarf bamboo (Sasa senanensis) population inferred from amplified fragment length polymorphism (AFLP) fingerprints. Mol. Ecol. 2000, 9, 901–906. [Google Scholar] [CrossRef] [PubMed]
- Isagi, Y.; Shimada, K.; Kushima, H.; Tanaka, N.; Nagao, A.; Ishikawa, T.; Onodera, H.; Watanabe, S. Clonal structure and flowering traits of a bamboo [Phyllostachys pubescens (Mazel) Ohwi] stand grown from a simultaneous flowering as revealed by AFLP analysis. Mol. Ecol. 2004, 13, 2017–2021. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Lou, Y.; Zhang, Y.; Yuan, X.; He, J.; Fang, W. Identification of genetic diversity among cultivars of Phyllostachys violascens using ISSR, SRAP and AFLP markers. Bot. Rev. 2011, 77, 223–232. [Google Scholar] [CrossRef]
- Isagi, Y.; Oda, T.; Fukushima, K.; Lian, C.; Yokogawa, M.; Kaneko, S. Predominance of a single clone of the most widely distributed bamboo species Phyllostachys edulis in East Asia. J. Plant Res. 2016, 129, 21–27. [Google Scholar] [CrossRef]
- Lai, C.; Hsiao, J. Genetic variation of Phyllostachys pubescens (Bambusoideae, Poaceae) in Taiwan based on DNA polymorphisms. Bot. Bull. Acad. Sin. 1997, 38, 145–152. [Google Scholar]
- Tang, D.-Q.; Lu, J.-J.; Fang, W.; Zhang, S.; Zhou, M.-B. Development, characterization and utilization of GenBank microsatellite markers in Phyllostachys pubescens and related species. Mol. Breed. 2010, 25, 299–311. [Google Scholar] [CrossRef]
- Peng, Z.; Lu, T.; Li, L.; Liu, X.; Gao, Z.; Hu, T.; Yang, X.; Feng, Q.; Guan, J.; Weng, Q. Genome-wide characterization of the biggest grass, bamboo, based on 10,608 putative full-length cDNA sequences. BMC Plant Biol. 2010, 10, 116. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Guan, Y.; Yang, L.; Yu, L.; Luo, S. Analysis of SSRs information and development of SSR markers from Moso bamboo (Phyllostachys edulis) ESTs. Acta Hortic. Sin. 2011, 38, 989–996. [Google Scholar]
- Lin, X.C.; Ruan, X.S.; Lou, Y.F.; Guo, X.Q.; Fang, W. Genetic similarity among cultivars of Phyllostachys pubescens. Plant Syst. Evol. 2009, 277, 67–73. [Google Scholar] [CrossRef]
- Zhou, M.-B.; Wu, J.-J.; Ramakrishnan, M.; Meng, X.-W.; Vinod, K.K. Prospects for the study of genetic variation among Moso bamboo wild-type and variants through genome resequencing. Trees 2018, 33, 371–381. [Google Scholar] [CrossRef]
- Chen, C.; Wang, W.; Wang, X.; Shen, D.; Wang, S.; Wang, Y.; Gao, B.; Wimmers, K.; Mao, J.; Li, K.; et al. Retrotransposons evolution and impact on lncRNA and protein coding genes in pigs. Mob. DNA 2019, 10, 19. [Google Scholar] [CrossRef] [PubMed]
- Li, S.-F.; Guo, Y.-J.; Li, J.-R.; Zhang, D.-X.; Wang, B.-X.; Li, N.; Deng, C.-L.; Gao, W.-J. The landscape of transposable elements and satellite DNAs in the genome of a dioecious plant spinach (Spinacia oleracea L.). Mob. DNA 2019, 10, 3. [Google Scholar] [CrossRef] [PubMed]
- McCue, A.D.; Slotkin, R.K. Transposable element small RNAs as regulators of gene expression. Trends Genet. 2012, 28, 616–623. [Google Scholar] [CrossRef]
- Gao, D.; Abernathy, B.; Rohksar, D.; Schmutz, J.; Jackson, S.A. Annotation and sequence diversity of transposable elements in common bean (Phaseolus vulgaris). Front. Plant Sci. 2014, 5, 339. [Google Scholar] [CrossRef] [Green Version]
- Gao, D.; Jiang, N.; Wing, R.A.; Jiang, J.; Jackson, S.A. Transposons play an important role in the evolution and diversification of centromeres among closely related species. Front. Plant Sci. 2015, 6, 216. [Google Scholar] [CrossRef] [Green Version]
- Bourque, G.; Burns, K.H.; Gehring, M.; Gorbunova, V.; Seluanov, A.; Hammell, M.; Imbeault, M.; Izsvák, Z.; Levin, H.L.; Macfarlan, T.S.; et al. Ten things you should know about transposable elements. Genome Biol. 2018, 19, 199. [Google Scholar] [CrossRef]
- Drongitis, D.; Aniello, F.; Fucci, L.; Donizetti, A. Roles of transposable elements in the different layers of gene expression regulation. IJMS 2019, 20, 5755. [Google Scholar] [CrossRef] [Green Version]
- Orozco-Arias, S.; Isaza, G.; Guyot, R. Retrotransposons in plant genomes: Structure, identification, and classification through bioinformatics and machine learning. IJMS 2019, 20, 3837. [Google Scholar] [CrossRef] [Green Version]
- Feschotte, C.; Pritham, E.J. DNA transposons and the evolution of eukaryotic genomes. Annu. Rev. Genet. 2007, 41, 331–368. [Google Scholar] [CrossRef] [Green Version]
- Finnegan, D.J. Eukaryotic transposable elements and genome evolution. Trends Genet. 1989, 5, 103–107. [Google Scholar] [CrossRef]
- Goodier, J.L. Restricting retrotransposons: A review. Mob. DNA 2016, 7, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wicker, T.; Gundlach, H.; Spannagl, M.; Uauy, C.; Borrill, P.; Ramirez-Gonzalez, R.H.; De Oliveira, R.; Mayer, K.F.X.; Paux, E.; Choulet, F. Impact of transposable elements on genome structure and evolution in bread wheat. Genome Biol. 2018, 19, 103. [Google Scholar] [CrossRef] [PubMed]
- Bennetzen, J.L.; Sanmiguel, P. Evidence that a recent increase in maize genome size was caused by the massive amplification of intergene retrotransposons. Ann. Bot. 1998, 82, 37–44. [Google Scholar] [CrossRef]
- Schnable, P.S.; Ware, D.; Fulton, R.S.; Stein, J.C.; Wei, F.; Pasternak, S.; Liang, C.; Zhang, J.; Fulton, L.; Graves, T.A.; et al. The B73 maize genome: Complexity, diversity, and dynamics. Science 2009, 326, 1112–1115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, M.-B.; Lu, J.-J.; Zhong, H.; Tang, K.-X.; Tang, D.-Q. Distribution and polymorphism of mariner-like elements in the Bambusoideae subfamily. Plant Syst. Evol. 2010, 289, 1–11. [Google Scholar] [CrossRef]
- Peng, Z.; Lu, Y.; Li, L.; Zhao, Q.; Feng, Q.; Gao, Z.; Lu, H.; Hu, T.; Yao, N.; Liu, K.; et al. The draft genome of the fast-growing non-timber forest species moso bamboo (Phyllostachys heterocycla). Nat. Genet. 2013, 45, 456–461. [Google Scholar] [CrossRef] [Green Version]
- Zhou, M.; Hu, B.; Zhu, Y. Genome-wide characterization and evolution analysis of long terminal repeat retroelements in moso bamboo (Phyllostachys edulis). Tree Genet. Genomes 2017, 13, 43. [Google Scholar] [CrossRef]
- Zhao, H.; Gao, Z.; Wang, L.; Wang, J.; Wang, S.; Fei, B.; Chen, C.; Shi, C.; Liu, X.; Zhang, H.; et al. Chromosome-level reference genome and alternative splicing atlas of moso bamboo (Phyllostachys edulis). GigaScience 2018, 7, giy115. [Google Scholar] [CrossRef]
- Bennetzen, J.L. Transposable element contributions to plant gene and genome evolution. Plant Mol. Biol. 2000, 42, 251–269. [Google Scholar] [CrossRef]
- Llorens, C.; Munoz-Pomer, A.; Bernad, L.; Botella, H.; Moya, A. Network dynamics of eukaryotic LTR retroelements beyond phylogenetic trees. Biol. Direct 2009, 4, 41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neumann, P.; Novák, P.; Hoštáková, N.; Macas, J. Systematic survey of plant LTR-retrotransposons elucidates phylogenetic relationships of their polyprotein domains and provides a reference for element classification. Mob. DNA 2019, 10, 1. [Google Scholar] [CrossRef] [PubMed]
- Bennetzen, J.L.; Wang, H. The contributions of transposable elements to the structure, function, and evolution of plant genomes. Annu. Rev. Plant Biol. 2014, 65, 505–530. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.; Benoit, M.; Catoni, M.; Drost, H.-G.; Brestovitsky, A.; Oosterbeek, M.; Paszkowski, J. Sensitive detection of pre-integration intermediates of long terminal repeat retrotransposons in crop plants. Nat. Plants 2019, 5, 26–33. [Google Scholar] [CrossRef]
- Griffiths, J.; Catoni, M.; Iwasaki, M.; Paszkowski, J. Sequence-independent identification of active LTR retrotransposons in Arabidopsis. Mol. Plant 2018, 11, 508–511. [Google Scholar] [CrossRef] [Green Version]
- Zhou, M.; Zhu, Y.; Bai, Y.; Hänninen, H.; Meng, X. Transcriptionally active LTR retroelement-related sequences and their relationship with small RNA in moso bamboo (Phyllostachys edulis). Mol. Breed. 2017, 37, 132. [Google Scholar] [CrossRef]
- Kaeppler, S.M.; Kaeppler, H.F.; Rhee, Y. Epigenetic aspects of somaclonal variation in plants. Plant Mol. Biol. 2000, 43, 179–188. [Google Scholar] [CrossRef]
- Miyao, A.; Nakagome, M.; Ohnuma, T.; Yamagata, H.; Kanamori, H.; Katayose, Y.; Takahashi, A.; Matsumoto, T.; Hirochika, H. Molecular spectrum of somaclonal variation in regenerated rice revealed by whole-genome sequencing. Plant Cell Physiol. 2012, 53, 256–264. [Google Scholar] [CrossRef] [Green Version]
- Paszkowski, J. Controlled activation of retrotransposition for plant breeding. Curr. Opin. Biotechnol. 2015, 32, 200–206. [Google Scholar] [CrossRef]
- Kalendar, R.; Grob, T.; Regina, M.; Suoniemi, A.; Schulman, A. IRAP and REMAP: Two new retrotransposon-based DNA fingerprinting techniques. Theor. Appl. Genet. 1999, 98, 704–711. [Google Scholar] [CrossRef]
- Kalendar, R.; Schulman, A.H. IRAP and REMAP for retrotransposon-based genotyping and fingerprinting. Nat. Protoc. 2007, 1, 2478. [Google Scholar] [CrossRef] [PubMed]
- Schulman, A.H.; Flavell, A.J.; Paux, E.; Ellis, T.H. The application of LTR retrotransposons as molecular markers in plants. Methods Mol. Biol. 2012, 859, 115–153. [Google Scholar] [CrossRef] [PubMed]
- Kalendar, R.; Amenov, A.; Daniyarov, A. Use of retrotransposon-derived genetic markers to analyse genomic variability in plants. Funct. Plant Biol. 2019, 46, 15–29. [Google Scholar] [CrossRef] [PubMed]
- Rahmani, M.-S.; Pijut, P.M.; Shabanian, N.; Nasri, M. Genetic fidelity assessment of in vitro-regenerated plants of Albizia julibrissin using SCoT and IRAP fingerprinting. Vitr. Cell. Dev. Biol. Plant 2015, 51, 407–419. [Google Scholar] [CrossRef]
- Vuorinen, L.A.; Kalendar, R.; Fahima, T.; Korpelainen, H.; Nevo, E.; Schulman, H.A. Retrotransposon-based genetic diversity assessment in wild emmer wheat (Triticum turgidum ssp. dicoccoides). Agronomy 2018, 8, 107. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.; Yin, H.; Li, L.; Song, Y.; Fan, L.; Zhang, S.; Wu, J. Evaluation of new IRAP markers of pear and their potential application in differentiating bud sports and other Rosaceae species. Tree Genet. Genomes 2015, 11, 25. [Google Scholar] [CrossRef]
- Branco, C.J.; Vieira, E.A.; Malone, G.; Kopp, M.M.; Malone, E.; Bernardes, A.; Mistura, C.C.; Carvalho, F.I.; Oliveira, C.A. IRAP and REMAP assessments of genetic similarity in rice. J. Appl. Genet. 2007, 48, 107–113. [Google Scholar] [CrossRef]
- Vukich, M.; Schulman, A.H.; Giordani, T.; Natali, L.; Kalendar, R.; Cavallini, A. Genetic variability in sunflower (Helianthus annuus L.) and in the Helianthus genus as assessed by retrotransposon-based molecular markers. Theor. Appl. Genet. 2009, 119, 1027–1038. [Google Scholar] [CrossRef]
- Fan, F.; Cui, B.; Zhang, T.; Ding, G.; Wen, X. LTR-retrotransposon activation, IRAP marker development and its potential in genetic diversity assessment of masson pine (Pinus massoniana). Tree Genet. Genomes 2014, 10, 213–222. [Google Scholar] [CrossRef]
- Lee, S.-I.; Kim, J.-H.; Park, K.-C.; Kim, N.-S. LTR-retrotransposons and inter-retrotransposon amplified polymorphism (IRAP) analysis in Lilium species. Genetica 2015, 143, 343–352. [Google Scholar] [CrossRef]
- Guo, Y.; Zhai, L.; Long, H.; Chen, N.; Gao, C.; Ding, Z.; Jin, B. Genetic diversity of Bletilla striata assessed by SCoT and IRAP markers. Hereditas 2018, 155, 35. [Google Scholar] [CrossRef] [PubMed]
- Hosid, E.; Brodsky, L.; Kalendar, R.; Raskina, O.; Belyayev, A. Diversity of LTR retrotransposon genome distribution in natural populations of the wild diploid wheat Aegilops speltoides. Genetics 2012, 190, 263–274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doyle, J.J.; Doyle, J.L. Isolation of plant DNA from fresh tissue. Focus 1990, 12, 39–40. [Google Scholar]
- Steinbiss, S.; Willhoeft, U.; Gremme, G.; Kurtz, S. Fine-grained annotation and classification of de novo predicted LTR retrotransposons. Nucleic Acids Res. 2009, 37, 7002–7013. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [Green Version]
- Saitou, N.; Nei, M. The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 1987, 4, 406–425. [Google Scholar] [CrossRef]
- De Riek, J.; Calsyn, E.; Everaert, I.; Van Bockstaele, E.; De Loose, M. AFLP based alternatives for the assessment of Distinctness, Uniformity and Stability of sugar beet varieties. Theor. Appl. Genet. 2001, 103, 1254–1265. [Google Scholar] [CrossRef]
- Jaccard, P. Nouvelles researches sur la distribution florale. Bull. Soc. Vaud. Sci. Natl. 1908, 223–270. [Google Scholar]
- Pavlicek, A.; Hrda, S.; Flegr, J. Free-Tree–freeware program for construction of phylogenetic trees on the basis of distance data and bootstrap/jackknife analysis of the tree robustness. Application in the RAPD analysis of genus Frenkelia. Folia. Biol. 1999, 45, 97–99. [Google Scholar]
- Page, R.D. TreeView: An application to display phylogenetic trees on personal computers. Comput. Appl. Biosci. 1996, 12, 357–358. [Google Scholar] [PubMed] [Green Version]
- Hammer, Ø.; Harper, D.; Ryan, P. PAST: Paleontological statistics software package for education and data analysis. Palaeontol. Electron. 2001, 4, 9. [Google Scholar]
- Legendre, P.; Legendre, L. Numerical Ecology, 2nd English edition; Elsevier: Amsterdam, The Netherlands, 1998. [Google Scholar]
- Jolliffe, I.T. Principal Component Analysis, 2nd ed.; Springer: New York, NY, USA, 2002. [Google Scholar]
- McGarigal, K.; Cushman, S.A.; Stafford, S. Multivariate Statistics for Wildlife and Ecology Research; Springer: New York, NY, USA, 2000. [Google Scholar]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar] [PubMed]
- Fraley, C.; Raftery, A.E. Model-based clustering, discriminant analysis, and density estimation. J. Am. Stat. Assoc. 2002, 97, 611–631. [Google Scholar] [CrossRef]
- Karandikar, R. On the markov chain monte carlo (MCMC) method. Sadhana 2006, 31, 81–84. [Google Scholar] [CrossRef] [Green Version]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software STRUCTURE: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peakall, R.; Smouse, P.E. GenAlEx 6.5: Genetic analysis in Excel. Population genetic software for teaching and research—An update. Bioinformatics 2012, 6, 2537–2589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalendar, R.; Flavell, A.J.; Ellis, T.H.N.; Sjakste, T.; Moisy, C.; Schulman, A.H. Analysis of plant diversity with retrotransposon-based molecular markers. Heredity 2010, 106, 520. [Google Scholar] [CrossRef] [Green Version]
- Kalendar, R.; Schulman, A.H. Transposon based tagging: IRAP, REMAP, and iPBS. Methods Mol. Biol. 2014, 1115, 233–255. [Google Scholar] [CrossRef] [Green Version]
- Zhao, H.; Peng, Z.; Fei, B.; Li, L.; Hu, T.; Gao, Z.; Jiang, Z. BambooGDB: A bamboo genome database with functional annotation and an analysis platform. Database 2014, 2014, bau006. [Google Scholar] [CrossRef] [Green Version]
- Paux, E.; Faure, S.; Choulet, F.; Roger, D.; Gauthier, V.; Martinant, J.P.; Sourdille, P.; Balfourier, F.; Le Paslier, M.C.; Chauveau, A.; et al. Insertion site-based polymorphism markers open new perspectives for genome saturation and marker-assisted selection in wheat. Plant Biotechnol. J. 2010, 8, 196–210. [Google Scholar] [CrossRef] [PubMed]
- Galindo-Gonzalez, L.; Mhiri, C.; Deyholos, M.K.; Grandbastien, M.A. LTR-retrotransposons in plants: Engines of evolution. Gene 2017, 626, 14–25. [Google Scholar] [CrossRef] [PubMed]
- Belyayev, A.; Kalendar, R.; Brodsky, L.; Nevo, E.; Schulman, A.H.; Raskina, O. Transposable elements in a marginal plant population: Temporal fluctuations provide new insights into genome evolution of wild diploid wheat. Mob. DNA 2010, 1, 6. [Google Scholar] [CrossRef] [Green Version]
- Simmonds, N.W. Monocarpy, calendars and flowering cycles in angiosperms. Kew Bulletin 1980, 35, 235–245. [Google Scholar] [CrossRef]
- Zhao, H.; Yang, L.; Peng, Z.; Sun, H.; Yue, X.; Lou, Y.; Dong, L.; Wang, L.; Gao, Z. Developing genome-wide microsatellite markers of bamboo and their applications on molecular marker assisted taxonomy for accessions in the genus Phyllostachys. Sci. Rep. 2015, 5, 8018. [Google Scholar] [CrossRef]
- Szućko, I.; Rogalska, S.M. Application of ISSR-PCR, IRAP-PCR, REMAP-PCR, and ITAP-PCR in the assessment of genomic changes in the early generation of triticale. Biol. Plant 2015, 59, 708–714. [Google Scholar] [CrossRef]
- Cheraghi, A.; Rahmani, F.; Hassanzadeh-Ghorttapeh, A. IRAP and REMAP based genetic diversity among varieties of Lallemantia iberica. Mol. Biol. Res. Commun. 2018, 7, 125–132. [Google Scholar] [CrossRef]
- Li, J.; Zhao, G.H.; Li, X.Y.; Chen, F.; Chen, J.B.; Zou, F.C.; Yang, J.F.; Lin, R.Q.; Weng, Y.B.; Zhu, X.Q. IRAP: An efficient retrotransposon-based electrophoretic technique for studying genetic variability among geographical isolates of Schistosoma japonicum. Electrophoresis 2011, 32, 1473–1479. [Google Scholar] [CrossRef]
- Li, J. Flora of China. Harv. Pap. Bot. 2007, 13, 301–303. [Google Scholar] [CrossRef]
- Khaleghi, E.; Sorkheh, K.; Chaleshtori, M.H.; Ercisli, S. Elucidate genetic diversity and population structure of Olea europaea L. germplasm in Iran using AFLP and IRAP molecular markers. 3 Biotech 2017, 7, 71. [Google Scholar] [CrossRef] [Green Version]
- Jiang, W.; Bai, T.; Dai, H.; Wei, Q.; Zhang, W.; Ding, Y. Microsatellite markers revealed moderate genetic diversity and population differentiation of moso bamboo (Phyllostachys edulis)—A primarily asexual reproduction species in China. Tree Genet. Genomes 2017, 13, 130. [Google Scholar] [CrossRef]
- Nachimuthu, V.V.; Muthurajan, R.; Duraialaguraja, S.; Sivakami, R.; Pandian, B.A.; Ponniah, G.; Gunasekaran, K.; Swaminathan, M.; Suji, K.K.; Sabariappan, R. Analysis of population structure and genetic diversity in rice germplasm using SSR markers: An initiative towards association mapping of agronomic traits in Oryza Sativa. Rice 2015, 8, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nilkanta, H.; Amom, T.; Tikendra, L.; Rahaman, H.; Nongdam, P. ISSR marker based population genetic study of Melocanna baccifera (Roxb.) Kurz: A commercially important bamboo of Manipur, North-East India. Scientifica 2017, 2017, 9. [Google Scholar] [CrossRef] [Green Version]
- Ma, Q.-Q.; Song, H.-X.; Zhou, S.-Q.; Yang, W.-Q.; Li, D.-S.; Chen, J.-S. Genetic structure in dwarf bamboo (Bashania fangiana) clonal populations with different genet ages. PLoS ONE 2013, 8, e78784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, J.; Zhang, Y.; Zhang, C.; Qi, F.; Li, X.; Mu, S.; Peng, Z. Characterization of the floral transcriptome of Moso bamboo (Phyllostachys edulis) at different flowering developmental stages by transcriptome sequencing and RNA-seq analysis. PLoS ONE 2014, 9, e98910. [Google Scholar] [CrossRef] [PubMed] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| S. No. | Phyllostachys Accessions | Source Location |
|---|---|---|
| 1. | Ph. aurea Carr. | Zhejiang, China |
| 2. | Ph. acutiligula G. H. Lai | Anhui, China |
| 3. | Ph. varioauriculata | Anhui, China |
| 4. | Ph. heteroclada | Sichuang, China |
| 5. | Ph. hispida | Zhejiang, China |
| 6. | Ph. hirtivagina | Anhui, China |
| 7. | Ph. edulis (Carrière) J. Houz | Jiangsu, China |
| 8. | Ph. edulis cv. Obliquinoda | Zhejiang, China |
| 9. | Ph. edulis cv. Pachyloen | Jiangxi, China |
| 10. | Ph. edulis cf. huamaoyhw (Wen.) Wen | Hunan, China |
| 11. | Ph. edulis cv. Viridisulcata | Zhejiang, China |
| 12. | Ph. edulis (Carr.) Matsumura | Zhejiang, China |
| 13. | Ph. edulis Mitford cv. | Zhejiang, China |
| 14. | Ph. edulis (Carr.) Mitford cv. Gracilis | Jiangsu, China |
| 15. | Ph. edulis cv. Tubaeformis S.Y. Wang | Hunan, China |
| 16. | Ph. incarnata T.H. Wen, Bull. Bot. Res | Zhejiang, China |
| 17. | Ph. nidularia | Italy |
| 18. | Ph. sulphurea (Carr)A. et C. Riv | Anhui, China |
| 19. | Ph. sulphurea var. viridis | Henan, China |
| 20. | Ph. mannii | Taiwan, China |
| 21. | Ph. virella | Zhejiang, China |
| 22. | Ph. yunhoensxian | Zhejiang, China |
| 23. | Ph. rubromarginata | Guangdong, China |
| 24. | Ph. elegans McClure | America |
| 25. | Ph. fimbriligula | Zhejiang, China |
| 26. | Ph. viridiglaucescens | Zhejiang, China |
| 27. | Ph. robustiramea | Zhejiang, China |
| 28. | Ph. flexuosa | Anhui, China |
| 29. | Ph. glauca | America |
| 30. | Ph. zhejiangensis | Zhejiang, China |
| 31. | Ph. angusta | Anhui, China |
| 32. | Ph. meyeri | Henan, China |
| 33. | Ph. vivax | Anhui, China |
| 34. | Ph. propinqua | Anhui, China |
| 35. | Ph. iridescens | Zhejiang, China |
| 36. | Ph. purpureociliata | Anhui, China |
| 37. | Ph. nuda | Jiangsu, China |
| 38. | Ph. primotina | Zhejiang, China |
| 39. | Ph. arcana | Jiangsu, China |
| 40. | Ph. verrucosa | Hunan, China |
| 41. | Ph. bissetii | Sichuang, China |
| 42. | Ph. aureosulcata | Zhejiang, China |
| 43. | Ph. longiciliata | Anhui, China |
| 44. | Ph. rutila | Zhejiang, China |
| 45. | Ph. bambusoides | Henan, China |
| 46. | Ph. bambusoides Sieb. et Zucc | Anhui, China |
| 47. | Ph. nigella | Zhejiang, China |
| 48. | Ph. dulcis | Zhejiang, China |
| 49. | Ph. aurita | Henan, China |
| 50. | Ph. funhuanensis | Zhejiang, China |
| 51. | Ph. platyglossa | Zhejiang, China |
| 52. | Ph. rubicunda | Zhejiang, China |
| 53. | Ph. atrovaginata | Zhejiang, China |
| 54. | Ph. parvifolia | Zhejiang, China |
| 55. | Ph. corrugata | Anhui, China |
| 56. | Ph. nigra | Hunan, China |
| 57. | Ph. nigra var. henonis | Jiangsu, China |
| 58. | Ph. violascens | Zhejiang, China |
| IRAP Marker | Primer Sequence (5′-3′) | IRAP Primer Location in LTR Retrotransposons (bp) | Allele Size Range (kb) | No. of Polymorphic Alleles | Total Number of Amplicons in the Population | Average Allele Frequency | PIC Value | Melting Temperature (Tm) °C |
|---|---|---|---|---|---|---|---|---|
| CL3-F | TATAAAGGTAGCTTTCGGGTATG | 428338_436487 | 0.65–2.7 | 13 | 231 | 0.306 | 0.350 | 52 |
| CL4-R | CTGGTATATAGCTGTTGAGCGACG | 82500_84292 | 0.6–2.4 | 13 | 192 | 0.255 | 0.351 | 57 |
| CL15-R | CTCGTGTATTCTCCCTTTGC | 221059_224205 | 0.43–2.5 | 12 | 227 | 0.326 | 0.400 | 53 |
| CL22-F | TGATCAGAGAAGAAAGGGGA | 49377_57704 | 0.42–2.7 | 13 | 125 | 0.166 | 0.256 | 52 |
| CL22-R | CACGCAGAGAGATTGACACG | 49377_57704 | 0.35–2.7 | 15 | 173 | 0.198 | 0.319 | 56 |
| CL34-F | GAACGATTACCTCACAGACA | 4137_7249 | 0.45–2.7 | 15 | 183 | 0.210 | 0.262 | 52 |
| CL34-R | GAGCAATAAAGAGAAGCCCG | 4137_7249 | 0.4–2.2 | 16 | 273 | 0.294 | 0.334 | 53 |
| CL37-F | AGATTGTTTGATTCGGGGGG | 153045_162321 | 0.3–2.5 | 12 | 183 | 0.263 | 0.325 | 54 |
| CL37-R | AGCGGCGTGGAGGAGTTACC | 153045_162321 | 0.5–2.7 | 13 | 234 | 0.310 | 0.376 | 61 |
| CL42-R | GCAACAACAAACCCTAAAAA | 18144_27070 | 0.3–2.7 | 14 | 230 | 0.283 | 0.369 | 50 |
| CL54-R | GCAAGAACATAAGAACAGAA | 56871_64575 | 0.3–2.0 | 8 | 209 | 0.450 | 0.336 | 55 |
| CL58-F | AAGGAATCGTCAGTCAACAA | 28670_35571 | 0.3–2.5 | 11 | 202 | 0.317 | 0.369 | 52 |
| CL59-F | TGTCAGACAGTACAGGTGCT | 1169_4536 | 0.2–2.7 | 14 | 204 | 0.251 | 0.329 | 55 |
| CL61-F | ATGACATAGGGCACACCAGA | 680968_684115 | 0.37–2.0 | 14 | 260 | 0.320 | 0.275 | 55 |
| CL62-F | TAAATAGGGAACGAGGAGCC | 178936_183841 | 0.5–2.7 | 15 | 190 | 0.218 | 0.306 | 56 |
| CL63-R | ACATTGTTTGATTCGGGGGG | 248490_260632 | 0.45–2.7 | 16 | 166 | 0.179 | 0.273 | 55 |
| Mean | - | - | - | 13.37 | 208.75 | 0.272 | 0.327 | - |
| Sub-Population Name | Phyllostachys Species Name | Membership No. | Average Similarity |
|---|---|---|---|
| PhSP1 | Ph. acutiligula G. H. Lai; Ph. heteroclada; Ph. hirtivagina; Ph. varioauriculata; Ph. hispida; Ph. rubromarginata; Ph. aurea Carr.; Ph. sulphurea (Carr)A. et C. Riv; Ph. virella; Ph. mannii; Ph. yunhoensxian. | 11 | 0.28 |
| PhSP2 | Ph. bissetii; Ph. aureosulcata; Ph. propinqua; Ph. verrucosa; Ph. vivax; Ph. nuda; Ph. arcana; Ph. meyeri; Ph. primotina; Ph. glauca; Ph. iridescens; Ph. angusta; Ph. sulphurea var. viridis; Ph. rutila; Ph. purpureociliata; Ph. zhejiangensis; Ph. fimbriligula; Ph. elegans McClure; Ph. viridiglaucescens. | 19 | 0.33 |
| PhSP3 | Ph. atrovaginata; Ph. parvifolia; Ph. rubicunda; Ph. funhuanensis; Ph. longiciliata; Ph. aurita; Ph. platyglossa; Ph. corrugata; Ph. dulcis; Ph. violascens; Ph. bambusoides; Ph. nigella; Ph. edulis (Carr.) Mitford cv. Gracilis; Ph. edulis cv. Tubaeformis S.Y. Wang; Ph. nigra; Ph. nigra var. henonis. | 16 | 0.29 |
| PhSP4 | Ph. edulis cf. huamaoyhw (Wen.) Wen; Ph. edulis cv. Pachyloen; Ph. bambusoides Sieb. et Zucc; Ph. edulis (Carrière) J. Houz; Ph. edulis cv. Viridisulcata; Ph. edulis Mitford cv.; Ph. edulis cv. Obliquinoda; Ph. edulis (Carr.) Matsumura. | 8 | 0.65 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, S.; Ramakrishnan, M.; Vinod, K.K.; Kalendar, R.; Yrjälä, K.; Zhou, M. Development and Deployment of High-Throughput Retrotransposon-Based Markers Reveal Genetic Diversity and Population Structure of Asian Bamboo. Forests 2020, 11, 31. https://doi.org/10.3390/f11010031
Li S, Ramakrishnan M, Vinod KK, Kalendar R, Yrjälä K, Zhou M. Development and Deployment of High-Throughput Retrotransposon-Based Markers Reveal Genetic Diversity and Population Structure of Asian Bamboo. Forests. 2020; 11(1):31. https://doi.org/10.3390/f11010031
Chicago/Turabian StyleLi, Shitian, Muthusamy Ramakrishnan, Kunnummal Kurungara Vinod, Ruslan Kalendar, Kim Yrjälä, and Mingbing Zhou. 2020. "Development and Deployment of High-Throughput Retrotransposon-Based Markers Reveal Genetic Diversity and Population Structure of Asian Bamboo" Forests 11, no. 1: 31. https://doi.org/10.3390/f11010031
APA StyleLi, S., Ramakrishnan, M., Vinod, K. K., Kalendar, R., Yrjälä, K., & Zhou, M. (2020). Development and Deployment of High-Throughput Retrotransposon-Based Markers Reveal Genetic Diversity and Population Structure of Asian Bamboo. Forests, 11(1), 31. https://doi.org/10.3390/f11010031