Microbial Taxa and Soil Organic Carbon Accumulation Driven by Tree Roots


Abstract
:1. Introduction
2. Materials and Methods
2.1. Experimental Setup and Sampling
2.2. Molecular and Bioinformatics Analyses
2.3. Calculations and Statistical Analyses
3. Results
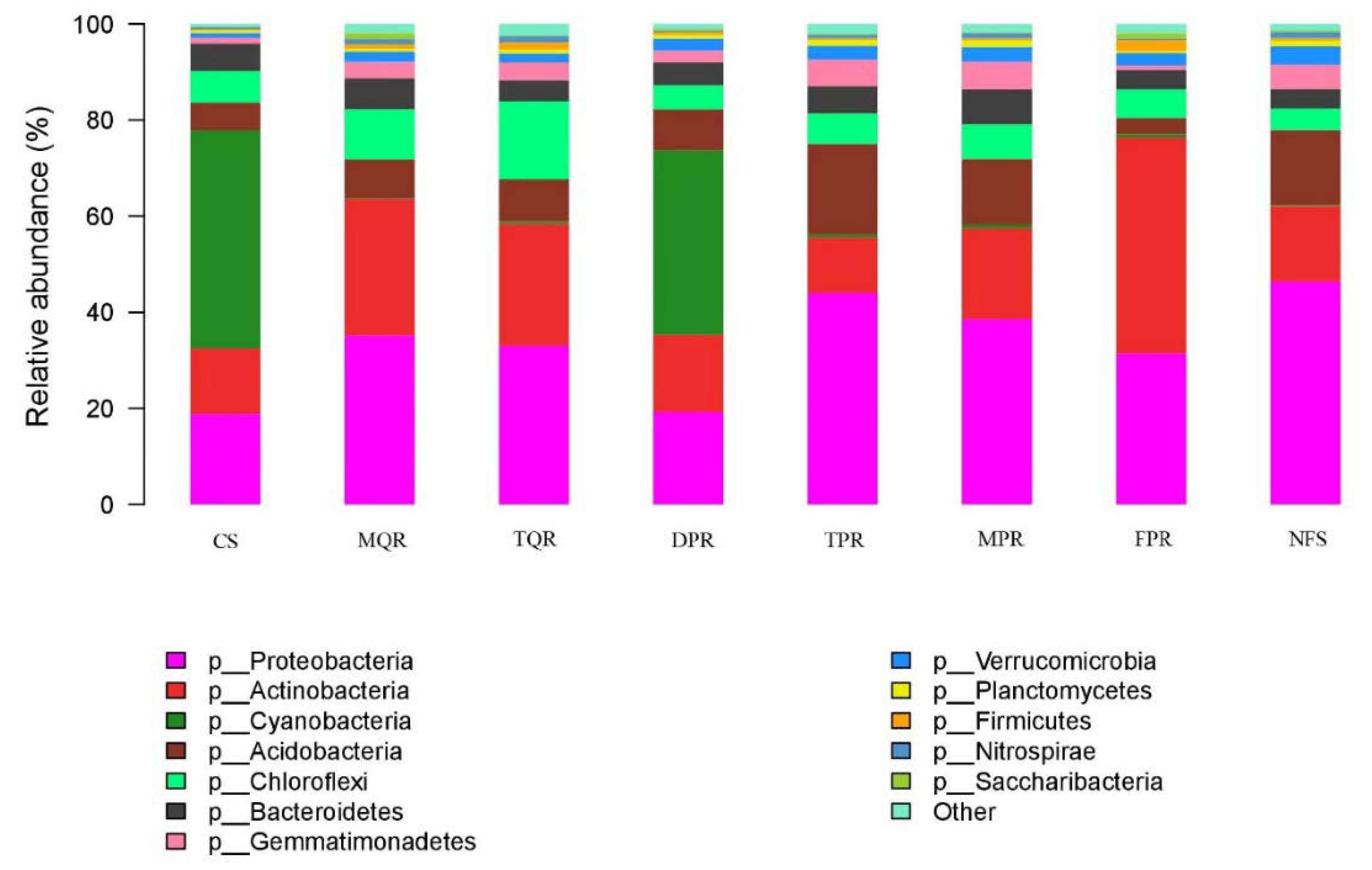
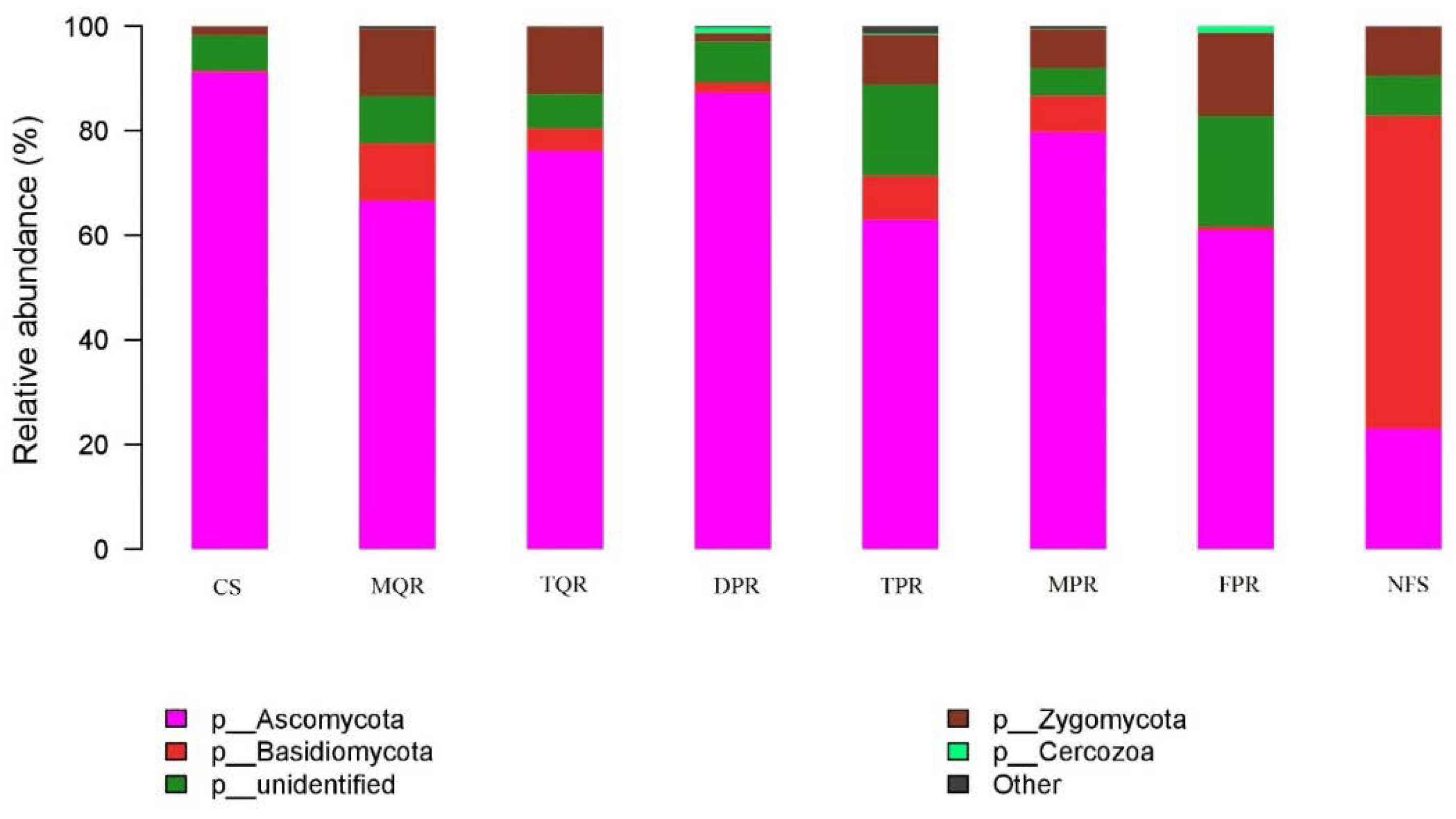
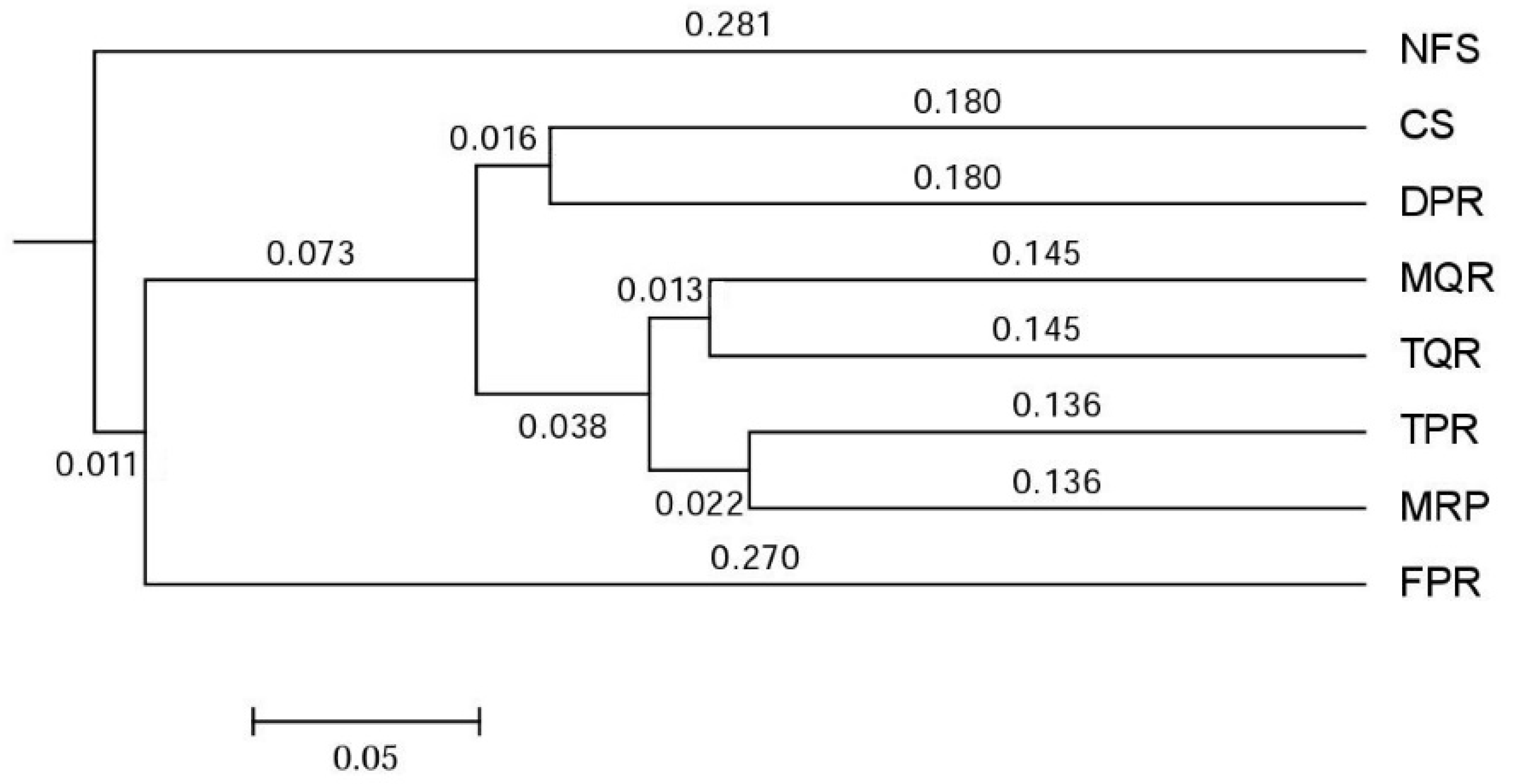
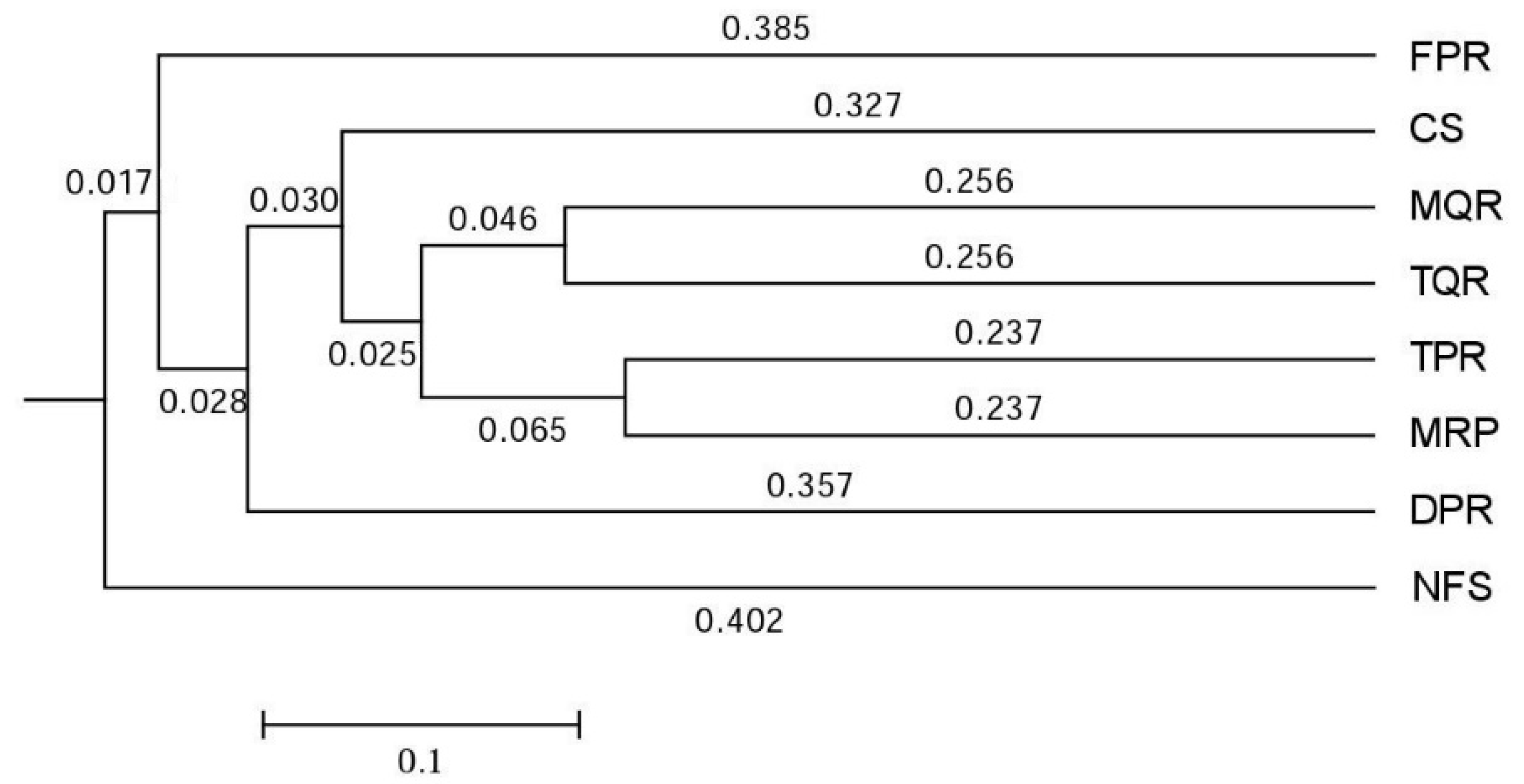
3.1. Changes in Soil Bacterial and Fungal Community Compositions
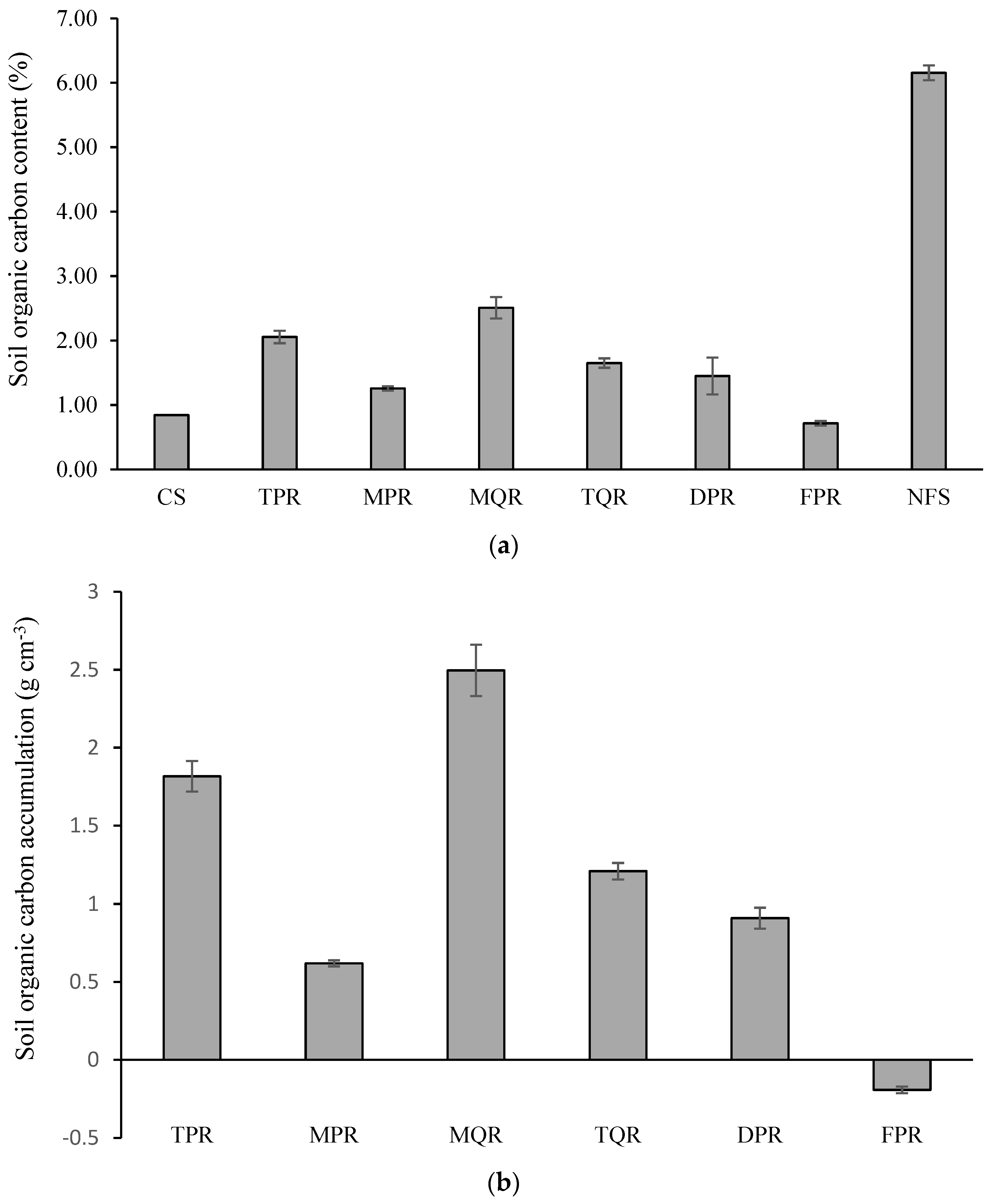
3.2. Changes in SOC Accumulation and Microbial Taxa
4. Discussion
4.1. Changes in Soil Bacterial and Fungal Communities
4.2. Influence on SOC Accumulation and Microbial Taxa
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Pausch, J.; Kuzyakov, Y. Carbon input by roots into the soil: Quantification of rhizodeposition from root to ecosystem scale. Glob. Chang. Biol. 2017, 24, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bond-Lamberty, B.; Thomson, A. Temperature-associated increases in the global soil respiration record. Nature 2010, 464, 579–584. [Google Scholar] [CrossRef] [PubMed]
- Fahey, T.J.; Woodbury, P.B.; Battles, J.J.; Goodale, C.L.; Hamburg, S.P.; Ollinger, S.V.; Woodall, C.W. Forest carbon storage: Ecology, management, and policy. Front. Ecol. Environ. 2010, 8, 245–252. [Google Scholar] [CrossRef]
- Treseder, K.K.; Holden, S.R. Fungal carbon sequestration. Science 2013, 339, 1528–1529. [Google Scholar] [CrossRef] [PubMed]
- Cheng, W.X.; Parton, W.J.; Gonzalez-Meler, M.A.; Phillips, R.; Asao, S.; Mc Nickle, G.G.; Brzostek, E.; Jastrow, J.D. Synthesis and modeling perspectives of rhizosphere priming. New Phytol. 2014, 201, 31–44. [Google Scholar] [CrossRef] [PubMed]
- Kuzyakov, Y. Priming effects: Interactions between living and dead organic matter. Soil Biol. Biochem. 2010, 42, 1363–1371. [Google Scholar] [CrossRef]
- Johnson, D.; Leake, J.R.; Read, D.J. Transfer of recent photosynthate into mycorrhizal mycelium of an upland grassland: Short-term respiratory losses and accumulation of 14C. Soil Biol. Biochem. 2002, 34, 1521–1524. [Google Scholar] [CrossRef]
- Rillig, M.C.; Mummey, D.L. Mycorrhizas and soil structure. New Phytol. 2006, 171, 41–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clemmensen, K.E.; Bahr, A.; Ovaskainen, O.; Dahlberg, A.; Ekblad, A.; Wallander, H.; Stenlid, J.; Finlay, R.D.; Wardle, D.A.; Lindahl, B.D. Roots and associated fungi drive long-term carbon sequestration in boreal forest. Science 2013, 339, 1615–1618. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.D.; Birdsey, R.A.; Fang, J.Y.; Houghton, R.; Kauppi, P.E.; Kurz, W.A.; Phillips, O.L.; Shvidenko, A.; Lewis, S.L.; Canadell, J.G. A large and persistent carbon sink in the world’s forests. Science 2011, 333, 988–993. [Google Scholar] [CrossRef] [PubMed]
- Deng, Q.; Cheng, X.X.; Hui, D.F.; Zhang, Q.; Li, M.; Zhang, Q.F. Soil microbial community and its interaction with soil carbon and nitrogen dynamics following afforestation in central China. Sci. Total Environ. 2016, 541, 230–237. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Liu, G.B.; Xue, S.; Wang, G. Soil bacterial community dynamics reflect changes in plant community and soil properties during the secondary succession of abandoned farmland in the Loess Plateau. Soil Biol. Biochem. 2016, 97, 40–49. [Google Scholar] [CrossRef]
- Song, W.C.; Tong, X.J.; Zhang, J.S.; Meng, P. Three-source partitioning of soil respiration by 13C natural abundance and its variation with soil depth in a plantation. J. For. Res. 2016, 27, 533–540. [Google Scholar] [CrossRef]
- Mooshammer, M.; Wanek, W.; Zechmeister-Boltenstern, S.; Richter, A. Stoichiometric imbalances between terrestrial decomposer communities and their resources: Mechanisms and implications of microbial adaptations to their resources. Front. Microbiol. 2014, 5, 22. [Google Scholar] [CrossRef] [PubMed]
- Song, W.C.; Tong, X.J.; Zhang, J.S.; Meng, P.; Li, J. Autotrophic and heterotrophic components of soil respiration caused by rhizosphere priming effects in a plantation. Plant Soil Environ. 2017, 63, 295–299. [Google Scholar]
- Khlifa, R.; Paquette, A.; Messier, C.; Reich, P.B.; Munson, A.D. Do temperate tree species diversity and identity influence soil microbial community function and composition? Ecol. Evol. 2017, 7, 7965–7974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, W.C.; Liu, Y.H.; Tong, X.J. Newly sequestrated soil organic carbon varies with soil depth and tree species in three forest plantations from northeastern China. For. Ecol. Manag. 2017, 400, 384–395. [Google Scholar] [CrossRef]
- Zhao, F.Z.; Kang, D.; Han, X.H.; Yang, G.H.; Feng, Y.Z.; Ren, G.X. Soil stoichiometry and carbon storage in long-term afforestation soil affected by understory vegetation diversity. Ecol. Eng. 2015, 74, 415–422. [Google Scholar] [CrossRef]
- Ren, C.J.; Zhao, F.Z.; Kang, D.; Yang, G.H.; Han, X.H.; Tong, X.G.; Feng, Y.Z.; Ren, G.X. Linkages of C:N:P stoichiometry and bacterial community in soil following afforestation of former farmland. For. Ecol. Manag. 2016, 376, 59–66. [Google Scholar] [CrossRef]
- Ren, C.; Chen, J.; Deng, J.; Zhao, F.; Han, X.; Yang, G.; Tong, X.; Feng, Y.; Shelton, S.; Ren, G. Response of microbial diversity to c:n:p stoichiometry in fine root and microbial biomass following afforestation. Biol. Fertil. Soils 2017, 53, 457–468. [Google Scholar] [CrossRef]
- Hannula, S.E.; Morriën, E.; Hollander, M.D.; Putten, W.H.V.D.; Veen, J.A.V.; Boer, W.D. Shifts in rhizosphere fungal community during secondary succession following abandonment from agriculture. ISME J. 2017, 11, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Laganière, J.; Boča, A.; Miegroet, H.V.; Paré, D. A tree species effect on soil that is consistent across the species’ range: The case of aspen and soil carbon in North America. Forests 2017, 8, 113. [Google Scholar] [CrossRef]
- Van der Heijden, M.G.; Bardgett, R.D.; van Straalen, N.M. The unseen majority: Soil microbes as drivers of plant diversity and productivity in terrestrial ecosystems. Ecol. Lett. 2008, 11, 296–310. [Google Scholar] [CrossRef] [PubMed]
- Kolaříková, Z.; Kohout, P.; Krüger, C.; Janoušková, M.; Mrnka, L.; Rydlová, J. Root-associated fungal communities along a primary succession on a mine spoil: Distinct ecological guilds assemble differently. Soil Biol. Biochem. 2017, 113, 143–152. [Google Scholar] [CrossRef]
- Porras-Alfaro, A.; Bayman, P. Hidden fungi, emergent properties: Endophytes and microbiomes. Annu. Rev. Phytopathol. 2011, 49, 291–315. [Google Scholar] [CrossRef] [PubMed]
- Jia, G.M.; Cao, J.; Wang, C.; Wang, G. Microbial biomass and nutrients in soil at the different stages of secondary forest succession in Ziwulin, northwest China. For. Ecol. Manag. 2005, 217, 117–125. [Google Scholar] [CrossRef]
- Noormets, A.; Epron, D.; Domec, J.C.; McNulty, S.G.; Fox, T.; Sun, G.; King, J.S. Effects of forest management on productivity and carbon sequestration: A review and hypothesis. For. Ecol. Manag. 2015, 355, 124–140. [Google Scholar] [CrossRef] [Green Version]
- Xiang, Y.; Cheng, M.; Huang, Y.; An, S.; Darboux, F. Changes in soil microbial community and its effect on carbon sequestration following afforestation on the Loess Plateau, China. Int. J. Environ. Res. Public Health 2017, 14, 948. [Google Scholar] [CrossRef] [PubMed]
- Zhao, F.Z.; Han, X.H.; Yang, G.H.; Feng, Y.Z.; Ren, G.X. Soil structure and carbon distribution in subsoil affectedby vegetation restoration. Plant Soil Environ. 2014, 60, 21–26. [Google Scholar]
- Liu, Z.; Yu, X.; Jia, G.; Jia, J.; Lou, Y.; Lu, W. Contrasting water sources of evergreen and deciduous tree species in rocky mountain area of Beijing, China. Catena 2017, 150, 108–115. [Google Scholar] [CrossRef]
- Taş, N.; Prestat, E.; McFarland, J.W.; Wickland, K.P.; Knight, R.; Berhe, A.A.; Jansson, J.K. Impact of fire on active layer and permafrost microbial communities and metagenomes in an upland Alaskan boreal forest. ISME J. 2014, 8, 1904–1919. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.T.; Peng, X.; Deng, G.H.; Sheng, H.F.; Wang, Y.; Zhou, H.W.; Tam, N.F. Illumina sequencing of 16s RNA tag revealed spatial variations of bacterial communities in a mangrove wetland. Microb. Ecol. 2013, 66, 96–104. [Google Scholar] [CrossRef] [PubMed]
- Jiang, D.M.; Cao, C.Y.; Zhang, Y.; Cui, Z.B.; Han, X.S. Plantations of native shrub species restore soil microbial diversity in the horqin sandy land, northeastern China. J. Arid Land 2014, 6, 445–453. [Google Scholar] [CrossRef]
- Singh, J.S.; Kumar, A.; Rai, A.N.; Singh, D.P. Cyanobacteria: A precious bio-resource in agriculture, ecosystem, and environmental sustainability. Front. Microbiol. 2016, 7, 529. [Google Scholar] [CrossRef] [PubMed]
- Prendergast-Miller, M.T.; Menezes, A.B.D.; Macdonald, L.M.; Toscas, P.; Bissett, A.; Baker, G.; Farrell, M.; Richardson, A.E.; Wark, T.; Thrall, P.H. Wildfire impact: Natural experiment reveals differential short-term changes in soil microbial communities. Soil Biol. Biochem. 2017, 109, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Chang, E.H.; Tian, G.; Chiu, C.Y. The effect of re-planting trees on soil microbial communities in a wildfire-induced subalpine grassland. Forests 2017, 8, 385. [Google Scholar] [CrossRef]
- Soong, J.L.; Dam, M.; Wall, D.H.; Cotrufo, M.F. Below-ground biological responses to pyrogenic organic matter and litter inputs in grasslands. Funct. Ecol. 2017, 31, 260–269. [Google Scholar] [CrossRef]
- López-Ballesteros, A.; Oyonarte, C.; Kowalski, A.S.; Serrano-Ortiz, P.; Sánchez-Cañete, E.P.; Moya, M.R.; Domingo, F. Can land degradation drive differences in the C exchange of two similar semiarid ecosystems? Biogeosciences 2018, 15, 1–25. [Google Scholar] [CrossRef]
- Kuzyakov, Y.; Xu, X. Competition between roots and microorganisms for nitrogen: Mechanisms and ecological relevance. New Phytol. 2013, 198, 656–669. [Google Scholar] [CrossRef] [PubMed]
- Briones, A.M.; Okabe, S.; Umemiya, Y.; Ramsing, N.B.; Reichardt, W.; Okuyama, H. Influence of different cultivars on populations of ammonia-oxidizing bacteria in the root environment of rice. Appl. Environ. Microb. 2002, 68, 67–75. [Google Scholar] [CrossRef]
- Herath, L.; Seneviratne, G.; Jayasinghe, J.W.W.; Senanayake, D.N. Microbial biofilms and mitigation of loss of agro-biodiversity in degraded soils. J. Natl. Sci. Found. Sri Lanka 2017, 45, 329–335. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Prot | Acti | Cyan | Acid | Chlo | Bact | Gemm | Verr | Plan | Firm | Nitr | Sacc | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| TPR | + ** | n | − ** | + ** | n | n | + ** | + * | n | n | n | n |
| MPR | + ** | n | − ** | + * | n | n | + ** | + * | n | n | n | n |
| MQR | + ** | + ** | − ** | n | + * | n | + * | n | n | n | n | n |
| TQR | + ** | + ** | − ** | n | + ** | n | + * | n | n | + * | + * | n |
| DPR | n | n | n | n | n | n | + * | + * | n | n | n | n |
| FPR | + * | n | n | + * | n | n | n | n | n | − * | n | − * |
| Ascomycota | Basidiomycota | Zygomycota | Cercozoa | Other | |
|---|---|---|---|---|---|
| TPR | − ** | + ** | + ** | n | + ** |
| MPR | − ** | + ** | + ** | n | n |
| MQR | − ** | + ** | + ** | n | n |
| TQR | − ** | + ** | + ** | n | n |
| DPR | n | n | n | n | n |
| FPR | − ** | n | + ** | n | n |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Song, W.; Liu, Y. Microbial Taxa and Soil Organic Carbon Accumulation Driven by Tree Roots. Forests 2018, 9, 333. https://doi.org/10.3390/f9060333
Song W, Liu Y. Microbial Taxa and Soil Organic Carbon Accumulation Driven by Tree Roots. Forests. 2018; 9(6):333. https://doi.org/10.3390/f9060333
Chicago/Turabian StyleSong, Wenchen, and Yanhong Liu. 2018. "Microbial Taxa and Soil Organic Carbon Accumulation Driven by Tree Roots" Forests 9, no. 6: 333. https://doi.org/10.3390/f9060333
APA StyleSong, W., & Liu, Y. (2018). Microbial Taxa and Soil Organic Carbon Accumulation Driven by Tree Roots. Forests, 9(6), 333. https://doi.org/10.3390/f9060333